Introduction
Protein O-Glucosyltransferase 1 (POGLUT1), also
known as Rumi, MDSRP or hCLP46(1–4), was
initially identified in CD34+ cells of patients with
acute myeloid leukemia that had transformed from myelodysplastic
syndrome. POGLUT1 contains a highly conserved domain termed CAP10,
as well as an endoplasmic reticulum retention signal motif, KTEL,
at the C-terminus and a hydrophobic signal peptide at its
N-terminus (5,6). Previous studies have reported that
BT474 human breast cancer cell growth increases in response to
POGLUT1 overexpression due to POGLUT1-induced inhibition of
transforming growth factor β1 (TGF-β1)-mediated induction of INK4a
gene expression (7,8). TGF-β1 is a multifunctional cytokine
with a central role in the regulation of numerous biological
processes, including cell proliferation, differentiation and the
modulation of immune responses (9).
TGF-β1 induces its various effects through serine/threonine kinase
transmembrane receptors and induces signaling from receptors to the
nucleus mediated through the phosphorylation of cytoplasmic
effector molecules of the Smad protein family (10). Phosphorylated (p)-Smad2 and Smad3
form heteromeric complexes with Smad4, which are then translocated
to the nucleus where they function as transcription factors
(11–13). TGF-β1 signaling has been reported to
increase during the inhibition of cell cycle progression, through
activating cyclin-dependent kinase inhibitors (CDKIs) and
inactivating c-Myc (14–16). A number of studies have investigated
the TGF-β1 signaling blockade inhibiting parathyroid
hormone-related protein secretion in breast cancer cells and bone
metastases development, as well as the regulatory role of TGF-β1 in
gastric cancer cell proliferation and differentiation (17–19).
POGLUT1 may have an important role in cellular
self-renewal and the development of various normal and malignant
tumor cells. Thus, investigations into the mechanism, interacting
molecules and regulation of POGLUT1 in tumor cells are required,
particularly in breast cancer which affects numerous females
worldwide. This may lead to an enhanced understanding of human
breast cancer occurrence and development.
It has been demonstrated that POGLUT1 stimulates the
proliferation of U937 human lymphoma cells and inhibits the
TGF-β-induced inhibition of U937 cell growth, suggesting that
POGLUT1 may be a cytokine which promotes and sustains tumor cell
malignant transformation (5). TGF-β
activates proteins in the Smad family through a membrane receptor,
and activated Smad proteins translocate from the cytoplasm to the
nucleus, to enhance the expression of the p16 and p15 target genes
(20). In cell cycle regulation,
CDKIs, CDKs and cyclin D, the cell cycle protein, form a
dynamically balanced system (21–23).
POGLUT1 may either downregulate the transcription of the p16 and
p15 genes or accelerate the degradation of the p16 and p15 proteins
through activating the intracellular proteolytic system.
The present study aimed to investigate the mechanism
and signal through which POGLUT1 antagonizes TGF-β1-induced p16
gene expression. In order to investigate the role of POGLUT1 in
tumor cell proliferation, a recombinant, Myc-labeled retroviral
vector, babe-puro-POGLUT1-Myc, was constructed and transduced into
BT474 human breast ductal adenocarcinoma cells to induce exogenous
POGLUT1 overexpression. The present study analyzed whether the
POGLUT1 gene was capable of antagonizing the activity of TGF-β1, an
important inhibitory factor in cell proliferation, thus promoting
BT474 cell proliferation. The present study aimed to elucidate the
targets of the signaling pathway through which the POGLUT1 gene
regulates TGF-β1, Smad3 and p16 activity.
Materials and methods
Cell culture and transfection
All of the cells were purchased from American Type
Culture Collection (Manassas, VA, USA). BT474 cells were grown in
RPMI-1640 (Invitrogen Life Technologies, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (FBS; Invitrogen Life
Technologies) and 293T cells were cultured in Dulbecco’s modified
Eagle’s medium (Invitrogen Life Technologies) with 10% FBS. Cells
were maintained at 37°C in humidified conditions containing 5%
CO2. Plasmid transfection was performed using
Lipofectamine® 2000 (Invitrogen Life Technologies)
according to the manufacturer’s instructions.
Recombinant retrovirus generation and
infection procedure
In order to transfer the POGLUT1 gene into the BT474
cells, a retroviral vector expressing POGLUT1 was constructed. The
plasmid vector pcDNA4/POGLUT1-myc contained a POGLUT1-myc-tagged
fusion protein for immunodetection. This construct was used to
generate a gene-transfer retroviral vector. The POGLUT1-myc
cassette amplified by polymerase chain reaction (PCR) and subcloned
into the pBabe-puro plasmid (Addgene, Cambridge, MA, USA) using
BamHI and SalI sites built into the primers. The
primer sequences were as follows: Forward,
5′-ATCCTCGAGCGTAGTTCAGTTTTCAA-3′ and reverse,
5′-ATCGTCGACCTACAGATCCTCTTCTGAGAT-3′. The recombinant retrovirus
vector pBabe-POGLUT1-Myc (pBaPM) was identified using
sequencing.
To generate a high-titer recombinant retrovirus,
pBaPM, pVSV and pHIT60 were contransfected into 293T cells using
Lipofectamine 2000. The virus stocks were collected 72 h after
transfection then filtered through a 0.45-μm filter and frozen at
below −70°C. BT474 human breast cancer cells were inoculated in
96-well culture plates and divided into four groups: pBaPM
retrovirus with TGF-β1 group; pBabe blank plasmid without TGF-β1;
pBabe blank plasmid with TGF-β1; and blank control group. BT474
cells were also infected with retrovirus stocks for 6 h, and washed
and cultured in fresh complete medium, with or without TGF-β1.
Cell proliferation assay
A colorimetric assay using MTT (Sigma-Aldrich, St.
Louis, MO, USA) was performed to assess cell growth and
proliferation. In brief, the BT474 cells were inoculated on 96-well
culture plates with 1×104 cells/well and 100 μl culture
medium per well. TGF-β1 (R&D Systems, Minneapolis, MN, USA) was
added to the cells at a final concentration of 100 pg/ml. Fresh
medium containing 10% MTT (5 mg/ml stock) was added to each well 72
h after infection. Plates were incubated at 37°C for 3 h then 300
μl dimethyl sulfoxide (Sigma-Aldrich) was added to each well and
shaken at room temperature for 10 min to dissolve the intracellular
MTT formazan crystals. Absorbance was then measured at 560 nm using
an enzyme microplate reader (SpectraMax® M5e; MDS Analytical
Technologies, Sunnyvale, CA, USA). Experiments were performed in
triplicate and repeated at least twice.
