Introduction
Steroid cell tumors (SCTs) of the ovary account for
<0.1% of all ovarian tumors and these tumors may present at any
age along with notable presentations as a result of the hormonal
activity and virilizing properties of the tumor (1). To the best of our knowledge, a small
number of cases of SCTs, not otherwise specified (NOS), have been
described (2–11). In the current report, a case of SCT,
NOS in a 59-year-old female with postmenopausal vaginal bleeding is
presented. The histopathological and clinical features of the SCT
are summarized and a review of the literature regarding this type
of tumor is presented. The study was approved by the ethics
committee of the Third Affliated Hospital of Sun Yet Sen University
(Guangzhou, China). The patient provided written informed
consent.
Case report
In August 2012, a 59-year-old female (gravida 3,
para 2) presented to the Department of Gynecology at The Third
Affiliated Hospital of Sun Yat-sen University following two months
of irregular vaginal bleeding, 12 years after menopause. A
transvaginal ultrasound scan identified an enlarged uterus with a
5-mm endometrium, a 22 × 19 mm left ovarian adnexal solid mass and
a small quantity of free fluid in the pelvis. The patient had
undergone diagnostic curettage at the Zhaoqing First People’s
Hospital (Zhaoqing, China) two weeks prior to attending The Third
Affiliated Hospital of Sun Yat-sen University and the pathological
result had shown a proliferative endometrium. The patient had a
history of hepatitis B and diabetes mellitus, however, the systemic
examination was unremarkable. A vaginal examination revealed a
small amount of blood in the vagina and a small uterus, while the
assessment of the adnexa of uterus was limited due to atrophy of
the ovaries following menopause. A transvaginal Doppler ultrasound
scan showed that the uterus was normal and the endometrium was 2-mm
thick. In addition, a 30×17-mm left ovarian adnexal solid mass was
observed as well as 20 mm free fluid in the pelvis. A pelvic
magnetic resonance imaging (MRI) scan showed a 20×15-mm left
adnexal cystic-solid mass. The patient’s liver enzyme levels were
marginally elevated, with alanine aminotransferase levels of 50 U/l
and a fasted blood glucose level of 7.27 mol/l. The level of cancer
antigen (CA)-125 was 95.6 U/l (normal range, 0–35 U/l) and other
