Introduction
Choriocarcinoma, which is a gestational
trophoblastic disease, is a highly malignant trophoblastic tumor.
It arises almost exclusively in the placenta of pregnant women and
may occur even after a normal pregnancy (1). The embryonic trophocyte loses its
original structure, and the abnormal tissue invades the muscular
layer of uterus and then further spreads to other organs through
the venous and lymphatic systems (2).
Transforming growth factor β (TGF-β), which belongs
to a growth factor super-family, is a potent regulator of tumor
growth (3). TGF-β is a
multifunctional polypeptide cytokine and it regulates a variety of
cellular processes, such as cell cycle arrest, differentiation,
proliferation, extracellular matrix (ECM) production, the promotion
of ECM formation and the suppression of immune response (4,5).
Previous studies have demonstrated that TGF-β1 has an inhibitory
effect at the first stage of tumorigenesis, but certain late-stage
tumor cells escape this cytostatic effect (6–8).
Compared with other tumor types, the role of the TGF-β/Smad
signaling pathway in the development and proliferation of placental
choriocarcinoma has rarely been investigated.
TGF-β exerts its biological function through the
TGF-β/Smad pathway, which initiates by binding to its serine and
threonine kinase receptors, the TGF-β receptor type II (TβRII) and
TβRI, on the cell membrane (9).
TβRII firstly phosphorylates and activates TβRI, then forms a
complex with TβRI. The receptor complex recruits and phosphorylates
the R-Smad proteins, Smad2/3, via phosphorylation at their
C-terminal serine residues (10).
Thus, the signal crosses the membrane to the inside of the cell.
Smad4 (Co-Smad) works as a mediator by transporting phosphorylated
R-Smad (Smad2 and Smad3) into the nucleus, where target genes are
processed (11,12). Any mutations of components in the
TGF-β signaling pathway contribute to the loss of TGF-β1 growth
control in cancer (13).
In addition to this Smad2/3 pathway, TGF-β has been
reported to activate other signaling molecules, such as
mitogen-activated protein kinases (MAPKs) (14). MAPKs consist of four distinct
groups: The extracellular signal-related kinases (ERKs), the c-jun
N-terminal kinases, the atypical MAPKs (ERK3, ERK5, and ERK8) and
the p38 MAPKs (15,16). The p38 pathway, like other MAPK
pathways, features a cascade of protein kinases, culminating in the
phosphorylation of p38 MAPK on specific threonine and tyrosine
residues (17). The phosphorylation
is mediated primarily by upstream MKK3 and MKK6. MKK3 and MKK6 in
turn are regulated by phosphorylation through upstream MAPK kinase
kinases (18,19). The p38 MAPK pathway is responsive to
environmental stresses (UV, ionizing radiation, oxidative stress
and FAS ligands) and inflammatory cytokines (20). Our previous study demonstrated that
blocking the TGF-β pathway by using a TGF-β receptor inhibitor
significantly reduces the expression levels of p38 and phospho-p38
in JEG-3 cells. Additionally, treatment of JEG-3 cells with a p38
MAPK inhibitor (SB 203580) attenuated TGF-β1-induced Smad3 protein
expression and suppressed the activation of Smad3 (21). These results suggested that there is
crosstalk between p38 MAPK and Smad3 through TGF-β signaling in
human choriocarcinoma.
TGF-β receptors are the gateways of the
intracellular signaling. The receptor complex is a central point
for protein interactions; post-translational modifications may have
key functions in the transduction of TGF-β signals (22). A previous study suggested that
inhibition of TβRI activity blocked TGF-β-induced MAPK activation
(23). According to a study by
Bandyopadhyay et al, differences in the level of TβRII
expression determined whether or not TGF-β activated or inhibited
ERK1/2, and TβRII alone was able to mediate TGF-β signaling to
ERK1/2 without participation of TβRI/Alk5 (5). TGF-β-mediated MAPK activation has been
reported to be associated with tyrosine phosphorylation of TGF-β
receptors (24). Therefore, the
present study aimed to investigate whether there is an association
between TGF-β receptors and p38 MAPK in choriocarcinoma. The study
focused on the interaction between the p38 MAPK signaling pathway
and TGF-β receptors through TGF-β1 stimulation in human
choriocarcinoma cells.
Materials and methods
Materials
The human placental choriocarcinoma JEG-3 cell line
was obtained from the State Key Laboratory of Reproductive Biology,
Institute of Zoology, Chinese Academy of Sciences (Beijing, China).
The study was approved by the ethics committee of the Natural
Science Foundation of Hebei Province and the Education Department
of Hebei Province, Hebei, China.
JEG-3 cell culture
JEG-3 cells were grown in an incubator, with 5%
CO2 at 37°C, in RPMI-1640 supplemented with 10% fetal
bovine serum (FBS; Hangzhou Sijiqing Biological Engineering
Materials Co., Ltd, Hangzhou, China), 100 mM sodium pyruvate, 200
mM glutamine, 100 μg/ml streptomycin and 100 U/ml penicillin. When
the cells reached ~80% confluency, they were subcultured with 0.25%
trypsin and 0.02% EDTA.