Western blot analysis
BT474 cells were infected with pBaPM and treated
with TGF-β1 (100 pmol/ml). After 48 h, the BT474 cells were
incubated in lysis buffer [150 mM NaCl, 1% NP40, 1 mM EDTA, 5 mM
benzamidine, 50 mM NaF and 20 mM Tris-HCl (pH 7.6)]. Cell
suspensions were vortexed briefly and the protein concentration was
determined using the DC Protein Assay (500–0112; Bio-Rad, Hercules,
CA, USA) according to the manufacturer’s instructions. Whole cell
lysates containing 50 μg total protein were boiled for 5 min in 1×
SDS buffer (Takara Bio, Inc., Shiga, Japan), resolved using 10%
SDS-PAGE and transferred to nitrocellulose membranes. The membranes
were blocked with TBST buffer [0.1 M Tris (pH 7.5), 0.9% NaCl and
0.05% Tween-20 containing 10% non-fat milk powder], then incubated
with the following primary monoclonal antibodies: Goat anti-human
p16, rabbit anti-human Smad3 and mouse anti-human p-Smad3 (Abcam
Cambridge, MA, USA). Membranes were then incubated with anti-goat
(rabbit or mouse) horseradish peroxidase-conjugated polyclonal
antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
Immunoreactive proteins were detected using an enhanced
chemiluminescence western blotting detection system (WesternBreeze®
Chromogenic Kits; Invitrogen Life Technologies). β-actin was used
as an endogenous control.
Fluorescence quantitative PCR (fqPCR)
analysis
BT474 cells were infected with pBaPM and treated
with TGF-β1 (100 pmol/ml). The cells were collected 48 h after
retrovirus infection. Total RNA was extracted using
TRIzol® reagent (Invitrogen Life Technologies) according
to the manufacturer’s instructions. Complementary (c)DNA was
synthesized using SuperScript® II reverse transcriptase
(Invitrogen Life Technologies). fqPCR analysis was performed using
SYBR® Green (Invitrogen Life Technologies) on an Applied
Biosystems 7900HT system (Applied Biosystems, Foster City, CA, USA)
to detect POGLUT1 gene transcription. GAPDH expression was used as
an endogenous control. The primer sequeneces were as follows:
Forward, 5′-GAT ATC ATG TAT CCT GCT TG-3′ and reverse, 5′-TTT TCC
ATG GCC ACT GTG GTC-3′ for POGLUT1; and forward, 5′-GGA AGG TGA AGG
TCG GAG TC-3′ and reverse, 5′-CGT TCT CAG CCT TGA CGG T-3′ for
GAPDH.
The cDNA from the infected BT474 cells was also used
to analyze p16 gene expression using fqPCR with TaqMan®
probes and the sequences were as follows: p16-F, 5′-CAT AGA TGC CGC
GGA AGG-3′; p16-R, 5′-AAG TTT CCC GAG GTT TCT CAG A-3′; and p16-T,
5′FAM-CCT CAG ACA TCC CCG ATT GAA AGA-3′TAMRA.
Statistical analysis
Statistical analysis was performed using SPSS,
version 16.0 (SPSS, Inc., Chicago, IL, USA). MTT assay data were
pooled and averaged. The statistical significance of the
differences between the control and target data sets was determined
using independent sample t-tests. P<0.05 was considered to
indicate a statistically significant difference.
Results
Cell proliferation assay
The BT474 cells were analyzed using an enzyme
microplate reader within 96 h of retrovirus infection, and the data
were analyzed to generate a cell growth curve. The BT474 cells that
were infected with a blank control plasmid with no TGF-β1
treatment, as well as those that were infected with the pBaPM
retrovirus and treated with TGF-β1, grew well. No significant
difference (P>0.05) in OD570 value was observed
between the two groups of cells. However, the BT474 cells that were
infected with the blank plasmid and treated with TGF-β1 exhibited a
lower OD570 value compared with those in the other two
groups, between 72 h and 96 h after infection. For example, after
72 h, the OD570 values of the cells in the pBaPM
retrovirus with TGF-β1 group and the blank plasmid without TGF-β1
group were 0.83 and 0.75, respectively, compared with 0.51 in the
cells in the blank plasmid with TGF-β1 group (P<0.01). Moreover,
after 96 h, the OD570 values of the cells in the pBaPM
retrovirus with TGF-β1 group and the blank plasmid without TGF-β1
group were 0.89 and 0.85, respectively, compared with 0.70 in the
cells in the blank plasmid with TGF-β1 group (P<0.01; Table I). These findings suggest that
TGF-β1 inhibits the proliferation of BT474 cells and that POGLUT1
overexpression promotes the growth of BT474 cells.
 | Table IBT474 cell growth following retrovirus
infection. |
Table I
BT474 cell growth following retrovirus
infection.
