Introduction
A description of ovarian steroid cell tumors was
first provided in 1979 by Scully (1). The tumors belong to the sex
cord-stromal tumors and account for <0.1% of all ovarian
neoplasms (2). According to their
cell of origin, these tumors have been divided into three subtypes:
Stromal luteomas, Leydig cell tumors and steroid cell tumors not
otherwise specified (NOS). The last group accounts for 60% of these
tumors (3), which often produce
steroids, particularly testosterone, and may present with
virilizing symptoms, including hirsutism, a deep voice, baldness
and amenorrhea. In total, ~25% of patients lack endocrine symptoms
(4). Malignant tumors are extremely
rare, with only ~31 reported cases found via a PubMed search of the
English literature (http://www.ncbi.nlm.nih.gov/pubmed) (5–17). The
present study reports a case of a malignant steroid cell tumor NOS
in a patient younger than the mean age of occurrence, with lower
back and leg pain as the initial symptoms, and with a lack of
endocrine symptoms. Due to the lack of specific symptoms, a correct
diagnosis was not formed pre-operatively, but by post-operative
pathological examination. Various aspects of the presentation,
diagnosis and treatment of this type of tumor are also discussed.
Patient provided written informed consent.
Case report
A 29-year-old female presented to the Second Xiangya
Hospital with complaints of lower back and leg pain that had
persisted for five months, and a history of a ovarian mass that had
been diagnosed upon presentation to the Hunan Provincial Tumor
Hospital with the same complaints two months previously. The
patient was admitted to the Second Xiangya Hospital for further
management of this condition. The patient history revealed that
menarche had occurred at 14 years old, and that regular menses
lasting 5 to 6 days had occurred every thirty days since then. The
family history was negative for any type of cancer. A pelvic
examination revealed a 5×5×6-cm solid right adnexal mass.
Laboratory workups revealed that the levels of sex and thyroid
hormones, and 12 types of serum tumor markers, including cancer
antigen (CA)-125 and CA19–9, were within the normal limits.
Gastroscopy and colonoscopy findings were also normal. A
transvaginal pelvic ultrasound showed a 57×36-mm solid-cystic mass
of the right ovary. No ascites or other abnormalities were present.
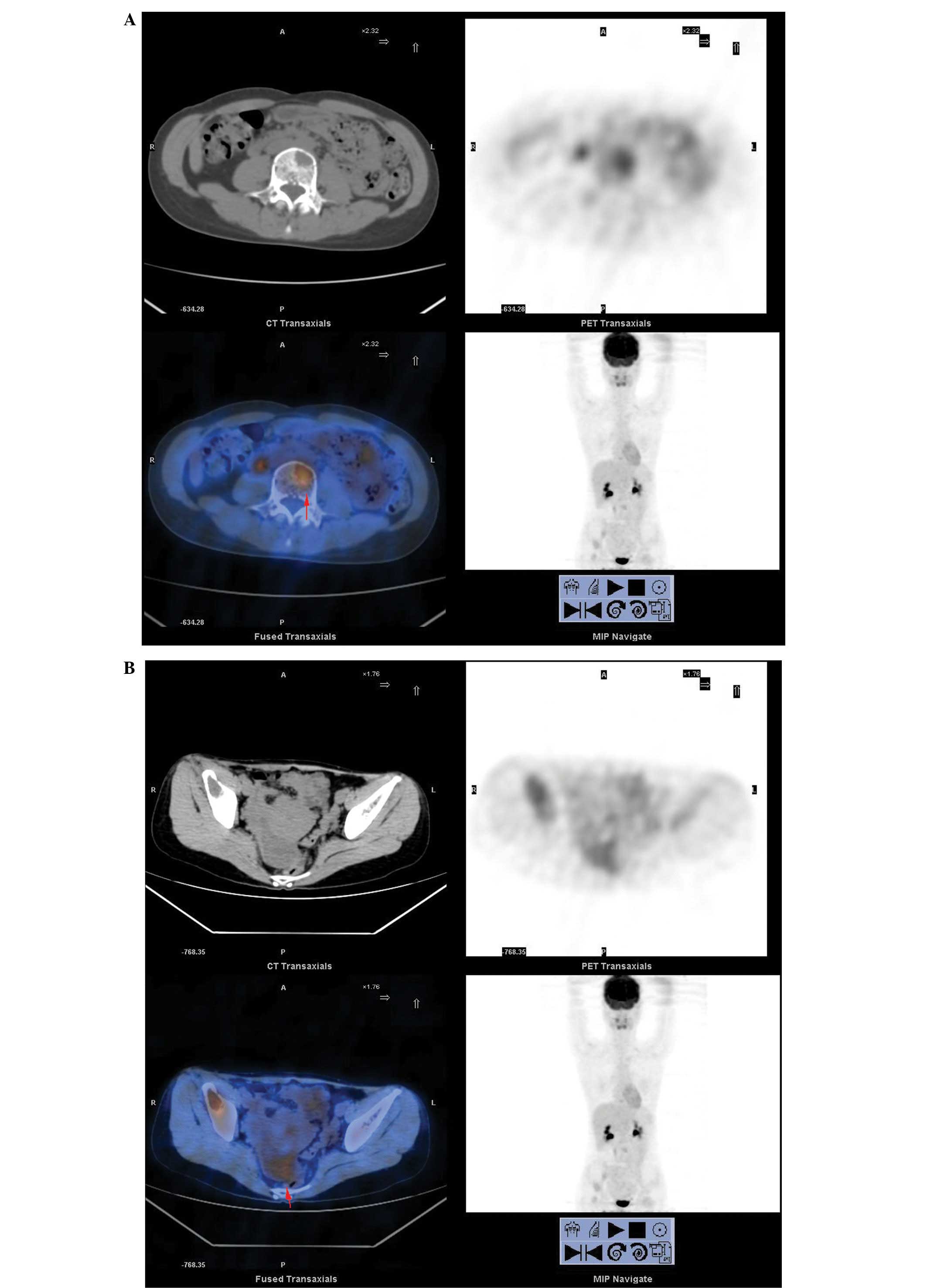
Whole body positron emission tomography (WBPET) and computed
tomography scans (performed at the Hunan Provincial Tumor Hospital,