tumor markers, including CA19-9, CA15-3, carcinoembryonic antigen
and α-fetoprotein (AFP) were within the normal limits. The
patient’s total serum testosterone level and estradiol (E2) level
were 22.28 nmol/l (normal range, 0.5–2.6 nmol/l) and 393.71 nmol/l
(normal value, <118.2 nmol/l for postmenopausal females),
respectively. Normal levels of luteinizing hormone (LH),
follicle-stimulating hormone (FSH) and dehydroepiandrosterone
(DHEA) were observed.
A laparotomy was undertaken and 100 ml fluid was
observed in the peritoneal cavity, in addition, a solid mass
measuring 3×2 cm was identified in the left ovary. The uterus, the
right ovary and the fallopian tubes appeared normal. A hysterectomy
and bilateral salpingo-oophorectomy were performed. The final
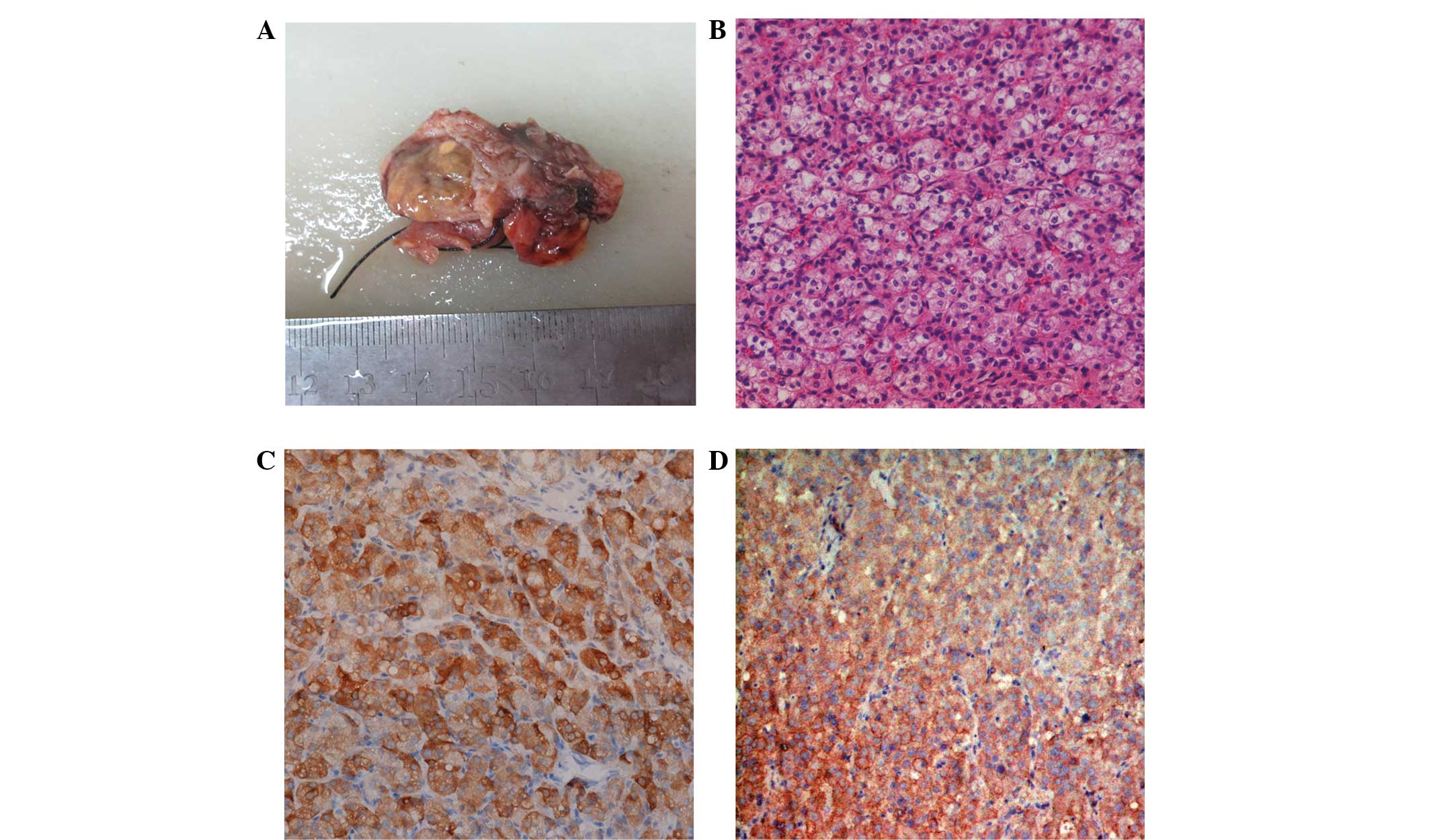
pathology result showed a well-circumscribed tumoral mass, size 3×3
cm. The cut surface of the solid neoplasm was homogeneous and
yellow. Microscopically, the tumor was predominantly composed of
granular eosinophilic or vacuolated cytoplasm. Reinke’s crystals,
prominent nucleoli and Call-Exner bodies were not observed, and no
mitotic figure was present. Immunohistochemistry of the neoplastic
cells exhibited positive staining for inhibin, however, was
negative for cytokeratin (CK; Fig.
1). There was weak staining for epithelial membrane antigen
(EMA), however, S-100, smooth muscle actin, CD68 and desmin were
negative; thus, a final diagnosis of SCT was established. By the
fourth postoperative day, the total testosterone and E2 levels fell
to 3.85 nmol/l and 282.26 nmol/l, respectively. After one month,
repeat assessments for total testosterone and E2 levels were
conducted in the clinic and revealed normal values. The patient is
currently being followed up at regular intervals.
Discussion
SCTs of the ovary are particularly rare and account
for <0.1% of all ovarian tumors (1). These tumors produce an excess of
steroid hormone, specifically testosterone, therefore they are
grouped into the category of sex cord-stromal tumors of the ovary.
They are divided into three further subtypes according to their
origins: Stromal luteoma, Leydig cell tumor and SCT, NOS, with the
latter being the most common of the three subtypes, accounting for
~60% of cases (2).
Ovarian SCTs, NOS, may occur at any age, however,
generally occur at younger ages compared with other SCTs and
occasionally prior to puberty (9).