| OD570 |
|---|
|
|
|---|
| Group | 24 h | 48 h | 72 h | 96 h |
|---|
| pBaPM retrovirus with
TGF-β1 | 0.22±0.04 | 0.30±0.05 | 0.83±0.11a | 0.89±0.13a |
| Blank plasmid without
TGF-β1 | 0.19±0.03 | 0.29±0.07 | 0.75±0.11a | 0.85±0.15a |
| Blank plasmid with
TGF-β1 | 0.22±0.04 | 0.27±0.06 | 0.51±0.09 | 0.70±0.11 |
POGLUT1 expression in infected BT474
cells
BT474 cells were collected 48 h after retrovirus
infection and first-strand cDNA was synthesized using reverse
transcription. fqPCR analysis was used to amplify the POGLUT1 gene
using SYBR Green. The melting curve for POGLUT1 amplification
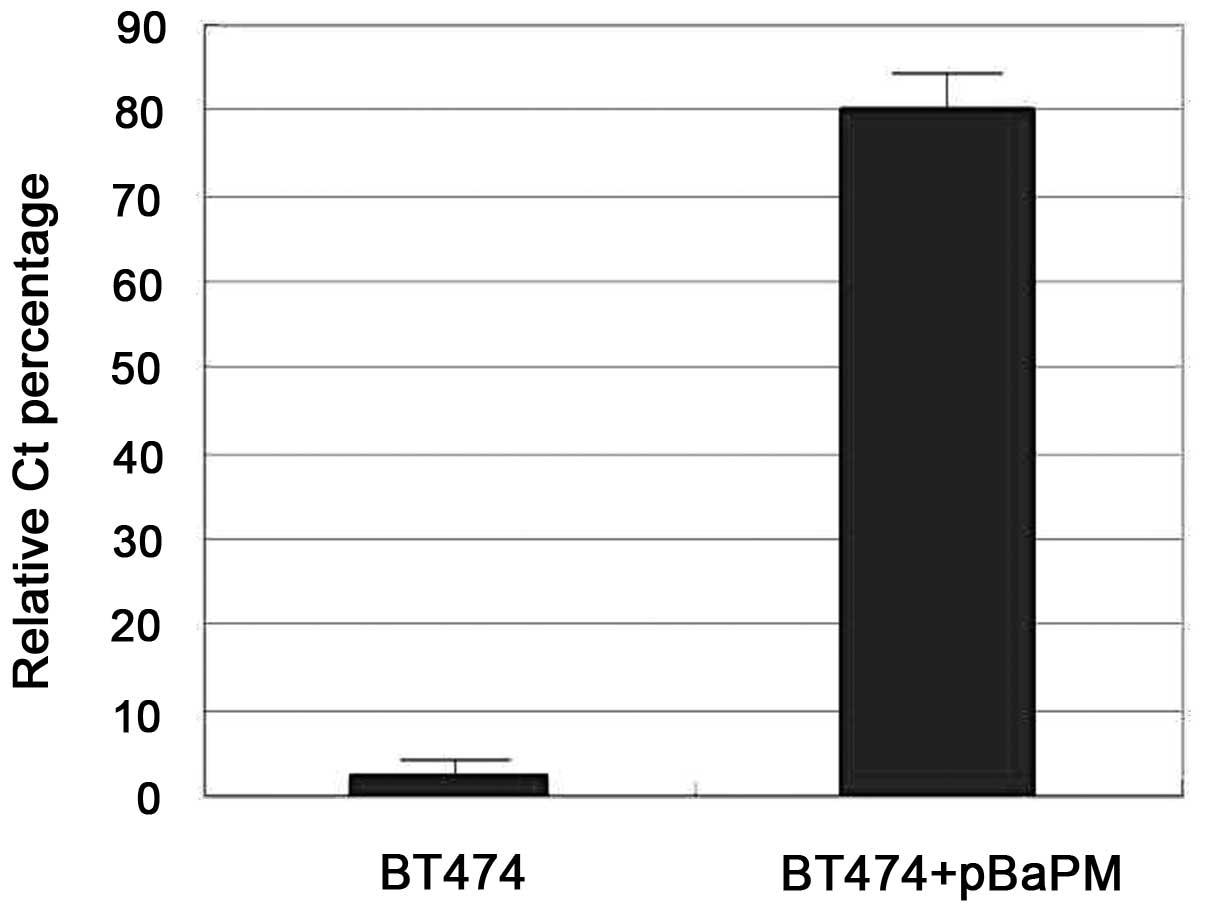
showed good specificity. fqPCR analysis revealed that the
expression of POGLUT1 in the BT474 cells infected with the pBaPM
retrovirus was increased compared with the control BT474 cells,
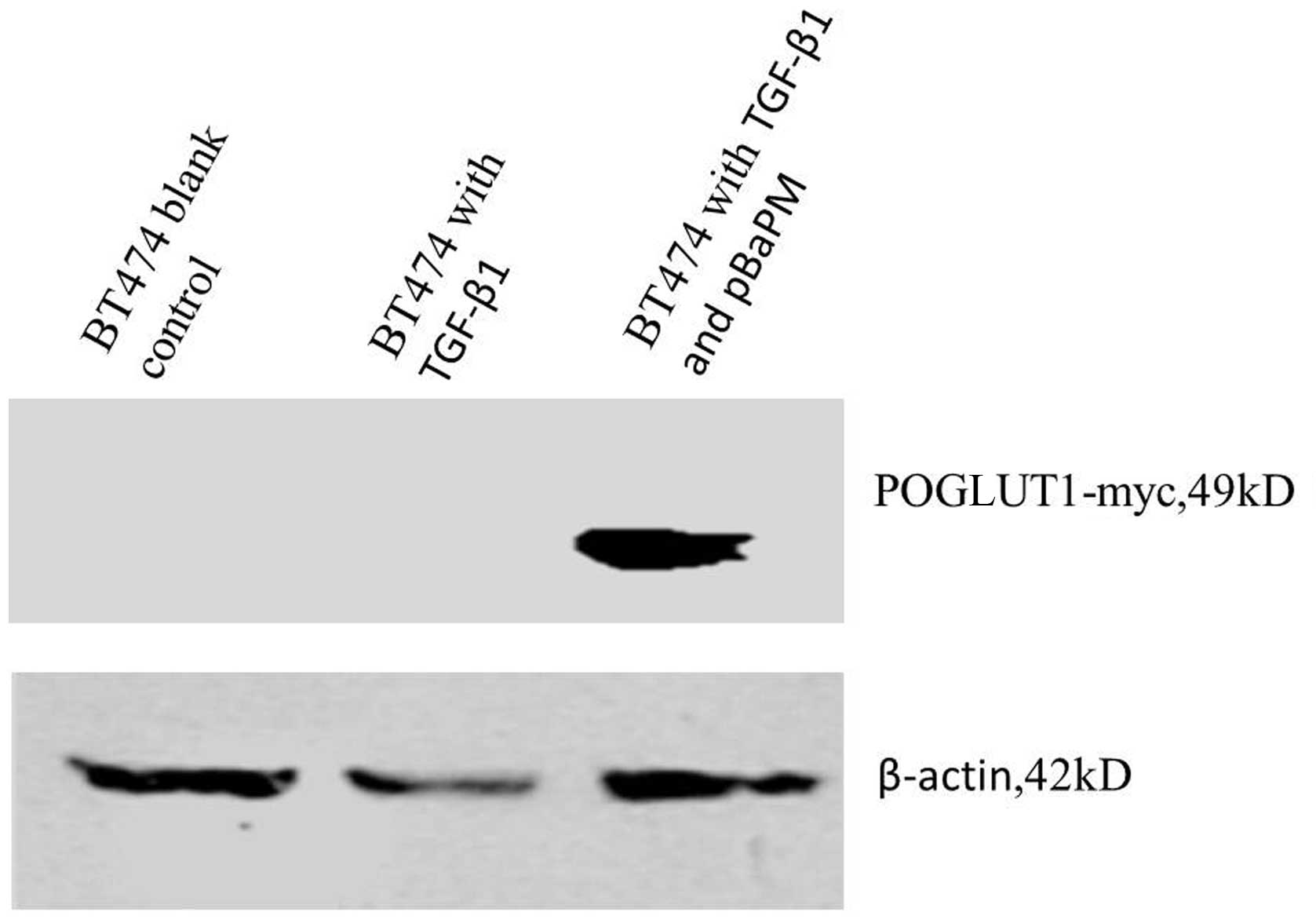
where little POGLUT1 expression was observed (Fig. 1). Furthermore, western blot analysis
revealed a specific band at ~49 kDa for the POGLUT1-myc protein,
while endogenous β-actin showed a band at ~42 kDa (Fig. 2).