Changsha, Hunan, China) found multiple areas of cystic destruction
of the bones, which were highly suspected to be caused by malignant
tumors (Fig. 1A). Previously,
Glaspy et al (18) reported
that PET imaging has the potential to demonstrate the biochemical
differences between normal and malignant tissues to reveal primary
or metastatic malignancy. A 33×64-mm solid-cystic mass was detected
on the right in the rear of the pelvis, indicating the possibility
of ovarian cancer (Fig. 1B). An
ovarian tumor was suspected. The patient underwent an exploratory
laparotomy; a small amount of yellowish ascites was observed
intraperitoneally, but the exploration of the liver, spleen, kidney
and greater omentum was normal. No lymphonodus was identified. The
bilateral fallopian tubes, left ovary and uterus were normal. A
well-encapsulated and solid 6×6-cm mass was located occupying 90%
of the right ovarian tissue, without any adhesions to the
surrounding structures. A right salpingo-oophorectomy and pelvic
washings for cytology were performed. A frozen section of the mass
revealed a malignant tumor, which could not be classified, with
large areas of necrotic tissue. Nitrogen mustard (2%) was used to
wash the abdominal cavity. The specimen was sent for
histopathological examination. Macroscopically, the mass measured
60×58×57 mm in size and had an intact and smooth external surface.
The cut surface was solid and yellow in color with multiple areas
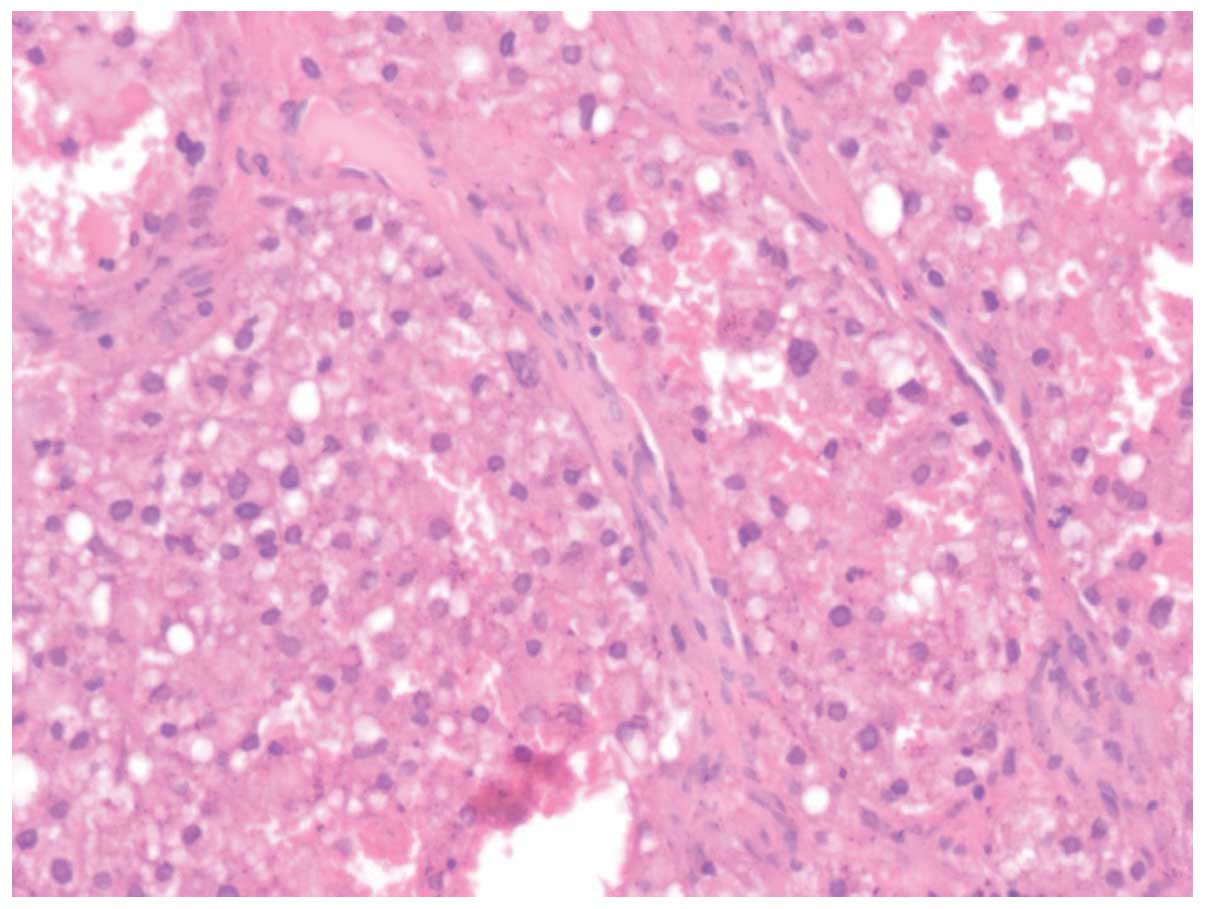
of necrosis. Upon microscopic examination, the tumor cells were
diffusely arranged, with the tumor areas exhibiting two types of
cells: i) Eosinophilic cells with abundant eosinophilic granular
cytoplasm, small to intermediate nuclei with small nucleoli and
distinct cell borders (Fig. 2); and
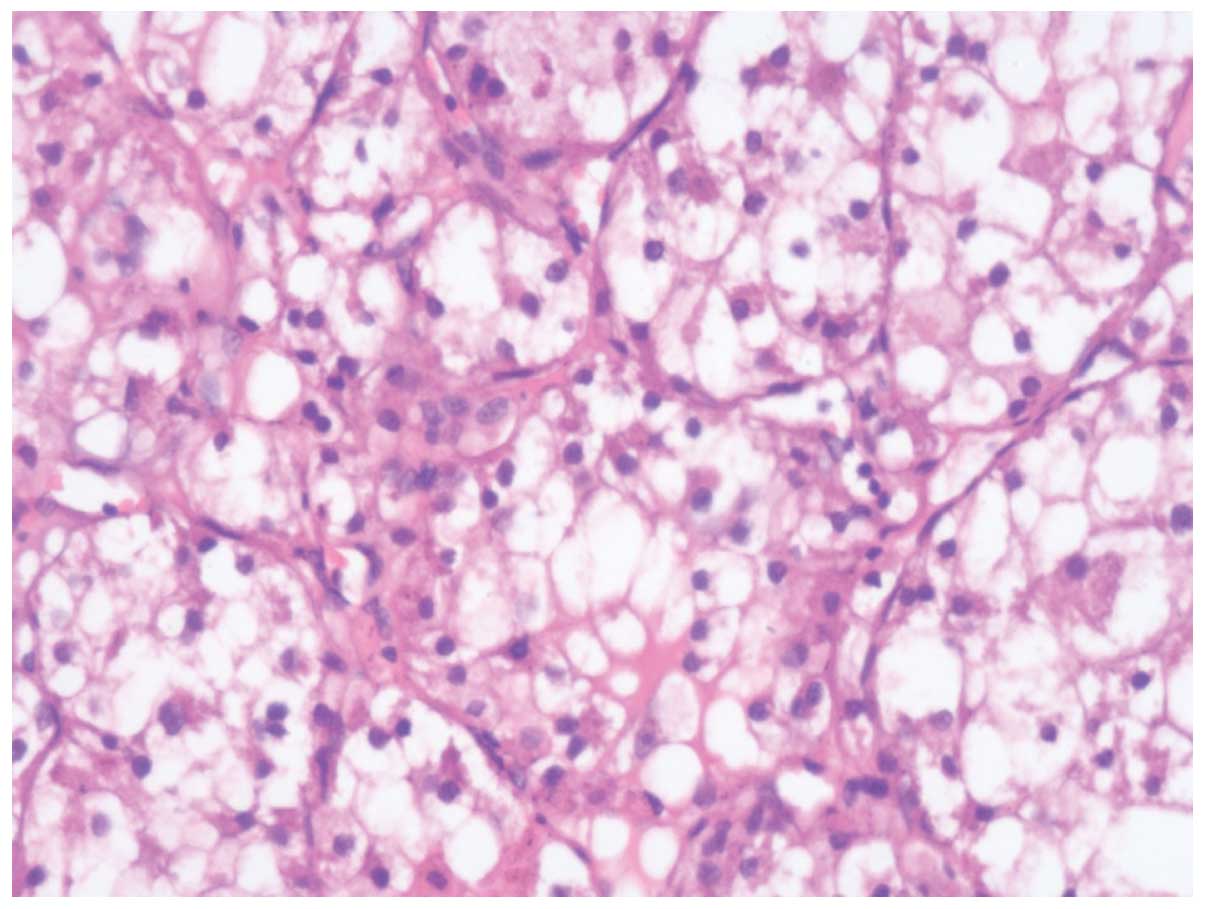
ii) clear cells rich in transparent cytoplasm, visible large
vacuole formation and small nuclei (Fig. 3). Regional cells were arranged in
cords or nests. Hemorrhagic necrosis, nuclear atypical and mitotic
figures were also apparent. Peritoneal fluid cytology did not
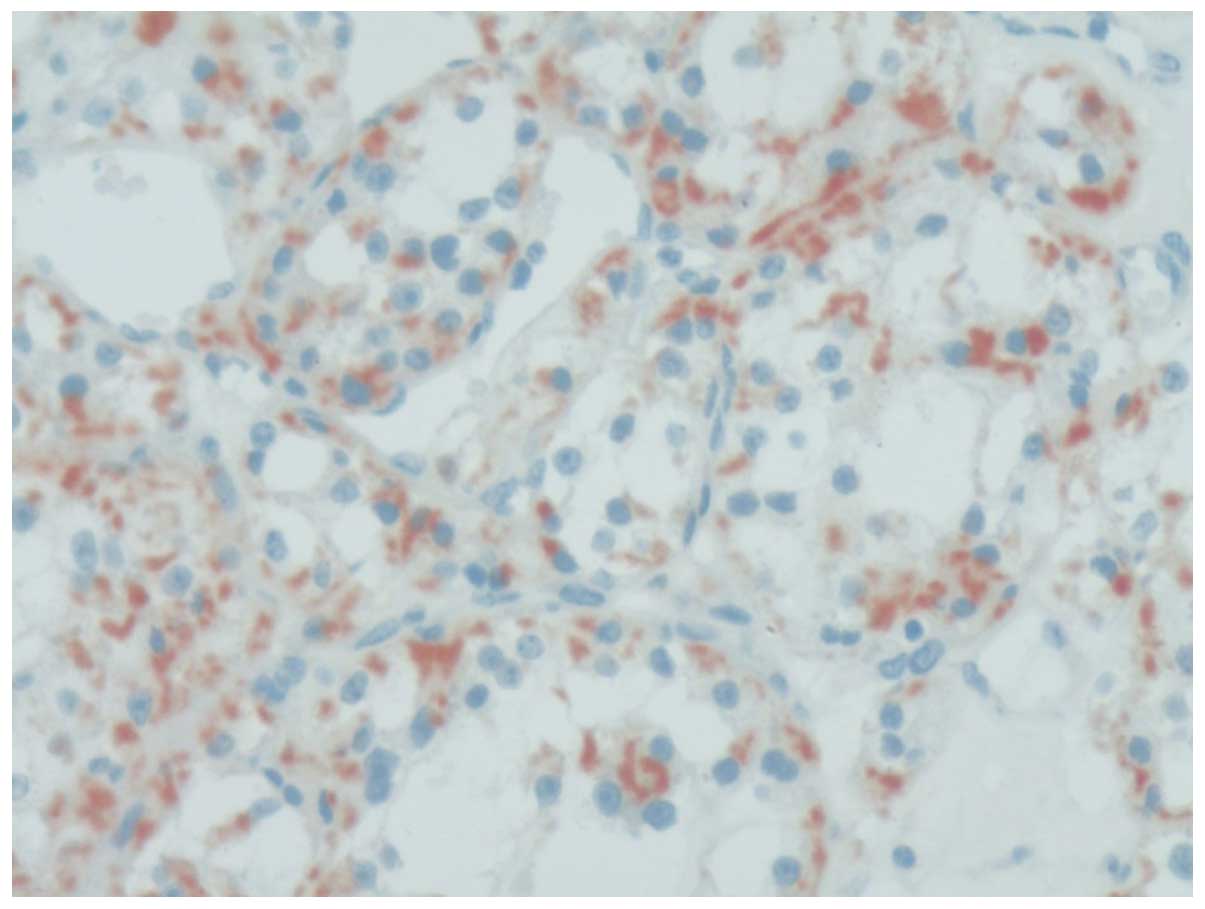
reveal any malignant cells. Immunohistochemical staining revealed
that neuron-specific enolase, chromogranin A (CgA; Fig. 4), cluster of differentiation (CD)68,
CD34, placental alkaline phosphatase, inhibin A and S100 were
positive, while smooth muscle actin (SMA), CD30, vimentin,
α-fetoprotein (AFP; Fig. 5),
epithelial membrane antigen (EMA) and cytokeratin were negative.