MTT assay
After reaching 80% confluency, cells were
trypsinized and cultured in 96-well plates with an initial
concentration of 3×104/ml in RPMI-1640 containing 10%
FBS per well. After 24 h culturing, the medium was changed to
RPMI-1640 without FBS, and cells were cultured for a further 12 h
to ensure cell synchronization. A total of 42 wells were divided
into seven groups as follows: Blank, control, and 1-, 2-, 6-, 12-
and 24-h groups. TGF-β1 (PeproTech, Inc., Rocky Hill, NJ, USA), at
a concentration of 5 ng/ml, was added to all wells of the plate,
with the exception of those for the blank and control groups. Cells
in the control group were cultured with RPMI-1640, while the wells
for the blank group were filled with phosphate-buffed saline only.
Each specimen was prepared in four replicates. After treatment with
TGF-β1 for 1, 2, 6, 12 and 24 h, the medium was removed from wells
in the 1-, 2-, 6-, 12- and 24-h groups, respectively. Next, 180 μl
RPMI-1640 supplemented with 20 μl of 5 ng/ml MTT (Sigma-Aldrich,
St. Louis, MO, USA) in phosphate buffered saline was added to each
of these wells, and the plate was incubated at 37°C for 4 h.
Following the incubation, the medium was removed and 150 μl of
dimethyl sulfoxide (Sigma-Aldrich) per dish was added. Following
agitation for 10 min at room temperature, the absorbance of each
group was assayed at 490 nm with an enzyme-linked immunosorbent
assay plate reader (Multiskan MK3; Thermo Labsystems Oy, Helsinki,
Finland).
Reverse transcription quantitative
real-time polymerase chain reaction (RT-qPCR)
JEG-3 cells were incubated in six-well plates with
an initial concentration of 5×104 cells/ml for 48 h.
Wells were divided into six groups as follows: Control, TGF-β1,
1-μM SB203580, 3-μM SB203580, 1-μM LY364947 and 3-μM LY364947
groups. Cells in the control group were cultured with RPMI-1640
only, while the cells in the TGF-β1 groups were treated with 5
ng/ml TGF-β1 and incubate for 2 h. When the cells reached ~80%
confluency, they were pretreated with the appropriate concentration
of TGF-β1 receptor inhibitor (LY36494; Sigma-Aldrich) and p38 MAPK
inhibitor (SB203580; Sigma-Aldrich), and cultured for 2 h. The
cells were cultured for 2 h as according to the MTT results
(Fig. 1), no obvious effect on
proliferation was identified with the treatment of TGF-β1 for 2 h,
ensuring that the expression changes of the TGF-β receptors and
Smad3 were not affected by changes in cell proliferation levels.
Subsequently, 5 ng/ml TGF-β1 (PeproTech, Inc.) was added to each
well, with the exception of those in the control group, and
incubation was continued for 2 h. Total RNA extraction of JEG-3
cells from each group was performed with TRIzol reagent (Invitrogen
Life Technologies, Carlsbad, CA, USA). RNA (1 μg) was reverse
transcribed using an M-MLV First-Strand cDNA Synthesis kit
(Invitrogen Life Technologies) and random oligodeoxynucleotide
primers.
qPCR amplifications were performed using SYBR premix
(Invitrogen Life Technologies) for TβRI, TβRII and Smad3. β-actin
mRNA was employed as an internal control. The primer sequences used
are listed in Table I. The cycling
conditions were as follows: 40 cycles of 95°C for 30 sec, 95°C for
5 sec, 95°C for 30 sec and 72°C for 30 sec, followed by 95°C for 1
min, 95°C for 30 sec and 75°C for 30 sec. The obtained results of
the mRNA copy number were recalculated per 1 μg of total RNA. Each
run was completed using melting curve analysis to confirm the
specificity of the amplification and the absence of the primer
dimers. All data were quantified by the use of the comparative
cycle threshold method, normalized to β-actin.
 | Table IPCR primers used in reaction. |
Table I
PCR primers used in reaction.
| Target | Primer | Sequence (5′-3′) | Length of amplicon
(bp) | Tm (°C) |
|---|
| TβRI | Forward |
GCAGTAAGACATGATTCAGCCACAG | 190 | 58.1 |
| Reverse |
CAATGGAACATCGTCGAGCAA | | |
| TβRII | Forward |
GAAATTCCCAGCTTCTGGCTCA | 143 | 57.2 |
| Reverse |
CTGTCCAGATGCTCCAGCTCAC | | |
| Smad3 |
| Forward |
CCAGGGCTTTGAGGCTGTCTA | 143 | 59.2 |
| Reverse |
GCAAAGGCCCATTCAGGTG | | |
| β-actin | Forward |
TGGCACCCAGCACAATGAA | 186 | 56.0 |
| Reverse |
CTAAGTCATAGTCCGCCTAGAAGCA | | |
Statistical analysis
All data are expressed as the mean ± standard
deviation. One-way analysis of variance was used to compare
differences among groups. The Student-Newman-Keuls test was
performed to assess the differences between pairs of groups.
Differences were considered statistically significant at values of
P<0.05. All statistical analysis was performed using SPSS 19.0
software (SPSS, Inc., Chicago, IL, USA).