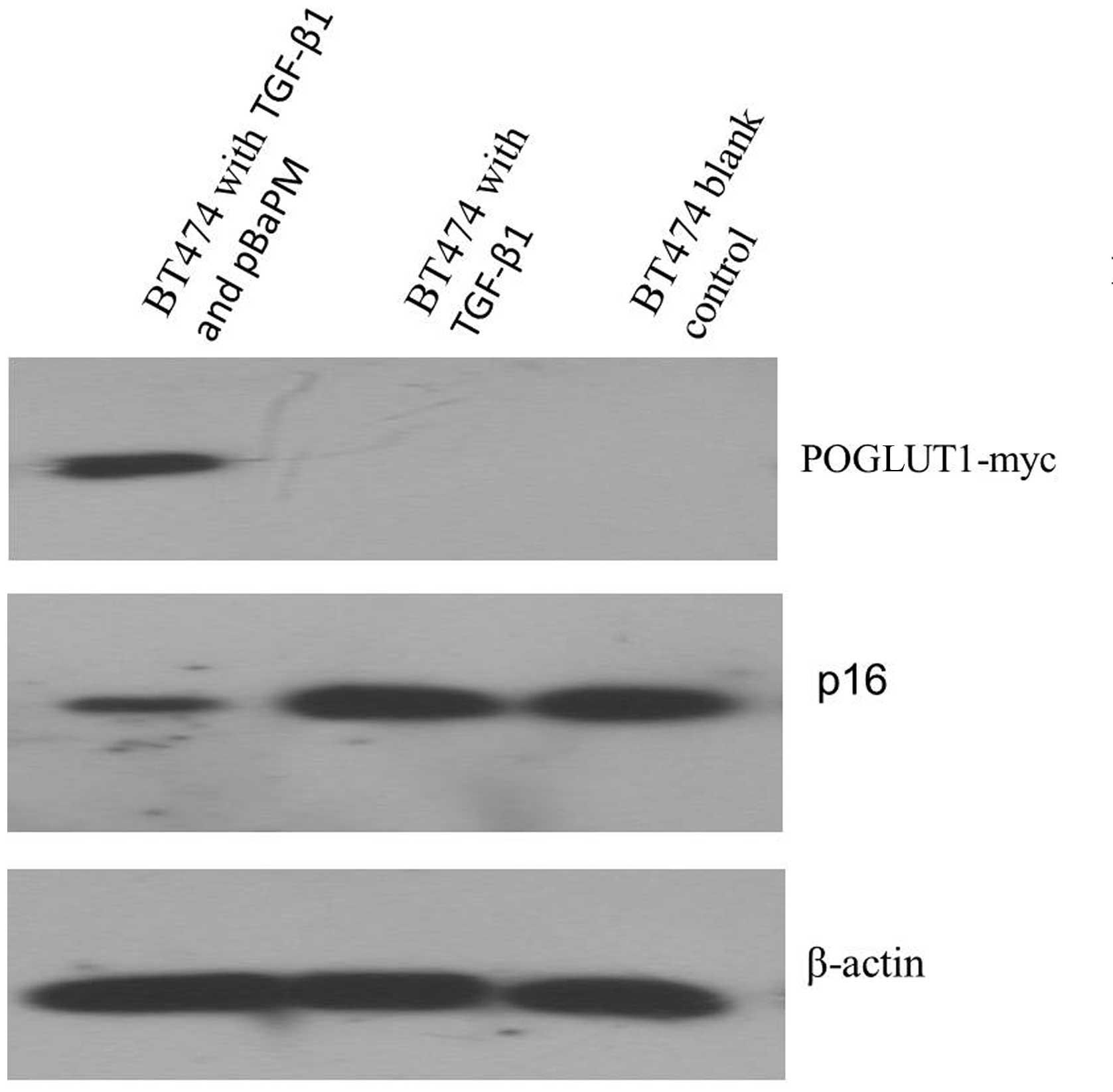
Detection of p16 expression in the
infected BT474 cells
p16 expression was found to be increased in the
presence of TGF-β1 compared with the untreated BT474 cells
(P<0.05). Following pBaPM retrovirus infection, exogenous
POGLUT1 was observed to be overexpressed and p16 expression was
found to be decreased in the absence of TGF-β1 to a lower degree
compared with the control cells (P<0.05). Moreover, in the
infected BT474 cells, p16 expression was found to be reduced to a
greater degree in the presence of TGF-β1 compared with the control
cells (P<0.01). Using the untreated BT474 cells as the baseline,
the relative quantity (RQ) value of p16 gene expression was found
to be ~76.13-fold lower in the POGLUT1+TGF-β1 BT474 cells
compared with the TGF-β1-stimulated BT474 cells (RQ=0.062 vs. 4.72;
P<0.01), while the RQ value of p16 expression was found to be
significantly decreased ~8.92-fold lower in the BT474 cells
overexpressing POGLUT1 compared with the TGF-β1-stimulated BT474
cells (RQ=0.53 vs. 4.72; P<0.01) (Table II).
| Table IIDetection of p16 expression using
TaqMan probes. |
Table II
Detection of p16 expression using
TaqMan probes.
| Groups | Ct-p16 | Ct-GAPDH | ΔCt | 2-ΔCt | ΔΔCt | RQ=2-ΔΔCt |
|---|
| POGLUT1 | 18.29±2.32 | 20.68±2.24 | −2.39 | 5.23a | 0.92 | 0.53a |
| POGLUT1+TGF-β1 | 25.80±3.07 | 25.10±2.98 | 0.70 | 0.61b | 4.02 | 0.062b |
| TGF-β1 | 17.52±2.54 | 23.07±2.61 | −5.55 | 46.83b | −2.24 | 4.72b |
| Control | 18.01±3.18 | 21.32±3.25 | −3.31 | 9.92 | 0.00 | 1.00 |
Effect of POGLUT1 overexpression on p16
protein expression
BT474 cells were infected with the pBaPM retrovirus,
cultured in the presence of TGF-β1 for 48 h and collected for the
analysis of p16 protein expression using western blot analysis. p16
protein expression was observed in the BT474 cells treated with
TGF-β1, as well as the 293T positive control cells. However, very
low p16 protein expression was detected in the
POGLUT1-overexpressing BT474 cells that were treated with TGF-β1
(Fig. 3).