The typical presentation, which occurs in premenopausal females
(mean age, 43 years), is virilization (2); >50% of cases are clinically
associated with androgenic changes. Ovarian SCT, NOS patients
exhibit symptoms, including hirsutism, hair loss, amenorrhea or
oligomenorrhea and ~25% of SCTs, NOS, do not produce any hormones
(3). It is uncommon for SCTs, NOS,
to produce hormones other than testosterone. However, the excess
production of estrogen, prolactin and prorenin has previously been
reported (8–10). The excess production of estrogen may
result in menorrhagia and postmenopausal bleeding; in particularly
rare cases, endometrial adenocarcinoma has also been documented
(3,11). Hyperandrogenism accompanied by
hyperestrogenism is uncommon in postmenopausal females. The
testosterone level of the patient in the present study was markedly
elevated and the E2 level was marginally elevated and, although the
patient exhibited an estrogenic manifestation, which presented as
postmenopausal bleeding and endometrial proliferation, the patient
did not demonstrate the associated androgenic manifestations.
However, it is possible that the changes were subtle and, thus, not
clinically evident.
Elevation of testosterone levels to >200 ng/dl is
the significant diagnostic threshold level for the discrimination
of androgen-secreting tumors and non-neoplastic lesions. Elevated
testosterone levels may result in adrenal, ovarian and iatrogenic
causes and therefore, must be determined in order to diagnose or
rule out disease/tumor. Elevated testosterone levels that present
with normal DHEA, LH, FSH and 17-hydroxyprogesterone levels warrant
the diagnosis of a virilizing ovarian tumor. MRI, endovaginal
ultrasound and color flow Doppler imaging also contribute to
differential diagnosis. Various other serum tumor markers
(including AFP and CA-125) facilitate the differential diagnosis of
ovarian adenocarcinoma (5).
The final diagnosis is usually made via the
histological examination of a surgically resected specimen. The
characteristic histological appearance includes the presence of
diffusely arranged cells with abundant granular eosinophilic or
vacuolated cytoplasm, which is often positive for fat stains, with
an absence of Reinke’s crystals and stromal hyperthecosis (3,6). In
addition to these microscopic features, immunohistochemistry is
particularly useful for the accurate diagnosis of an SCT. Numerous
immunohistochemical markers have been identified for the
differential diagnosis of sex cord-stromal tumors, and inhibin was
found to be particularly useful in differentiating sex cord-stromal
from non-sex cord-stromal tumors (12,13).
Inhibin, a hormonal polypeptide, is present in ovarian granulosa
and lutein cells, as well as testicular (Sertoli’s) and Leydig
cells, and suppresses the production of pituitary gonadotropins,
particularly FSH. The majority of SCTs are positive for inhibin.
CD99, the MIC2 gene product best known for its presence in Ewing’s
sarcoma and primitive neuroectodermal tumors, is another marker
that is expressed by sex cord-stromal tumors; furthermore, the CD99
antibody reacts with normal granulosa and Sertoli’s cells. Sex
cord-stromal tumors are predominantly negative for CK and the
majority of sex cord-stromal tumors are negative for EMA, although
a small subset of tumors may be positive for EMA. Focal EMA
positivity was reported in a previous case of SCT (14). In the present study, a diagnosis of
sex cord SCT was made on the basis of microscopic images as well as
the observation of positive immune reactivity to inhibin. Negative
staining for CK7 supported a diagnosis of sex cord-stromal tumor
and focal EMA positivity, which was observed in the present case,
was consistent with other reported cases (14).
The majority of SCTs are benign and unilateral. In a
series of 63 cases from the Massachusetts General Hospital (Boston,
MA, USA), 94% of the tumors were found to be unilateral (3). However, malignancy has been reported
in as high as 28.6–43% of cases and pathologic evaluation is
considered to be essential for the diagnosis of a malignancy. Hayes
and Scully (3) identified five
pathological features of tumors that are highly associated with
malignancy: i) More than two mitoses per 10 high-power field; ii)
presence of necrosis; iii) diameter, ≥7 cm; iv) hemorrhaging; and
v) grade 2 or 3 nuclear atypia. For the patient in the current
study, the pathology result did not demonstrate any obvious signs
of malignancy.
The management of SCTs is surgical removal of the
tumor, with an excision of the primary lesion by unilateral
oophorectomy, and is generally considered to be adequate for the
treatment of young patients. As the frequency of bilateral
occurrence is only 6%, and when future fertility is not an issue,
hysterectomy (removal of the contralateral ovary) and complete
surgical staging are recommended. However, subsequent monitoring of
hormone levels is required as part of the patient’s postoperative
follow-up. To the best of our knowledge, there are no reports
concerning the efficacy of radiation or chemotherapy. In recent
years attempts have been made to describe the use of
gonadotropin-releasing hormone analogues to induce the suppression
of secretions and apoptosis, which may lead to a non-surgical cure,
however, these approaches have primarily been attempted with
inoperable cases or patients with recurrent disease.