The clinical findings, histomorphology and immunohistochemistry of
the mass revealed a diagnosis of malignant ovarian steroid cell
tumor NOS. The patient was administered chemotherapy (docetaxel 120
mg and nedaplatin 80 mg) on post-operative day 6. The patient
received 8 cycles of the same chemotherapy regimen. The patient
lived a relatively healthy life and there was no deterioration of
the condition for a year and a half. However, following this, the
disease progressed and the patient succumbed 6 months later from
brain metastases.
Discussion
It has been reported that ovary steroid cell tumors
are rare and account for 0.1% of ovarian tumors (2). Steroid cell tumor NOS is a subtype of
steroid cell tumors; their cell lineage is not defined and they
cannot be categorized as either stromal luteomas or Leydig cell
tumors. The tumors occur at any age, but with an mean age of 43
years (4), which is younger than
that for other steroid cell tumors. Usually unilateral, only 6% of
cases are found to be bilateral. The majority are benign, with
25–40% of patients with malignant tumors. The clinical
manifestations of the tumor are associated with its hormonal
activity and virilizing properties. Among the patients affected
with the tumors, 56–77% have virilizing symptoms, including
hirsutism, acne, clitoral enlargement, a deep voice and alopecia,
and 6–23% have estrogenic manifestations, such as menorrhagia,
postmenopausal bleeding or even endometrial cancer. Cushing’s
syndrome occurs in only 6–10% of the cases, while gynecological
examinations or surgery reveal that ~25% are not associated with
hormonal disturbances (2,11,19–23).
The patient in the present study was 29 years old, younger than the
average age of occurrence, with an apparent absence of androgenic
manifestations. Lower back and leg pain were the first symptoms,
and a gynecological examination, transvaginal pelvic ultrasound and
WBPET/CT located a right adnexal mass. Clinically, NOS tumors can
present as abdominal pain, abdominal distention and bloating,
however, to the best of our knowledge, metastatic bone pain as the
first symptom has not previously been reported.
Pathological examination is an important method to
diagnose steroid cell tumors. The gross specimen shows a tumor with
a clear boundary, almost always well circumscribed and solid, with
an enveloped, lobulated or nodular appearance (24,25). A
total of 94% of cases are unilateral, while 6% are bilateral
(2). The average diameter of the
tumor is 8.5 cm, and the cut surface is yellow or orange (25), with occasional bleeding or cystic
degeneration. Microscopically, the tumor cells are round or
polygonal, and medium-to-large in size with distinct cell borders.
The tumor cells often have central nuclei and prominent nucleoli.
The neoplastic cells are usually of two types; eosinophilic and
clear cells. The two types of cells differ only in their
cytoplasmic appearance. The eosinophilic cells contain
eosinophilic, slightly granular cytoplasm, while the clear cells
have abundant vacuolated clear cytoplasm which is often positive
for fat stains (2,16,20,25–27).
In contrast to Leydig cells, the clear cells lack crystals of
Reinke in the cytoplasm (26,27).
The tumor cells are arranged predominantly in a diffuse pattern,
but may be arranged in nests, clusters, columns or cords, separated
by rich vascular characteristics. In the present case, the gross
and microscopic findings were consistent with the characteristics
of the ovarian steroid cell tumor NOS. The malignant cells show
marked atypia, mitoses, necrosis and hemorrhage. The literature
indicates that 25–43% of steroid cell tumor cases are clinically
malignant, with 20% of cases found to exhibit metastases outside of
the ovary during surgery (2,7). If
metastasis is present, ~20% of metastatic lesions usually occur
within the peritoneal cavity and rarely occur at distant sites.