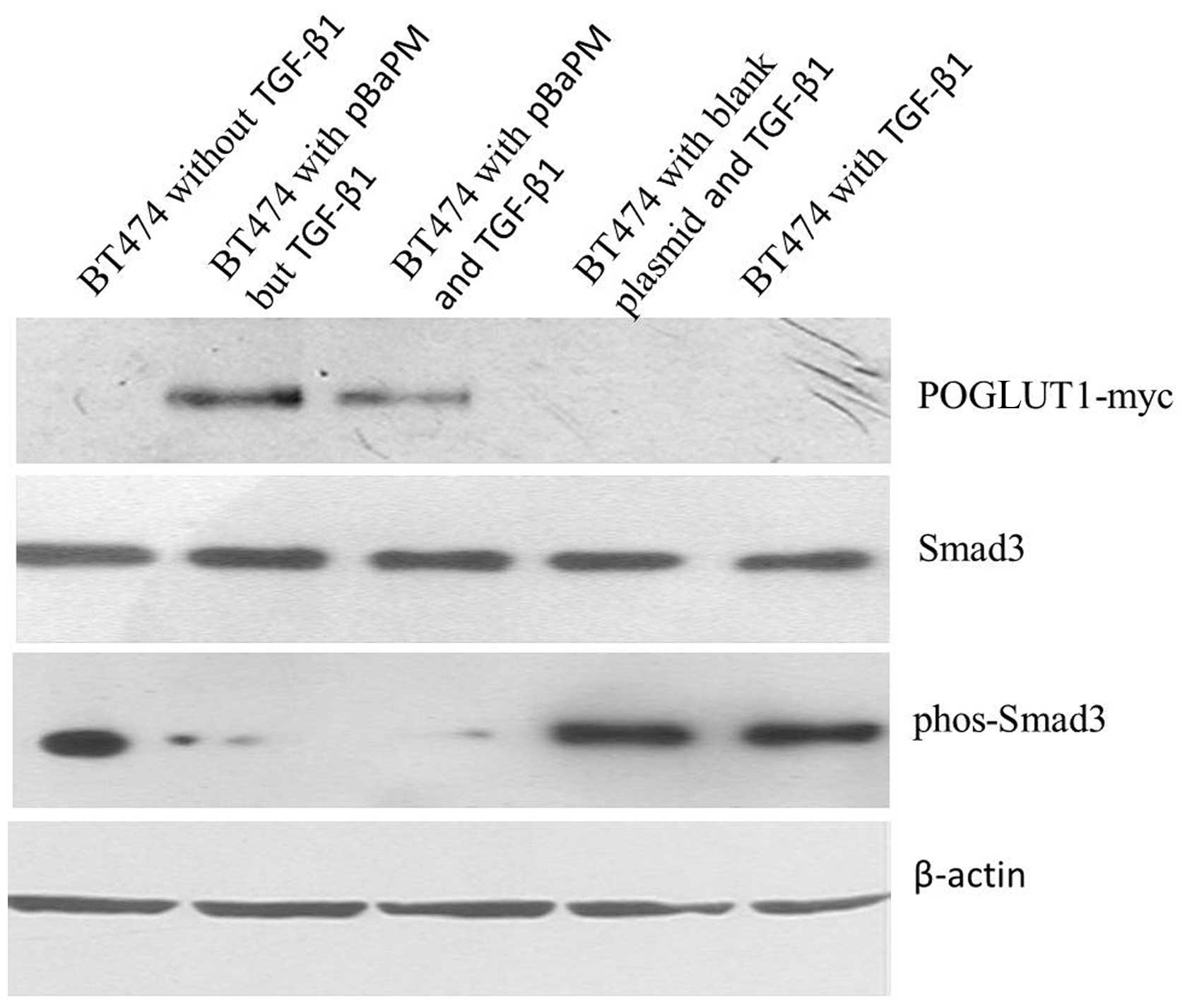
Effect of POGLUT1 overexpression on
p-Smad3 protein expression
The infected BT474 cells were cultured with TGF-β1
for 48 h, then collected for Smad3 and p-Smad3 protein detection
using western blot analysis. The overexpression of POGLUT1 was
found to inhibit the expression of the p-Smad3 protein in the BT474
cells. The Smad3 protein and the internal control β-actin were
observed to be expressed to the same extent in the POGLUT1
overexpression group, blank plasmid group and control group.
However, p-Smad3 expression was found to be markedly increased in
the two control groups and markedly decreased in the POGLUT1
overexpression group (Fig. 4).
Discussion
In the present study, pBabe-POGLUT1-myc(pBaPM)
retrovirus was recombined, which overexpresses exogenous POGLUT1 in
human breast cancer BT474 cells. In addition, the overexpression of
exogenous POGLUT1 may repress p16 expression and inhibit the
p-Smad3 protein expression in the presence of TGF-β1. First, the
recombinant retrovirus vector pBabe-puro-POGLUT1-Myc was
constructed in vitro, then transformed and packaged into an
integral virus, which was used to transfer the POGLUT1 gene into
BT474 human breast cancer cells in order to investigate the
function and mechanism of POGLUT1.
To identify the activity of the
pBabe-puro-POGLUT1-Myc recombinant retrovirus, it was transformed
into 293T human embryo kidney package cells. The live retrovirus
was then collected and cultured with the BT474 target cells for 48
h and POGLUT1 expression was detected. fqPCR analysis revealed high
POGLUT1 mRNA expression in the BT474 cells infected with the pBaPM
virus and low POGLUT1 expression in the control BT474 cells.
Furthermore, western blot analysis revealed high POGLUT1 protein
expression in the infected BT474 cells. These findings demonstrate
that a pBaPM retrovirus with biological activity was successfully
generated and could be used in the subsequent analyses.
In the present study, POGLUT1 overexpression was
found to promote BT474 cell proliferation. A previous study induced
the recombinant plasmid pcDNA3.1-POGLUT1 into BT474 cells with
persistent TGF-β1 in the supernatant and found that POGLUT1
overexpression promoted BT474 cell proliferation detected using MTT
assay (5). In the present study,
BT474 cells were infected with the pBaPM recombinant retrovirus and
a significant increase in cell proliferation was also observed
(P<0.01). These findings suggest that POGLUT1 overexpression
promotes BT474 cell proliferation.