Results
Effect of TGF-β on choriocarcinoma
cellular proliferation
JEG-3 cells were treated with the same concentration
of TGF-β1 at 5 ng/ml, and then cellular proliferation was
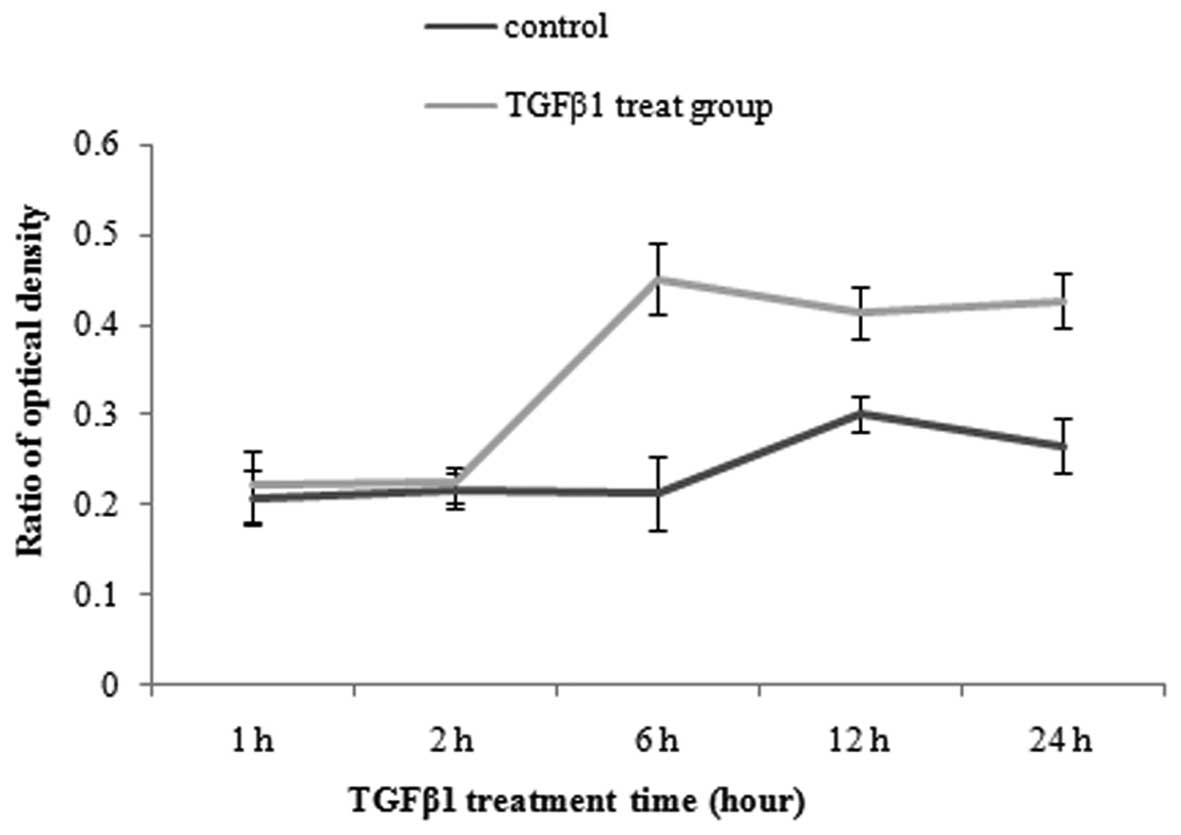
determined by MTT assay at different time points. As shown in
Fig. 1, JEG-3 cells were not
significantly affected by the presence of TGF-β at 1 and 2 h,
compared with control group. This suggested that TGF-β did not
promote the proliferation of choriocarcinoma before 2 h. By the
time of 6 h, TGF-β1 exhibited a marked effect on JEG-3 cell
proliferation. Additionally, the viability of JEG-3 cell
proliferation was gradually decreased by TGF-β1 at 12 and 24
h(Fig. 1).
Effect of p38 MAPK inhibition on the
transcriptional levels of Smad3 in the JEG-3 cell line
To examine the roles of Smad3 in the TGF-β pathway
and p38 MAPK, the transcriptional levels of Smad3 were examined
using the p38 MAPK inhibitor (SB203580) and TGF-β1 receptor
inhibitor (LY36494). Following this, 5 ng/ml TGF-β1 was added into
each well, except those of the control group, and incubation was
continued for 2 h. Previous results from our laboratory indicated
that p38 MAPK inhibitors can attenuate TGF-β1-induced Smad3 protein
expression (21); thus; we further
examined the effect of p38 inhibitors on the transcriptional levels
of Smad3 in response to TGF-β1. Compared with the control group,
the mRNA expression levels of Smad3 were significantly elevated in
TGF-β1 group (P<0.05) (Fig. 2).
The results revealed that TGF-β1 promotes Smad3 transcription. With
an increasing concentration of inhibitors, the Smad3
transcriptional levels in the LY364947 and SB203580 groups
gradually reduced compared with the other two groups (P<0.05)
(Fig. 2).