In conclusion, ovarian SCTs, NOS (grouped under sex
chord-stromal tumors) are a particularly rare type of ovarian
tumor. They are usually benign, unilateral and are characterized by
hyperandrogenism and virilization. However, in a rare case, such as
the present patient, hyperandrogenism and hyperestrogenism were
present and the clinical manifestation was not considered to be
typical. The primary treatment method was surgical extirpation of
the primary lesion. We recommend that any patient who presents with
high testosterone levels should be investigated systematically in
order to determine whether the origin is adrenal or ovarian. An
awareness of this entity will extend the appreciation of NOS
ovarian steroid cell tumors.
References
|
1
|
Young RH and Shully RE: Steroid cell
tumors of the ovary. Obstetric & Gynecological Pathology. Fox H
and Wells M: Churchill Livingstone; Edinburgh, Spain: pp. 845–856.
2003
|
|
2
|
Singh P, Deleon F and Anderson R: Steroid
cell ovarian neoplasm, not otherwise specified: a case report and
review of the literature. Case Rep Obstet Gynecol.
2012:2531522012.
|
|
3
|
Hayes MC and Scully RE: Ovarian steroid
cell tumors (not otherwise specified). A clinicopathological
analysis of 63 cases. Am J Surg Pathol. 11:835–845. 1987.
|
|
4
|
Reedy MB, Richards WE, Ueland F, Uy K, Lee
EY, Bryant C and van Nagell JR Jr: Ovarian steroid cell tumors, not
otherwise specified: a case report and literature review. Gynecol
Oncol. 75:293–297. 1999.
|
|
5
|
Kim YT, Kim SW, Yoon BS, Kim SH, Kim JH,
Kim JW and Cho NH: An ovarian steroid cell tumor causing
virilization and massive ascites. Yonsei Med J. 48:142–146.
2007.
|
|
6
|
Liu AX, Sun J, Shao WQ, Jin HM and Song
WQ: Steroid cell tumors, not otherwise specified (NOS), in an
accessory ovary: a case report and literature review. Gynecol
Oncol. 97:260–262. 2005.
|
|
7
|
Zhang X and Lü B: Ovarian steroid cell
tumor, not otherwise specified (NOS): an unusual case with
myelolipoma. Int J Gynecol Pathol. 30:460–465. 2011.
|
|
8
|
Lee SH, Kang MS, Lee GS and Chung WY:
Refractory hypertension and isosexual pseudoprecocious puberty
associated with renin-secreting ovarian steroid cell tumor in a
girl. J Korean Med Sci. 26:836–838. 2011.
|
|
9
|
Sawathiparnich P, Sitthinamsuwan P,
Sanpakit K, Laohapensang M and Chuangsuwanich T: Cushing’s syndrome
caused by an ACTH-producing ovarian steroid cell tumor, NOS, in a
prepubertal girl. Endocrine. 35:132–135. 2009.
|
|
10
|
Elhadd TA, Connolly V, Cruickshank D and
Kelly WF: An ovarian lipid cell tumour causing virilization and
Cushing’s syndrome. Clin Endocrinol (Oxf ). 44:723–725. 1996.
|
|
11
|
Luk WT, Lee N, Chang TC and Chu KK: Lipid
cell tumor of the ovary associated with endometrial adenocarcinoma
- a case report. Changgeng Yi Xue Za Zhi. 12:244–248. 1989.
|
|
12
|
Rabban JT and Zaloudek CJ: A practical
approach to immunohistochemical diagnosis of ovarian germ cell
tumours and sex cord-stromal tumours. Histopathology. 62:71–88.
2013.
|
|
13
|
Zhao C, Vinh TN, McManus K, Dabbs D,
Barner R and Vang R: Identification of the most sensitive and
robust immunohistochemical markers in different categories of
ovarian sex cord-stromal tumors. Am J Surg Pathol. 33:354–366.
2009.
|
|
14
|
Amneus MW and Natarajan S: Pathologic quiz
case: a rare tumor of the ovary. Arch Pathol Lab Med. 127:890–892.
2003.
|
|
15
|
Murhekar K, Louis R and Majhi U: A rare
occurrence of a steroid cell tumor of the pelvic mesentery: a case
report. J Med Case Rep. 5:5172011.
|