TGF-βs are negative regulators of cell
proliferation. TGF-β receptor II (TβRII) is a constitutively active
protein kinase that is autophosphorylated upon TGF-β ligand
binding. Phosphorylated TβRII propagates the signal through
phosphorylating receptor-regulated Smad proteins, inducing their
accumulation in the nucleus where they participate in the
transcriptional regulation of target genes and anti-oncogenes in
order to inhibit cell proliferation (24–26).
In the present study, a cell growth curve was generated and showed
that BT474 cell growth increased in the absence of TGF-β1, compared
with in the presence of it. Furthermore, the growth curve showed
that the BT474 cells overexpressing POGLUT1 rapidly grew when
treated with TGF-β1 compared with the blank control cells. These
findings show that POGLUT1 overexpression promotes BT474 cell
proliferation.
During the regulation of the cell cycle, there are
two essential checkpoints at G1/S and G2/M
phase. The present study aimed to investigate which checkpoint is
targeted by POGLUT1 in order to promote BT474 cell proliferation.
Variations in the cell cycle were assessed in BT474 cells
overexpressing POGLUT1. In total, 81.39% of the BT474 cells were
found to stay in G0/G1 phase in the presence
of TGF-β1 and 14.57% were found to stay in S phase, which was lower
than the percentage in the control group (19.95%). However, the
percentage of POGLUT1-overexpressing BT474 cells in S phase was
found to be 25.80%, which was higher than that in the control group
(14.57%). No significant differences were observed in any of the
other cell cycle phases. These findings suggest that POGLUT1
functions primarily at the G1/S-phase of the cell
cycle.
In the present study, POGLUT1 overexpression was
found to inhibit the upregulation of the CDKI p16 by TGF-β1, as p16
expression was significantly reduced in the presence of TGF-β1 in
the infected BT474 cells compared with the control cells
(P<0.01). The molecular mechanism of cell cycle regulation
involves cell cycle proteins (cyclins), enzymes which are activated
by cyclins (CDKs) and CDK suppression proteins(CDKIs), which affect
the expression and regulation of CDKs. During different phases,
cyclins and their corresponding CDKs combine to form cyclin-CDK
complexes, which leads to the activation of CDKs (26–28). A
number of CDK suppression proteins compete with cyclins to bind to
CDKs or cyclin-CDK complexes, inhibiting CDK activity. The first
key step in the cell cycle is the initiation of G1
phase. Thus, much research has focused on the G1/S
phase. In the G1 phase, cyclin D and CDK4 combine to
activate CDK4, which causes retinoblastoma (Rb)-sensitive proteins
to become phosphorylated resulting in the loss of the suppression
of the E2F transcription factor, which may initiate DNA synthesis
to induce cell cycle progression from G1 into S phase
(29–32).
The overexpression of POGLUT1 in BT474 cells may
counteract the inhibition of TGF-β1 to promote cell proliferation,
suggesting that POGLUT1 may be a potential factor in the early
stage of abnormal hemopoietic stem cell differentiation. Thus,
further investigations are required regarding the association
between the CDKIs p15 and p16, and POGLUT1.
The Taqman probe method of fqPCR analysis was used
to detect p15 and p16 expression with β-actin as an internal
control probe. p16 expression was observed to increase in the
presence of TGF-β1, while exogenous POGLUT1 overexpression markedly
suppressed p16 expression in the presence of TGF-β1. However, there
was no detectable p15 expression in the BT474 cells with or without
exogenous POGLUT1 expression.
These findings demonstrate that TGF-β significantly
enhances p16 expression in BT474 cells and that POGLUT1
overexpression significantly reduces p16 expression in BT474 cells
in the presence of TGF-β1 (P<0.01). In addition, these findings
suggest that no detectable p15 is expressed in BT474 cells. For
further confirmation, western blot analysis was used to assess p15
and p16 protein expression, and the findings were in accordance
with those from the fqPCR analysis.
The present study also aimed to investigate the
effect of POGLUT1 overexpression on p-Smad3 and to analyze the
target of POGLUT1 in the suppression of p16 expression. Variations
in the Smad3 protein, which acts downstream in the TGF-β signaling
pathway, were assessed. Western blot analysis revealed that the
overexpression of POGLUT1 inhibited the expression of p-Smad3 in
BT474 cells. The protein expression of Smad3 and the internal
control β-actin were observed to be expressed to the same extent in
the POGLUT1 overexpression group, blank plasmid group and control
group. However, p-Smad3 expression increased markedly in the two
control groups and decreased markedly in the POGLUT1 overexpression
group. These findings demonstrate that the overexpression of
POGLUT1 may inhibit the expression of p-Smad3.
The findings of the present study suggest that
POGLUT1 overexpression may inhibit p16 upregulation through TGF-β1.
CDKs, cyclin D and CDKIs, including p15, p16 and p27, form a
dynamic equilibrium system which maintains normal cell
proliferation (33). Moreover,
cyclin D and CDK4 act together to activate CDK4. However, the
functional products of the p15 and p16 genes, which are p15INK4b
and p16INK4a, respectively, compete with CDK4/CDK6 for cyclin D in
order to suppress CDK4/cyclin D or CDK6/cyclin D complex formation
and block the CDK/pRb pathway to initiate G1-phase
arrest (34). The balance of the
two pathways is a key factor in cell proliferation.
In tumor cells with abnormal proliferation, the
equilibrium in the G1 phase is impaired, with CDKIs,
including p15 and p16, becoming deactivated through certain
modifications. This leads to inhibition of the CDK/pRb pathway and
the release excessive E2F protein, which shortens the G1
phase and subsequently promotes an increase in tumor cell
proliferation (35). In the present
study, p16 expression was detected in BT474 cells.