Although malignancy is often associated with metastasis, a study by
Hayes and Scully (2) identified
five pathological features predictive of malignancy: i) Two or more
mitoses per 10 high-power fields (92% malignant); ii) a tumor
diameter of >7 cm (78% malignant); iii) necrosis (86%
malignant); iv) hemorrhage (77% malignant); and v) grade 2 or 3
nuclear atypia (64% malignant) (1).
However, tumors with a benign histomorphology may be clinically
malignant (17). The tumor of the
current case was ~6 cm in diameter and hemorrhagic necrosis,
nuclear atypia and mitotic figures were apparent. WBPET and CT
scans found multiple areas of cystic destruction of the bones,
which was highly suspected to be the result of malignant tumors.
Due to these characteristics, the diagnosis of malignant steroid
cell tumor NOS was determined. The most unusual feature in the
present case was the bone metastasis, as it occurs infrequently in
steroid cell tumors. Multiple bone metastases, without pelvic and
lymph node metastasis, has not been previously reported. Another
unusual feature is the apparent absence of endocrine
manifestations; the patient’s hormone levels were normal. Other
than the aforementioned microscopic features, immunohistochemical
analysis aids the formation of a correct diagnosis. A number of
immunohistochemical markers can be applied. The majority of steroid
cell tumors are positive for calretinin and inhibin (28), while 75% of cases are
vimentin-positive. The presence of HMB45 is inconsistent, however,
other markers, such as EMA, cytokeratin, CD99 and S-100, have been
reported to be positive, while CgA, LeuM1, AFP, carcinoembryonic
antigen and periodic acid-Schiff have been reported to be negative
(29). Positivity for CgA has never
previously been reported in the English literature (4), However, CgA was found to be positive
in the present case. This is a novel finding. In this case, the
gross and microscopic findings were consistent with an ovarian
steroid cell tumor NOS. Positivity for inhibin and negativity for
SMA, AFP and EMA supported this diagnosis. The multiple metastases
in the bone, the large areas of necrosis and the nuclear mitoses
were indicative of a malignant nature.
The differential diagnosis of steroid cell tumor NOS
includes stromal luteomas, Leydig cell tumors, luteinized thecomas,
luteinized granulosa cell tumors, pregnancy luteomas and
carcinomas, primary clear cell carcinomas, metastatic renal cell
carcinomas and adrenocortical carcinomas. All these were excluded
prior to reaching the diagnosis of a steroid cell tumor NOS.
The treatment of ovarian steroid cell tumors is
primarily surgical. In a young patient, unilateral
salpingo-oophorectomy is adequate for the treatment of a stage Ia
tumor. Older females who do not want to preserve their fertility
should undergo a total abdominal hysterectomy, bilateral
salpingo-oophorectomy and complete surgical staging. As
pathologically benign steroid cell tumors can behave in a
clinically malignant manner, those patients who have high hormone
levels prior to surgery require measurements of the sex hormone
levels throughout. Patients with malignant tumors should
immediately undergo debulking surgery, with post-operative
chemotherapy or radiotherapy. In the present case, considering the
patient had bone metastasis and that the tumor was confined to the
ovary, with no pelvic metastases, a right salpingo-oophorectomy was
performed. Due to the rarity of this type of tumor, little focus
has been assigned to its study, and as the majority of these tumors
are diagnosed in an early stage and do not recur or metastasize,
the therapeutic value of chemotherapy and radiotherapy is poorly
understood (30). Adjuvant
chemotherapy regimens currently recommended for treatment are as
follows: Bleomycin, etoposide and cisplatin; cisplatin, doxorubicin
and cyclophosphamide; taxane and platinum; and bleomycin,
vinblastine and cisplatin (31–34).
The most effective regimen has not yet been found, with the
majority reporting poor efficacy. In the present study, with
treatment using a docetaxel and nedaplatin regimen, the patient
survived for two years with multiple bone metastases. In recent
years, the application of gonadotropin releasing hormone analogue
has been attempted in order to treat this disease (35); gonadotropin releasing hormone
agonist can restrain hormone secretion and induce cell apoptosis.
This therapy has been attempted in patients who cannot tolerate
surgery, or in the case of recurrence. This treatment method is
only considered as an experimental therapy. However, it could
provide a feasible treatment method and is worth further
research.