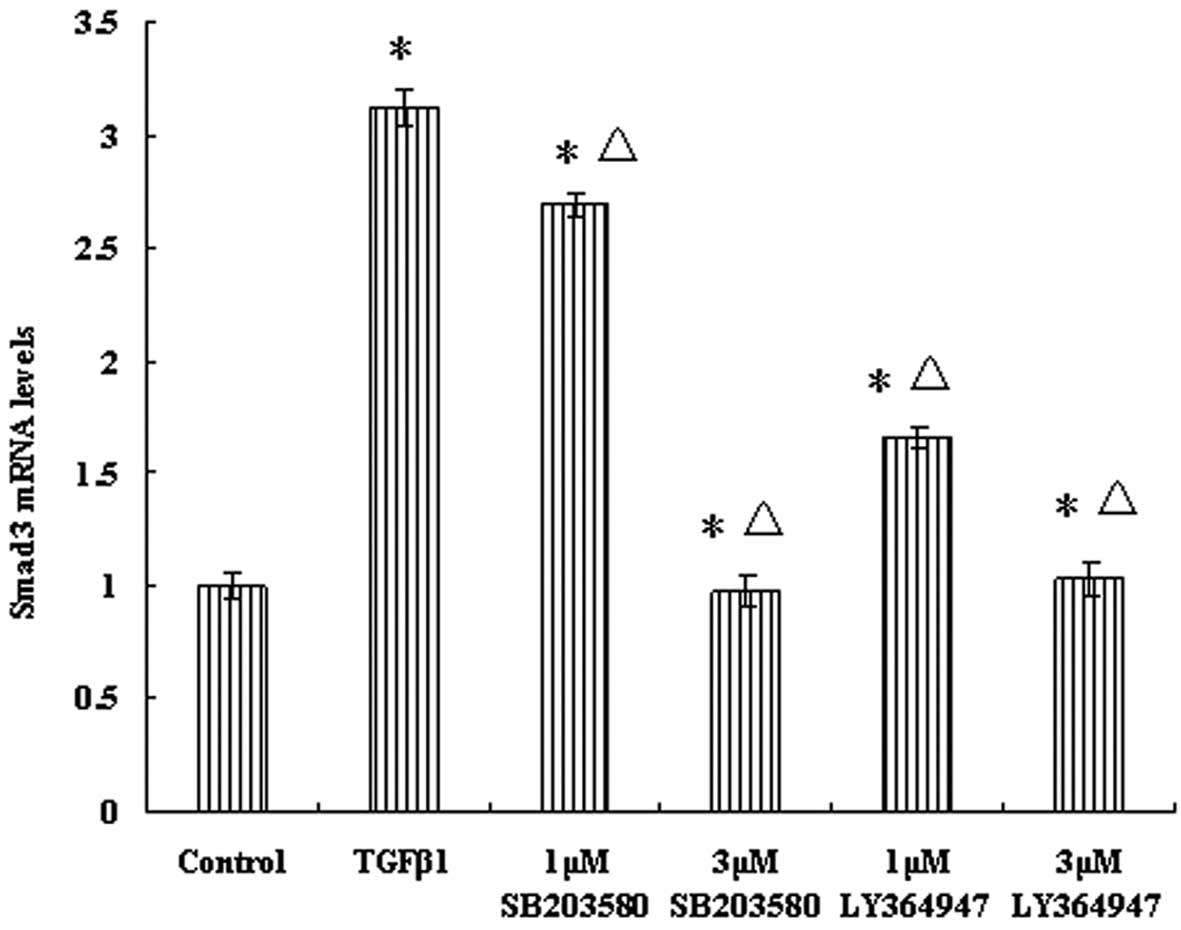 | Figure 2Effect of p38 MAPK and TGF-β1 receptor
inhibitors (SB203580 and LY36494, respectively) on the
transcriptional levels of Smad3 in the JEG-3 cell line. Cells were
divided into six groups: Control group, TGF-β1 group, 1-μM
SB203580, 3-μM SB203580, 1-μM LY364947 and 3-μM LY364947 groups.
Cells were pretreated with different concentrations of p38 MAPK and
TGF-β1 receptor inhibitors, and cultured for 2 h. Subsequently, 5
ng/ml TGF-β1 was added to cells in all groups except the control
group, and incubation was continued for 2 h. The mRNA expression
levels of Smad3 were determined by reverse transcription
quantitative real-time polymerase chain reaction. The data are
presented as the mean ± SD (P<0.05). The results are
representative of at least three independent experiments. MAPK,
mitogen-activated protein kinase; TGF-β1, transforming growth
factor β1. *P<0.05 vs. control group;
ΔP<0.01 vs. TGF-β1 group. |
Effect of p38 MAPK inhibition on TβRI and
TβRII transcriptional levels in the JEG-3 cell line
In order to examine the roles of TβRI and TβRII in
the regulation of p38 MAPK, the TβRI and TβRII transcriptional
levels were examined by blocking p38 MAPK using a p38 MAPK
inhibitor, SB203580. The mRNA expression levels of TβRI and TβRII
in the TGF-β1 group were both increased compared with those in the
control group (P<0.05). Pretreatment with LY36494, a TGF-β-1
inhibitor, resulted in significant decrease (P<0.05) in the mRNA
levels of TβRI, in a dose-dependent manner, compared with those of
the TGF-β1 group. In the groups treated with SB203580, the trend of
TβRI transcriptional levels was similar to that in the
LY36494-treated groups, which indicated that the p38 MAPK inhibitor
downregulates the TβRI transcriptional level (P<0.05) (Fig. 3).