The TGF-β signaling pathway has a central role in
cell cycle regulation. TGF-β may combine with cell membrane
acceptors in order to activate Smad3 through phosphorylation, which
then forms a complex with Smad2 which enters the cell nucleus with
the assistance of Smad4 in order to suppress cell proliferation
through blocking its target genes. Smad3 activation has a very
important role in the entire signal passage (36). Studies have shown that Smad3 is one
of two CDK4/6 phosphorylation substrates. In normal cells,
CDK/Smad3/p16 forms a dynamic equilibrium system, through negative
feedback adjustment. Moreover, CDK4 activates Smad3 through
phosphorylation, then phosphorylated Smad3 suppresses Id-1
expression. Furthermore, Id-1 may suppress Ets1 and Ets2
expression, which are located upstream of p16 and promote
transcription. Thus the suppression of Id-1 increases p16
expression (35). The increase in
p16 expression upon CDK4/6 inhibition decreases CDK4/6-induced
Smad3 phosphorylation and the upregulation of p16 reduces through
negative feedback activity (37),
maintaining the dynamic balance. However, in the presence of
exogenous TGF-β1, Smad3 phosphorylation no longer relies on CDK4/6
activity and the CDK/Smad3/p16 equilibrium is impaired. Exogenous
TGF-β1 may strengthen the phosphorylation function of Smad3, then
upregulate the expression of p16, as well as increase the
inhibition of p16 on CDK4/6. Thus, cell proliferation is inhibited
as the cells remain in the G1 phase.
The findings of the present study suggest that
p-Smad3 expression is reduced in BT474 cells in the absence of
TGF-β1, and that POGLUT1 overexpression reduces the endogenous
phosphorylation of the Smad3 protein in order to decrease p16
expression at the gene and protein levels. In the presence of
TGF-β1, p-Smad3 expression was found to markedly increase
(P<0.05) and the expression of p16 also increased at the gene
and protein levels. POGLUT1 overexpression was observed to decrease
the phosphorylation of Smad3 and the expression of p16 was found to
decrease to a level even lower than the background level, as the
normal negative feedback regulation mechanism is already impaired
in tumor cells. The present study found that the signaling pathway
of POGLUT1 in BT474 human breast cancer cells involves a
POGLUT1/Smad3/p16/CDK/pRb pathway, and the signal is increased by
TGF-β1. However, further investigations on how POGLUT1 interacts
with these proteins and how POGLUT1 affects Smad3 phosphorylation
levels are required in order to understand the specific mechanism
of POGLUT1 in cancer.
References
|
1
|
Takeuchi H, Fernández-Valdivia RC, Caswell
DS, Nita-Lazar A, Rana NA, Garner TP, et al: Rumi functions as both
a protein O-glucosyltransferase and a protein O-xylosyltransferase.
Proc Natl Acad Sci USA. 108:16600–16605. 2011.
|
|
2
|
Fernandez-Valdivia R, Takeuchi H,
Samarghandi A, Lopez M, Leonardi J, Haltiwanger RS and Jafar-Nejad
H: Regulation of mammalian Notch signaling and embryonic
development by the protein O-glucosyltransferase Rumi. Development.
138:1925–1934. 2011.
|
|
3
|
Sethi MK, Buettner FF, Ashikov A, Krylov
VB, Takeuchi H, Nifantiev NE, et al: Molecular cloning of a
xylosyltransferase that transfers the second xylose to
O-glucosylated epidermal growth factor repeats of notch. J Biol
Chem. 287:2739–2748. 2012.
|
|
4
|
Ma W, Du J, Chu Q, Wang Y, Liu L, Song M
and Wang W: hCLP46 regulates U937 cell proliferation via Notch
signaling pathway. Biochem Biophys Res Commun. 408:84–88. 2011.
|
|
5
|
Teng Y, Liu Q, Ma J, Liu F, Han Z, Wang Y
and Wang W: Cloning, expression and characterization of a novel
human CAP10-like gene hCLP46 from CD34(+) stem/progenitor cells.
Gene. 371:7–15. 2006.
|
|
6
|
Wang Y, Chang N, Zhang T, Liu H, Ma W, Chu
Q, et al: Overexpression of human CAP10-like protein 46 KD in
T-acute lymphoblastic leukemia and acute myelogenous leukemia.
Genet Test Mol Biomarkers. 14:127–133. 2010.
|
|
7
|
Pierelli L, Marone M, Bonanno G, Mozzetti
S, Rutella S, Morosetti R, et al: Modulation of bcl-2 and p27 in
human primitive proliferating hematopoietic progenitors by
autocrine TGF-beta1 is a cell cycle-independent effect and
influences their hematopoietic potential. Blood. 95:3001–3009.
2000.
|
|
8
|
Cipriano R, Kan CE, Graham J, Danielpour
D, Stampfer M and Jackson MW: TGF-beta signaling engages an
ATM-CHK2-p53-independent RAS-induced senescence and prevents
malignant transformation in human mammary epithelial cells. Proc
Natl Acad Sci USA. 108:8668–8673. 2001.
|
|
9
|
Bardeesy N, Morgan J, Sinha M, Signoretti
S, Srivastava S, Loda M, et al: Obligate roles for p16(Ink4a) and
p19(Arf)-p53 in the suppression of murine pancreatic neoplasia. Mol
Cell Biol. 22:635–643. 2002.
|
|
10
|
Liu P, Zhang C, Feng JB, Zhao YX, Wang XP,
Yang JM, et al: Cross talk among Smad, MAPK, and integrin signaling
pathways enhances adventitial fibroblast functions activated by
transforming growth factor-beta1 and inhibited by Gax. Arterioscler
Thromb Vasc Biol. 28:725–731. 2008.
|
|
11
|
Bran GM, Sommer UJ, Goessler UR, Hörmann
K, Riedel F and Sadick H: TGF-β1 antisense impacts the SMAD
signalling system in fibroblasts from keloid scars. Anticancer Res.
30:3459–3463. 2010.
|
|
12
|
Janknecht R, Wells NJ and Hunter T:
TGF-beta-stimulated cooperation of smad proteins with the
coactivators CBP/p300. Genes Dev. 12:2114–2119. 1998.
|
|
13
|
Kim RH, Wang D, Tsang M, Martin J, Huff C,
de Caestecker MP, et al: A novel smad nuclear interacting protein,
SNIP1, suppresses p300-dependent TGF-beta signal transduction.
Genes Dev. 14:1605–1616. 2000.
|
|
14
|
Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y
and Wang XF: Transforming growth factor beta induces the
cyclin-dependent kinase inhibitor p21 through a p53-independent
mechanism. Proc Natl Acad Sci USA. 92:5545–5549. 1995.
|
|
15
|
McConnell BB, Gregory FJ, Stott FJ, Hara E
and Peters G: Induced-expression of p16(INK4a) inhibits both CDK4-
and CDK2-associated kinase activity by reassortment of
cyclin-CDK-inhibitor complexes. Mol Cell Biol. 19:1981–1989.