In conclusion, ovarian steroid cell tumors NOS are
rare, accounting for 60% of ovarian steroid cell tumors. The
majority are benign, unilateral and usually characterized by
virilizing symptoms. Pathological evaluation is essential for the
diagnosis of malignancy, and immunohistochemical testing also aids
in the formation of an accurate diagnosis. Surgery is the main
treatment method and little is known about the response of the
tumors to therapies such as chemotherapy or radiation. The present
case is unique, as the patient was young, with an apparent absence
of endocrine manifestations. The pain caused by bone metastasis as
the first symptom has not been reported previously. Another unusual
finding in this case is that the tumor was confined to the ovary,
with no pelvic and lymph node metastasis, but with multiple bone
metastases found by PET/CT. Additionally, CgA staining was found to
be positive in this case, which was not consistent with the
reported literature. The patient underwent a right
salpingo-oophorectomy and chemotherapy of carboplatin and
paclitaxel regimen for 8 cycles, and subsequently survived for two
years. The patient lived a relatively healthy life until the last
six months, when the disease progressed and the patient succumbed
to brain metastasis. This case indicates that post-operative
chemotherapy can achieve a good effect and prolong the survival of
patients, even in those with multiple bone metastases.
References
|
1
|
Scully RE: Tumors of the ovary and
maldeveloped gonads. Atlas of Tumor Pathology. (Second Series
Fascicle 16). Armed Forces Institute of Pathology; Washington, DC,
USA: pp. 215–220. 1979
|
|
2
|
Hayes MC and Scully RE: Ovarian steroid
cell tumors (not otherwise specified). A clinicopathological
analysis of 63 cases. Am J Surg Pathol. 11:835–845. 1987.
|
|
3
|
Kim YT, Kim SW, Yoon BS, et al: An ovarian
steroid cell tumor causing virilization and massive ascites. Yonsei
Med J. 48:142–146. 2007.
|
|
4
|
Amneus MW and Natarajan S: Pathologic quiz
case: a rare tumor of the ovary. Arch Pathol Lab Med. 127:890–892.
2003.
|
|
5
|
Novoa-Vargas A, Sánchez-Bautista K and
Coudillo-Luna I: Malignant arrhenoblastoma. Case report and
literature review. Ginecol Obstet Mex. 79:45–51. 2011.(In
Spanish).
|
|
6
|
Galinier P, Carfagna L, Delsol M, et al:
Ovarian torsion. Management and ovarian prognosis: a report of 45
cases. J Pediatr Surg. 44:1759–1765. 2009.
|
|
7
|
Sawathiparnich P, Sitthinamsuwan P,
Sanpakit K, et al: Cushing’s syndrome caused by an ACTH-producing
ovarian steroid cell tumor, NOS, in a prepubertal girl. Endocrine.
35:132–135. 2009.
|
|
8
|
Saida T, Tanaka YO and Minami M: Steroid
cell tumor of the ovary, not otherwise specified: CT and MR
findings. AJR Am J Roentqenol. 188:W393–W394. 2007.
|
|
9
|
Powell JL, Dulaney DP and Shiro BC:
Androgen-secreting steroid cell tumor of the ovary. South Med J.
93:1201–1204. 2000.
|
|
10
|
Brewer CA and Shevlin D: Encouraging
response of an advanced steroid-cell tumor to GnRH agonist therapy.
Obstet Gynecol. 92:661–663. 1998.
|
|
11
|
Wang PH, Chao HT, Lee RC, et al: Steroid
cell tumors of the ovary: clinical, ultrasonic, and MRI diagnosis -
a case report. Eur J Radiol. 26:269–273. 1998.
|
|
12
|
Wang PH, Chao HT and Lee WL: Use of a
long-acting gonadotropin-releasing hormone agonist for treatment of
steroid cell tumors of the ovary. Fertil Steril. 69:353–355.
1998.
|
|
13
|
Wang PH, Chao HT, Liu RS, Cho YH, Ng HT
and Yuan CC: Diagnosis and localization of testosterone-producing
ovarian tumors: imaging or biochemical evaluation. Gynecol Oncol.
83:596–598. 2001.
|
|
14
|
Dowdy SC, O’Kane DJ, Keeney GL, Boyd J and
Podratz KC: Telomerase activity in sex cord-stromal tumors of the
ovary. Gynecol Oncol. 82:257–260. 2001.
|
|
15
|
Jiang W, Tao X, Fang F, Zhang S and Xu C:
Benign and malignant ovarian steroid cell tumors, not otherwise
specified: case studies, comparison, and review of the literature.
J Ovarian Res. 6:532013.
|
|
16
|
Murhekar K, Louis R and Majhi U: A rare
occurrence of a steroid cell tumor of the pelvic mesentery: a case
report. J Med Case Rep. 5:5172011.
|
|
17
|
Mehdi G, Ansari HA, Sherwani RK, Rahman K
and Akhtar N: Ovarian steroid cell tumor: correlation of
histopathology with clinicopathologic features. Patholog Res Int.