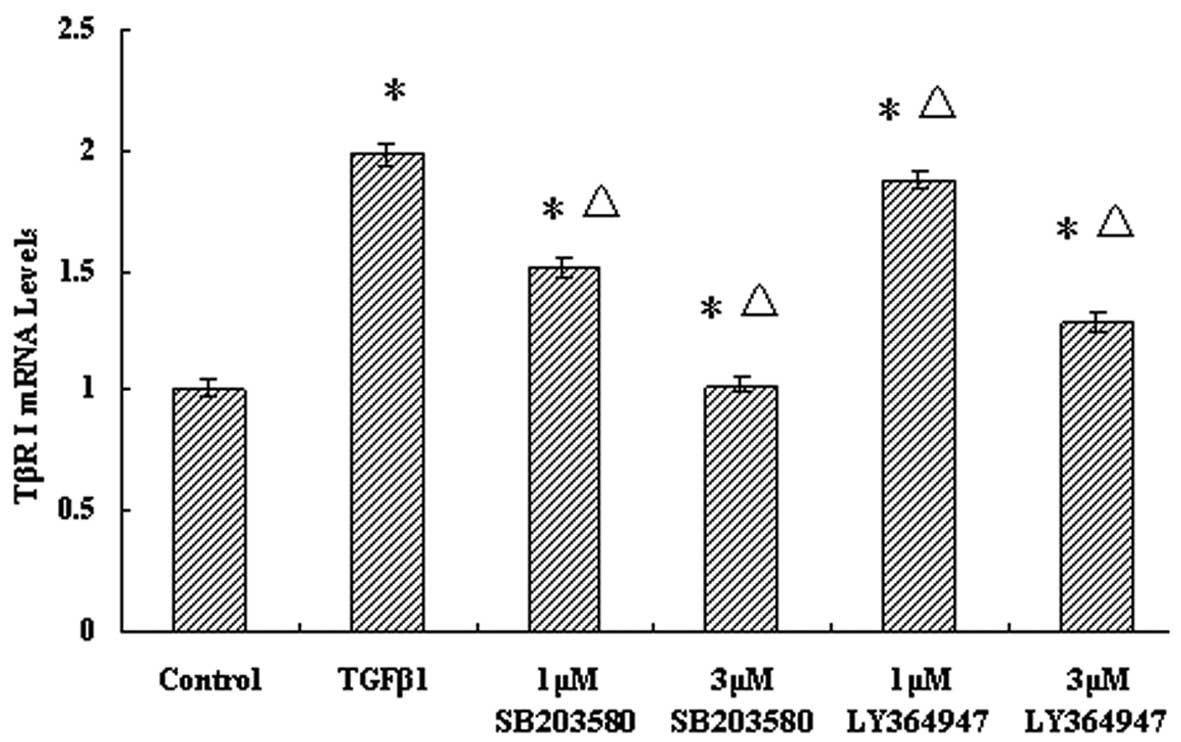 | Figure 3Effect of p38 MAPK inhibition on TβRI
transcriptional levels in the JEG-3 cell line. Cells were divided
into six groups: Control group, TGF-β1 group, 1-μM SB203580, 3-μM
SB203580, 1-μM LY364947 and 3-μM LY364947 groups. Cells were
pretreated with different concentrations of p38 MAPK and TGF-β1
receptor inhibitors, and cultured for 2 h. Next, 5 ng/ml TGF-β1 was
added to cells in all groups except the control group, and
incubation was continued for 2 h. The mRNA expression levels of
TβRI were determined by reverse transcription quantitative
real-time polymerase chain reaction. Results are presented as the
mean ± SD from at least three independent experiments (P<0.05).
MAPK, mitogen-activated protein kinase; TGF-β1, transforming growth
factor β1; TβRI, TGF-β receptor type I. *P<0.05 vs.
control group; ΔP<0.01 vs. TGF-β1 group. |
The TβRII mRNA expression results were similar to
those for TβRI (Fig. 4). The mRNA
levels of TβRII were reduced in both the LY36494- and
SB203580-treated groups. As the concentration of inhibitor
increased, the mRNA levels of TβRII gradually decreased (Fig. 4). The results revealed that blocking
the p38 MAPK pathway can modulate the TβRII transcriptional level
induced by TGF-β1 in JEG-3 cells.
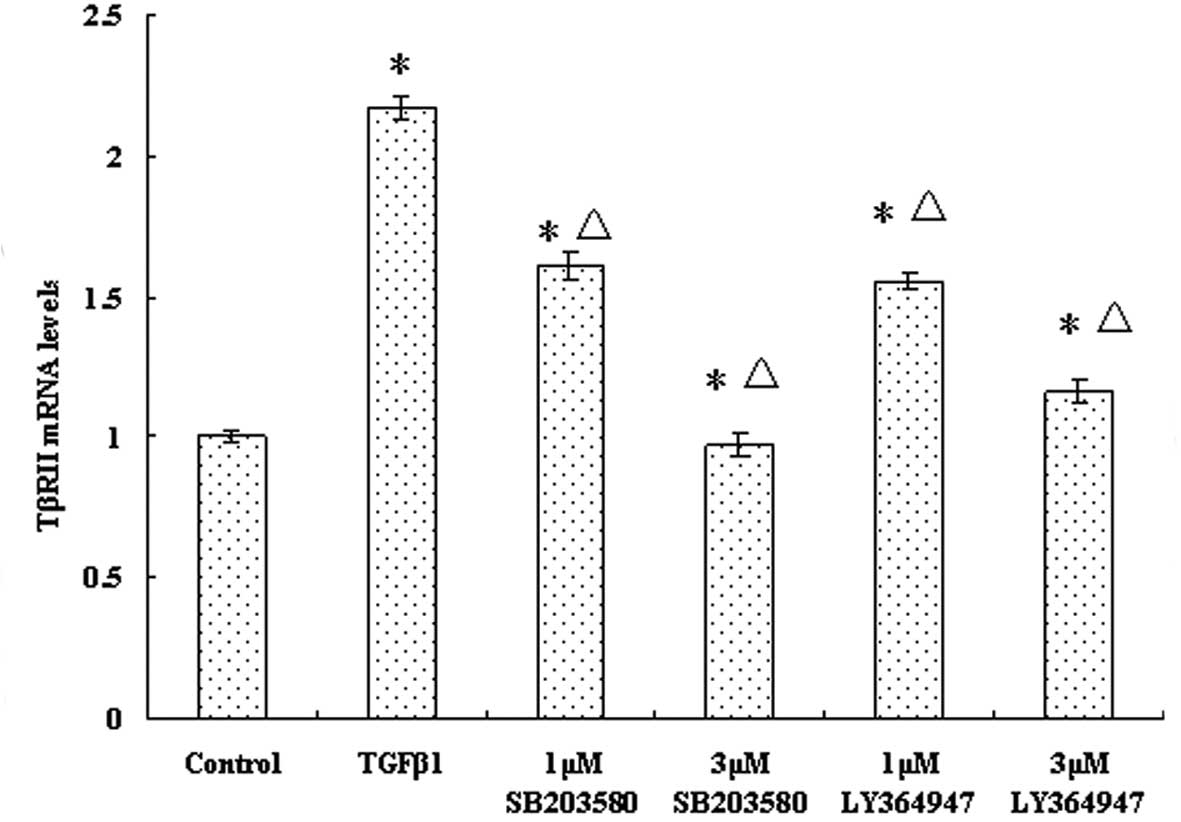 | Figure 4Effect of p38 MAPK inhibition on TβRII
transcriptional levels in the JEG-3 cell line. Cells were divided
into six groups: Control group, TGF-β1 group, 1-μM SB203580, 3-μM
SB203580, 1-μM LY364947 and 3-μM LY364947 groups. Cells were
pretreated with different concentrations of p38 MAPK and TGF-β1
receptor inhibitors, and cultured for 2 h,. Following this, 5 ng/ml
TGF-β1 was added to cells in all groups except the control group,
and incubation was continued for 2 h. The mRNA expression levels of
TβRII were determined by reverse transcription quantitative
real-time polymerase chain reaction. Results are presented as the
mean ± SD from at least three independent experiments (P<0.05).
MAPK, mitogen-activated protein kinase; TGF-β1, transforming growth
factor β1; TβRI, TGF-β receptor type II. *P<0.05 vs.
control group; ΔP<0.01 vs. TGF-β1 group. |
Discussion
Choriocarcinoma is a fast-growing and highly
malignant tumor. Although the availability of chemotherapy has made