1999.
|
|
16
|
Aprelikova O, Xiong Y and Liu ET: Both p16
and p21 families of cyclin-dependent kinase (CDK) inhibitors block
the phosphorylation of cyclin-dependent kinases by the
CDK-activating kinase. J Biol Chem. 270:18195–18197. 1995.
|
|
17
|
Bockstaele L, Kooken H, Libert F, Paternot
S, Dumont JE, de Launoit Y, et al: Regulated activating Thr172
phosphorylation of cyclin-dependent kinase 4(CDK4): its
relationship with cyclins and CDK ‘inhibitors’. Mol Cell Biol.
26:5070–5085. 2006.
|
|
18
|
Noh SJ, Li Y, Xiong Y and Guan KL:
Identification of functional elements of p18INK4C essential for
binding and inhibition of cyclin-dependent kinase (CDK) 4 and CDK6.
Cancer Res. 59:558–564. 1999.
|
|
19
|
Omura-Minamisawa M, Diccianni MB, Chang
RC, Batova A, Bridgeman LJ, Schiff J, et al: p16/p14(ARF) cell
cycle regulatory pathways in primary neuroblastoma: p16 expression
is associated with advanced stage disease. Clin Cancer Res.
7:3481–3490. 2001.
|
|
20
|
Yao J, Pollock RE, Lang A, Tan M, Pisters
PW, Goodrich D, et al: Infrequent mutation of the p16/MTS1 gene and
overexpression of cyclin-dependent kinase 4 in human primary
soft-tissue sarcoma. Clin Cancer Res. 4:1065–1070. 1998.
|
|
21
|
Kubo A, Nakagawa K, Varma RK, Conrad NK,
Cheng JQ, Lee WC, et al: The p16 status of tumor cell lines
identifies small molecule inhibitors specific for cyclin-dependent
kinase 4. Clin Cancer Res. 5:4279–4286. 1999.
|
|
22
|
Stein GH, Drullinger LF, Soulard A and
Dulić V: Differential roles for cyclin-dependent kinase inhibitors
p21 and p16 in the mechanisms of senescence and differentiation in
human fibroblasts. Mol Cell Biol. 19:2109–2117. 1999.
|
|
23
|
Akervall J, Bockmühl U, Petersen I, Yang
K, Carey TE and Kurnit DM: The gene ratios c-MYC:cyclin-dependent
kinase (CDK)N2A and CCND1:CDKN2A correlate with poor prognosis in
squamous cell carcinoma of the head and neck. Clin Cancer Res.
9:1750–1755. 2003.
|
|
24
|
Mihira H, Suzuki HI, Akatsu Y, Yoshimatsu
Y, Igarashi T, Miyazono K and Watabe T: TGF-β-induced mesenchymal
transition of MS-1 endothelial cells requires Smad-dependent
cooperative activation of Rho signals and MRTF-A. J Biochem.
151:145–156. 2012.
|
|
25
|
Zhang SJ, Endo S, Ichikawa T, Washiyama K
and Kumanishi T: Frequent deletion and 5′ CpG island methylation of
the p16 gene in primary malignant lymphoma of the brain. Cancer
Res. 58:1231–1237. 1998.
|
|
26
|
Alcorta DA, Xiong Y, Phelps D, Hannon G,
Beach D and Barrett JC: Involvement of the cyclin-dependent kinase
inhibitor p16 (INK4a) in replicative senescence of normal human
fibroblasts. Proc Natl Acad Sci USA. 93:13742–13747. 1996.
|
|
27
|
Dreyling MH, Bullinger L, Ott G,
Stilgenbauer S, Müller-Hermelink HK, Bentz M, et al: Alterations of
the Cyclin D1/p16-pRB Pathway in mantle cell lymphoma. Cancer Res.
57:4608–4614. 1997.
|
|
28
|
FitzGerald MG, Harkin DP, Silva-Arrieta S,
MacDonald DJ, Lucchina LC, Unsal H, et al: Prevalence of germ-line
mutations in p16, p19ARF, and CDK4 in familial melanoma: analysis
of a clinic-based population. Proc Natl Acad Sci USA. 93:8541–8545.
1996.
|
|
29
|
Cai D, Byth KF and Shapiro GI: AZ703, an
imidazo[1,2-a]pyridine inhibitor of cyclin-dependent kinases 1 and
2, induces E2F-1-dependent apoptosis enhanced by depletion of
cyclin-dependent kinase 9. Cancer Res. 66:435–444. 2006.
|
|
30
|
Park DS, Morris EJ, Bremner R, Keramaris
E, Padmanabhan J, Rosenbaum M, et al: Involvement of retinoblastoma
family members and E2F/DP complexes in the death of neurons evoked
by DNA damage. J Neurosci. 20:3104–3114. 2000.
|
|
31
|
Bartkova J, Lukas J, Guldberg P, Alsner J,
Kirkin AF, Zeuthen J and Bartek J: The p16-cyclin D/Cdk4-pRb
pathway as a functional unit frequently altered in melanoma
pathogenesis. Cancer Res. 56:5475–5483. 1996.
|
|
32
|
Tsukiyama-Kohara K, Toné S, Maruyama I,
Inoue K, Katsume A, Nuriya H, et al: Activation of the
CKI-CDK-Rb-E2F pathway in full genome hepatitis C virus-expressing
cells. J Biol Chem. 279:14531–14541. 2004.
|
|
33
|
Guo K and Walsh K: Inhibition of
myogenesis by multiple cyclin-Cdk complexes. Coordinate regulation
of myogenesis and cell cycle activity at the level of E2F. J Biol
Chem. 272:791–797. 1997.
|
|
34
|
Ojima H, Saito K, Yamauchi H, Yamaki E,
Idetu A, Hosouchi Y, Nishida Y, et al: P16 protein abnormality in
Epstein-Barr virus-associated gastric carcinomas. Anticancer Res.
26:933–937. 2006.
|
|
35
|
Alani RM, Young AZ and Shifflett CB: Id1
regulation of cellular senescence through transcriptional
repression of p16/Ink4a. Proc Natl Acad Sci USA. 98:7812–7816.
2001.
|
|
36
|
Yuen PW, Man M, Lam KY and Kwong YL:
Clinicopathological significance of p16 gene expression in the
surgical treatment of head and neck squamous cell carcinomas. J
Clin Pathol. 55:58–60. 2002.
|
|
37
|
Singh RP, Agarwal C and Agarwal R:
Inositol hexaphosphate inhibits growth, and induces G1 arrest and
apoptotic death of prostate carcinoma DU145 cells: modulation of
CDKI-CDK-cyclin and pRb-related protein-E2F complexes.
Carcinogenesis. 24:555–563. 2003.
|