2011:9878952011.
|
|
18
|
Glaspy JA, Hawkins RA, Hoh CK and Phelps
ME: Use of positron emission tomography in oncology. Oncology
(Williston Park. 7:41–46. 1993.
|
|
19
|
Taylor HB and Norris HJ: Lipoid cell
tumors of the ovary. Cancer. 20:1953–1962. 1967.
|
|
20
|
Donovan JT, Otis CN, Powell JL and
Cathcart HK: Cushing’s syndrome secondary to malignant lipoid cell
tumor of the ovary. Gynecol Oncol. 50:249–253. 1993.
|
|
21
|
Anderson PW, D’Abling G III, Penny R,
Sherrod A and Do YS: Secretion of prorenin by virilizing ovarian
tumor. Gynecol Oncol. 45:58–61. 1992.
|
|
22
|
Reedy MB, Richards WE, Ueland F, et al:
Ovarian steroid cell tumors, not otherwise specified: a case report
and literature review. Gynecol Oncol. 75:293–297. 1999.
|
|
23
|
Lee SH, Kang MS, Lee GS and Chung WY:
Refractory hypertension and isosexual pseudoprecocious puberty
associated with rennin-secreting ovarian steroid cell tumor in a
girl. J Korean Med Sci. 26:836–838. 2011.
|
|
24
|
Boyraz G, Selcuk I, Yusifli Z, et al:
Steroid cell tumor of the ovary in an adolescent: a rare case
report. Case Rep Med. 2013:5276982013.
|
|
25
|
Tsai HJ, Chen SC, Wei HY and Chen GD:
Hypothyroidism and hyperlipidemia with a virilizing ovarian steroid
cell tumor, not otherwise specified. Gynecol Endocrinol. 23:69–71.
2007.
|
|
26
|
Zhang X and Lü B: Ovarian steroid cell
tumor, not otherwise specified (NOS): an unusual case with
myelolipoma. Int J Gynecol Pathol. 30:460–465. 2011.
|
|
27
|
Kim YT, Kim SW, Yoon BS, et al: An ovarian
steroid cell tumor causing virilization and massive ascites. Yonsei
Med J. 48:142–146. 2007.
|
|
28
|
Sielert L, Liu C, Nagarathinam R and Craig
LB: Androgen-producing steroid cell ovarian tumor in a young woman
and subsequent spontaneous pregnancy. J Assist Reprod Genet.
30:1157–1160. 2013.
|
|
29
|
Jones MW, Harri R, Dabbs DJ and Carter GJ:
Immunohistochemical profile of steroid cell tumor of the ovary: a
study of 14 cases and a review of the literature. Int J Gynecol
Pathol. 29:315–320. 2010.
|
|
30
|
Ye L, Wu XL, Xu L, Huang Q, et al: Ovarian
steroid cell tumor, not otherwise specified: a clinicopathologic
study. Zhonghua Bing Li Xue Za Zhi. 36:516–520. 2007.(In
Chinese).
|
|
31
|
Gershenson DM, Copeland LJ, Kavanagh JJ,
et al: Treatment of metastatic stromal tumors of the ovary with
cisplatin, doxorubicin, and cyclophosphamide. Obstet Gynecol.
70:765–769. 1987.
|
|
32
|
Homesley HD, Bundy BN, Hurteau JA and Roth
LM: Bleomycin, etoposide, and cisplatin combination therapy of
ovarian granulosa cell tumors and other stromal malignancies: A
Gynecologic Oncology Group Study. Gynecol Oncol. 72:131–137.
1999.
|
|
33
|
Brown J, Shvartsman HS, Deavers MT, et al:
The activity of taxanes compared with bleomycin, etoposide, and
cisplatin in the treatment of sex cord-stromal ovarian tumours.
Gynecol Oncol. 97:489–496. 2005.
|
|
34
|
Pecorelli S, Wagenaar HC, Vergote IB, et
al: Cisplatin (P), vinblastine (V) and bleomycin (B) combination
chemotherapy in recurrent or advanced granulose(-theca) cell
tumours of the ovary. An EORTC Gynaecological Cancer Cooperative
Group study. Eur J Cancer. 35:1331–1337. 1999.
|
|
35
|
Pascale MM, Pugeat M, Roberts M, et al:
Androgen suppressive effect of GnRH agonist in ovarian
hyperthecosis and virilizing tumors. Clin Endocrinol (Oxf).
41:571–576. 1994.
|