the prognosis highly favorable (25), numerous patients cannot tolerate the
toxicity and side effects. Therefore, there is an urgent
requirement to explore the mechanism of development of
choriocarcinoma for new molecular target therapy. TGF-β appears to
be a key factor in the development of choriocarcinoma. Any
mutations that occur in the components of the TGF-β/Smad signaling
pathway can cause the formation of tumors. For example, TβRII has
been found to be overexpressed in a bladder cancer cell line,
concomitant with point mutations, particularly the Glu269 to Lys
mutation (G to A) (26). The
mutations of TβRII, which promoted tumor cell growth, did not
affect Smad2/3 binding.
In the process of tumor transformation, TGF-β plays
two conflicting roles of a tumor suppressor and a tumor promoter.
In the early stage of cancer development, TGF-β is
anti-proliferative or works as a tumor suppressor, whereas in the
late stage it functions as a tumor promoter, involved in metastasis
(27–29). In the present study, MTT assay was
used to examine the effects of TGF-β1 on JEG-3 cell proliferation.
The viability of JEG-3 cell proliferation was tested at 1, 2, 6, 12
and 24 h, and the results demonstrated that TGF-β-1 was able to
promote the proliferation of choriocarcinoma. It was also found
that, following treatment with TGF-β1, the transcriptional levels
of Smad3, TβRI and TβRII were all elevated compared with the
control group (Figs. 2–4). This suggested that TGF-β1 can also
activate the TGF-β/Smad signaling pathway in choriocarcinoma and
the extracellular signal is successfully transmitted into
cytoplasm.
TGF-β not only transmit its signal via Smad
proteins, but can also activate other signaling molecules such as
p38 MAPK (30). A study by Daroqui
et al demonstrated that p38 MAPK and MEK contribute to TGF-β
stimulation of cell motility and invasion by analyzing signal
transduction mediators. Additionally, both the MAPK-dependent and
-independent pathways are necessary for TGF-β-induced effects
(31). According to a study by Gui
et al, the prolonged and sustained activation of the p38
MAPK pathway requires Smad signaling, which is observed in
hepatocytes, osteoblasts and pancreatic carcinoma cells. Smad
activation induces the expression of GADD45β, an upstream activator
of MKK4, and thus promotes the prolonged activation of p38 MAPK
(32).
The TGF-β receptors are gateways to the TGF-β/Smad
pathway, which aids the extracellular signal to cross the membrane
into the cytoplasm (33). According
to a study by Huang and Chen, TβRII alone is able to mediate TGF-β
signaling to ERK1/2, and differences in the level of TβRII
expression determine whether or not TGF-β activates or inhibits
ERK1/2 (6). It has been suggested
that inhibiting the activity of TGF-β receptor blocks TGF-β-induced
MAPK activation. Dalliher et al demonstrated that TGFβII
mutants in breast cancer cells completely abrogated p38 MAPK
activation induced by TGF-β, but failed to affect TGF-β stimulation
of Smad2/3 (34). A study by
Ohshima showed that mutant TGFβI did not affect activation of the
Smad pathway, but retained signaling via the MAP kinase pathway
(35). The study also suggested
that TGF-β receptor-activated p38 is involved in TGF-β-induced
apoptosis but not growth arrest in mouse mammary gland epithelial
cells. Based on these observations, the current study attempted to
investigate the effect of crosstalk between TGF-β/Smad and p38 MAPK
signaling on the expression of TGF-β receptors and Smad3 in
choriocarcinoma (JEG-3) cells. Cells were pretreated with different
concentrations of TGF-β1 receptor inhibitor and p38 MAPK inhibitor,
respectively, and incubated for 2 h. Subsequently, 5 ng/ml TGF-β1
was added to the cells and cultured for 2 h. According to the MTT
assay results, no obvious effect on proliferation was observed with
treatment of TGF-β1 for 2 h, so 2 h was selected as the duration of
TGF-β1 treatment to ensure that the expression changes in TGF-β
receptors and Smad3 were not affected by changes in cell
proliferation levels. The expression of TβRI, TβRII and Smad3 was
reduced in a dose-dependent manner in the 1- and 3-μM LY364947
groups, compared with the control group. This indicated that the
TGF-β receptor inhibitor was able to inhibit the TGF-β/Smad
signaling pathway to an extent. Additionally, in the 1- and 3 μM
SB203580 groups, the trends of TβRI, TβRII and Smad3
transcriptional levels were consistent with those in the LY364947
treatment groups. p38 MAPK inhibitors can attenuate TGF-β1-induced
TβRI, TβRII and Smad3 transcriptional levels. These results
revealed that the TGF-β/Smad signaling pathway may be affected by
the p38 MAPK pathway, and blockade of the p38 MAPK pathway can
downregulate the activated TβRI, TβRII and Smad3. This suggests
that diverse biological responses regulated by TGF-β are mediated
not only via Smad proteins, but also by different downstream
R-Smad-independent signaling pathways. Any changes that occur in
these downstream signaling pathways may have an effect on the
genesis or progression of choriocarcinoma. Further clarification of
the mechanisms of the crosstalk between the TGF-β and p38 MAPK
pathways in cell models may offer novel breakthroughs and potential
applications in the field of therapeutic approaches.
References
|
1
|
Alifrangis C and Seckl MJ: Genetics of
gestational trophoblastic neoplasia: an update for the clinician.
Future Oncol. 6:1915–1923. 2010.
|
|
2
|
Braunstein GD: Endocrine changes in
pregnancy. Williams Textbook of Endocrinology. 12th edition. Melmed
S, Polonsky KS, Larsen PR and Kronenberg HM: Saunders Elsevier;
Philadelphia, Pa: pp. 221–230. 2011
|
|
3
|
Lin KW, Yakymovych I, Jia M, Yakymovych M
and Souchelnytskyi S: Phosphorylation of eEF1A1 at Ser300 by TβR-I
results in inhibition of mRNA translation. Curr Biol. 20:1615–1625.
2010.
|
|
4
|
Sawai H, Yasuda A, Ochi N, et al: TGF-β
regulates invasive behavior of human pancreatic cancer cells by
controlling Smad expression. Arch Med Sci. 3:185–191. 2007.
|
|
5
|
Bandyopadhyay B, Han A, Dai J, et al:
TbetaRI/Alk5-independent TbetaRII signaling to ERK1/2 in human skin
cells according to distinct levels of TbetaRII expression. J Cell
Sci. 124(Pt 1): 19–24. 2011.
|
|
6
|
Huang F and Chen YG: Regulation of TGF-β
receptor activity. Cell Bios. 2:9–19. 2012.
|
|
7
|
Calone I and Souchelnytskyi S: Inhibition
of TGFβ signaling and its implications in anticancer treatments.
Exp Oncol. 34:9–16. 2012.
|
|
8
|
Galliher-Beckley AJ and Schiemann WP: Grb2
binding to Tyr284 in TbetaR-II is essential for mammary tumor
growth and metastasis stimulated by TGF-beta. Carcinogenesis.
29:244–251. 2008.
|
|
9
|
Gao Jin and Laurence JW: Potential
regeneration capacity of periodontal ligament with autocrine
production of transforming growth factor-beta 1 and its receptors.
Int J Dent Clin. 3:5–8. 2011.
|
|
10
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003.
|
|
11
|
Lu L, Wang J, Zhang F, et al: Role of SMAD
and non-SMAD signals in the development of Th17 and regulatory T
cells. J Immunol. 84:4295–4306. 2010.
|
|
12
|
Konrad L, Scheiber JA, Völck-Badouin E, et
al: Alternative splicing of TGF-betas and their high-affinity
receptors T beta RI, T beta RII and T beta RIII (betaglycan) reveal
new variants in human prostatic cells. BMC Genomics. 8:3182007.
|
|
13
|
Shinto O, Yashiro M, Kawajiri H, Shimizu
K, Shimizu T, Miwa A and Hirakawa K: Inhibitory effect of a TGFbeta
receptor type-I inhibitor, Ki26894, on invasiveness of scirrhous
gastric cancer cells. Br J Cancer. 102:844–851. 2010.
|
|
14
|
Bakkebø M, Huse K, Hilden VI, Smeland EB
and Oksvold MP: TGF-β-induced growth inhibition in B-cell lymphoma
correlates with Smad1/5 signalling and constitutively active p38
MAPK. BMC Immunol. 11:572010.
|
|
15
|
Corrêa SA and Eales KL: The role of p38
MAPK and its substrates in neuronal plasticity and
neurodegenerative disease. J Signal Transduct. 2012:6490792012.
|
|
16
|
Whitmarsh AJ: A central role for p38 MAPK
in the early transcriptional response to stress. BMC Biol.
8:472010.
|
|
17
|
Wood CD, Thornton TM, Sabio G, Davis RA
and Rincon M: Nuclear localization of p38 MAPK in response to DNA
damage. Int J Biol Sci. 5:428–437. 2009.
|
|
18
|
Gangwal RP, Bhadauriya A, Damre MV, Dhoke
GV and Sangamwar AT: p38 mitogen-activated protein kinase
inhibitors: a review on pharmacophore mapping and QSAR studies.
Curr Top Med Chem. 13:1015–1035. 2013.
|
|
19
|
Krementsov DN, Thornton TM, Teuscher C and
Rincon M: The emerging role of p38 mitogen-activated protein kinase
in multiple sclerosis and its models. Mol Cell Biol. 33:3728–3734.
2013.
|
|
20
|
Mavropoulos A, Orfanidou T, Liaskos C, et
al: p38 MAPK signaling in pemphigus: implications for skin
autoimmunity. Autoimmune Dis. 2013:7285292013.
|
|
21
|
Xu Q, Tan Y, Zhang K and Li Y: Crosstalk
between p38 and Smad3 through TGF-β1 in JEG-3 choriocarcinoma
cells. Int J Oncol. 43:1187–1193. 2013.
|
|
22
|
Mincione G, Di Marcantonio MC, Artese L,
et al: Loss of expression of TGF-beta1, TbetaRI and TbetaRII
correlates with differentiation in humanoral squamous cell
carcinomas. Int J Oncol. 32:323–331. 2008.
|
|
23
|
Zhang YE: Non-Smad pathways in TGF-β
signaling. Cell Res. 19:128–139. 2009.
|
|
24
|
Chapnick DA, Warner L, Bernet J, Rao T and
Liu X: Partners in crime: TGFβ and MAPK pathways in cancer
progression. Cell Biosci. 1:422011.
|
|
25
|
Liu X, Gu W and Li X: HLA-G regulates the
invasive properties of JEG-3 choriocarcinoma cells by controlling
STAT3 activation. Placenta. 34:1044–1052. 2013.
|
|
26
|
Bian J, Li B, Zeng X, et al: Mutation of
TGF-β receptor II facilitates human bladder cancer progression
through altered TGF-β1 signaling pathway. Int J Oncol.
43:1549–1559. 2013.
|
|
27
|
Zhang P, Nakatsukasa H, Tu E and Kasagi S:
PARP-1 regulates expression of TGF-β receptors in T cells. Blood.
122:2224–2232. 2013.
|
|
28
|
Braunger BM, Pielmeier S, Demmer C, et al:
TGF-β signaling protects retinal neurons from programmed cell death
during the development of the mammalian eye. J Neurosci.
33:14246–14258. 2013.
|
|
29
|
Schedlich LJ, Yenson VM and Baxter RC:
TGF-β-induced expression of IGFBP-3 regulates IGF1R signaling in
human osteosarcoma cells. Mol Cell Endocrinol. 377:56–64. 2013.
|
|
30
|
Bhowmick NA, Zent R, Ghiassi M, McDonnell
M and Moses HL: Integrin beta 1 signaling is necessary for
transforming growth factor-beta activation of p38 MAPKand
epithelial plasticity. J Biol Chem. 276:46707–46713. 2001.
|
|
31
|
Daroqui MC, Vazquez P, Bal de Kier Joffé
E, Bakin AV and Puricelli LI: TGF-β autocrine pathway and MAPK
signaling promote cell invasiveness and in vivo
mammaryadenocarcinoma tumor progression. Oncol Rep. 28:567–575.
2012.
|
|
32
|
Gui T, Sun Y, Shimokado A and Muragaki Y:
The roles of mitogen-activated protein kinase pathways in
TGF-β-induced epithelial-mesenchymal transition. J Signal
Transduct. 2012:2892432012.
|
|
33
|
Jachec W, Foremny A, Domal-Kwiatkowska D,
et al: Expression of TGF-beta1 and its receptor genes (TbetaR I,
TbetaR II, and TbetaR III-betaglycan) in peripheral blood
leucocytes in patients with idiopathic pulmonary arterial
hypertension and Eisenmenger’s syndrome. Int J Mol Med. 21:99–107.
2008.
|
|
34
|
Galliher AJ and Schiemann WP: Src
phosphorylates Tyr284 in TGF-beta type II receptor and regulates
TGF-beta stimulation of p38 MAPK during breast cancer cell
proliferation and invasion. Cancer Res. 67:3752–3758. 2007.
|
|
35
|
Ohshima T and Shimotohno K: Transforming
growth factor-beta-mediated signaling via the p38 MAP kinase
pathway activates Smad-dependent transcription through SUMO-1
modification of Smad4. J Biol Chem. 278:50833–50842. 2003.
|