Introduction
Primary lymphoma of the bone (PLB) is an extranodal
lymphoma that arises from the medullary cavity and manifests as a
localized, solitary lesion, which represents ~3% of all primary
malignant bone tumors and 1% of all malignant lymphomas (1). PLB was first described by Oberling in
1928 (2) and is generally an
extremely rare condition. The cause of PLB is not well-known and
any part of the skeleton can be involved (3). The cell subtype of PLB varies and the
molecular features have not been well studied (4). Staging varies with different
diagnosing criteria at different times (5). Imaging features are usually
non-specific (6). As PLB is a
highly curable disease, it is important for it to be differentiated
from other causes of lytic bone lesions, such as carcinomas and
other primary bone tumors. The prognosis of PLB improves following
chemotherapy and radiotherapy. The present study reports one case
of PLB of the bone and a review of the literature with regard to
PLB to elucidate the clinical manifestation, imaging features,
staging, diagnosis and differential diagnosis, optimal treatment
and prognosis of this unique disease. Patient provided written
informed consent.
Case report
A 73-year-old female presented to the Internal
Department of Oncology, Shandong Cancer Hospital and Institute
(Jinan, China) with pain in the left hip that had persisted for two
months. Plain X-rays showed no abnormalities of the pelvic bones,
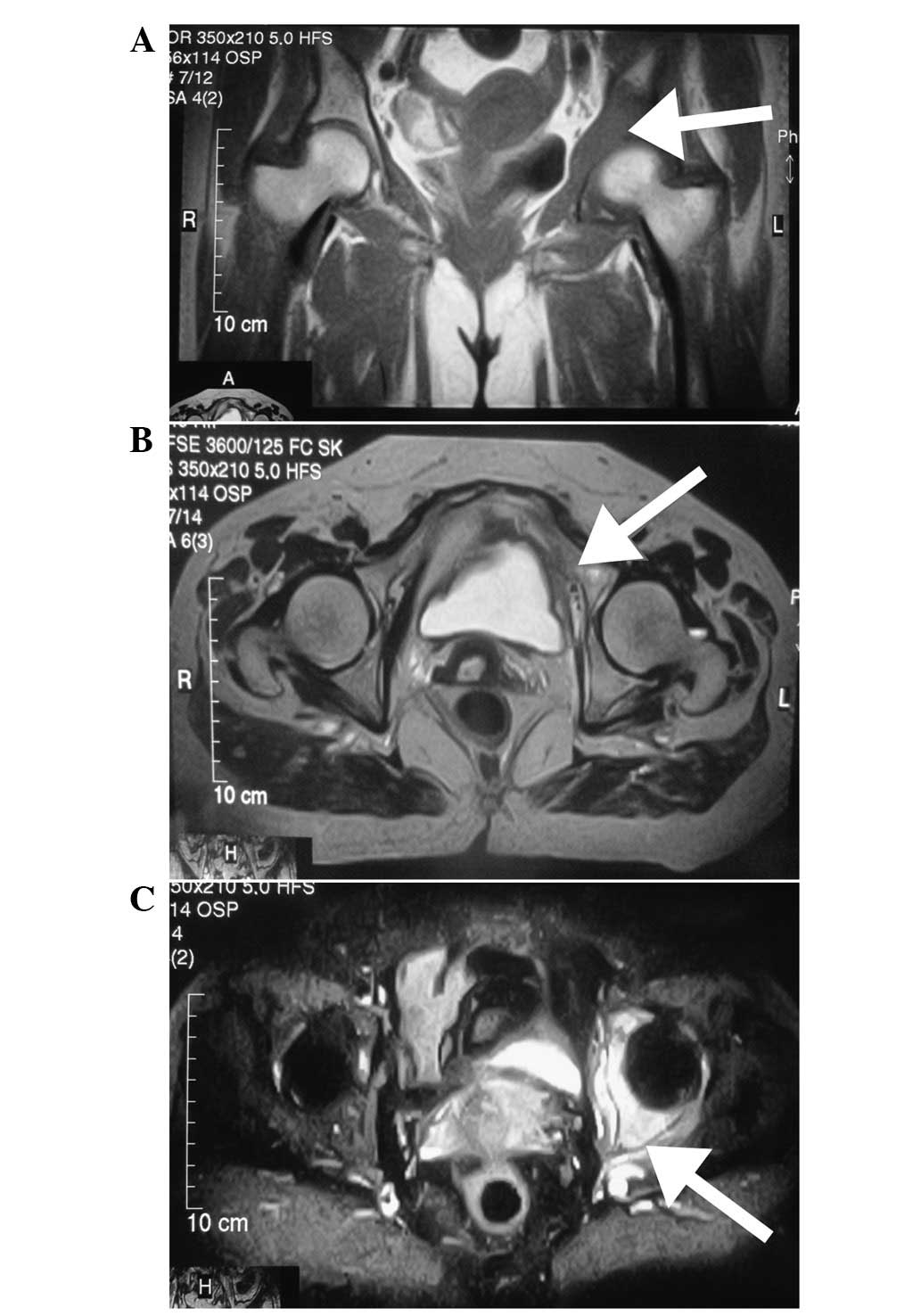
however, magnetic resonance imaging (MRI) of the left hip was
performed and showed abnormal signals involving the left
innominatum, with soft tissue formation. The signal intensity was
low on T1-weighted images (Fig.
1A), high or isointense on T2-weighted images (Fig. 1B) and hyperintense on short TI
inversion recovery (Fig. 1C).
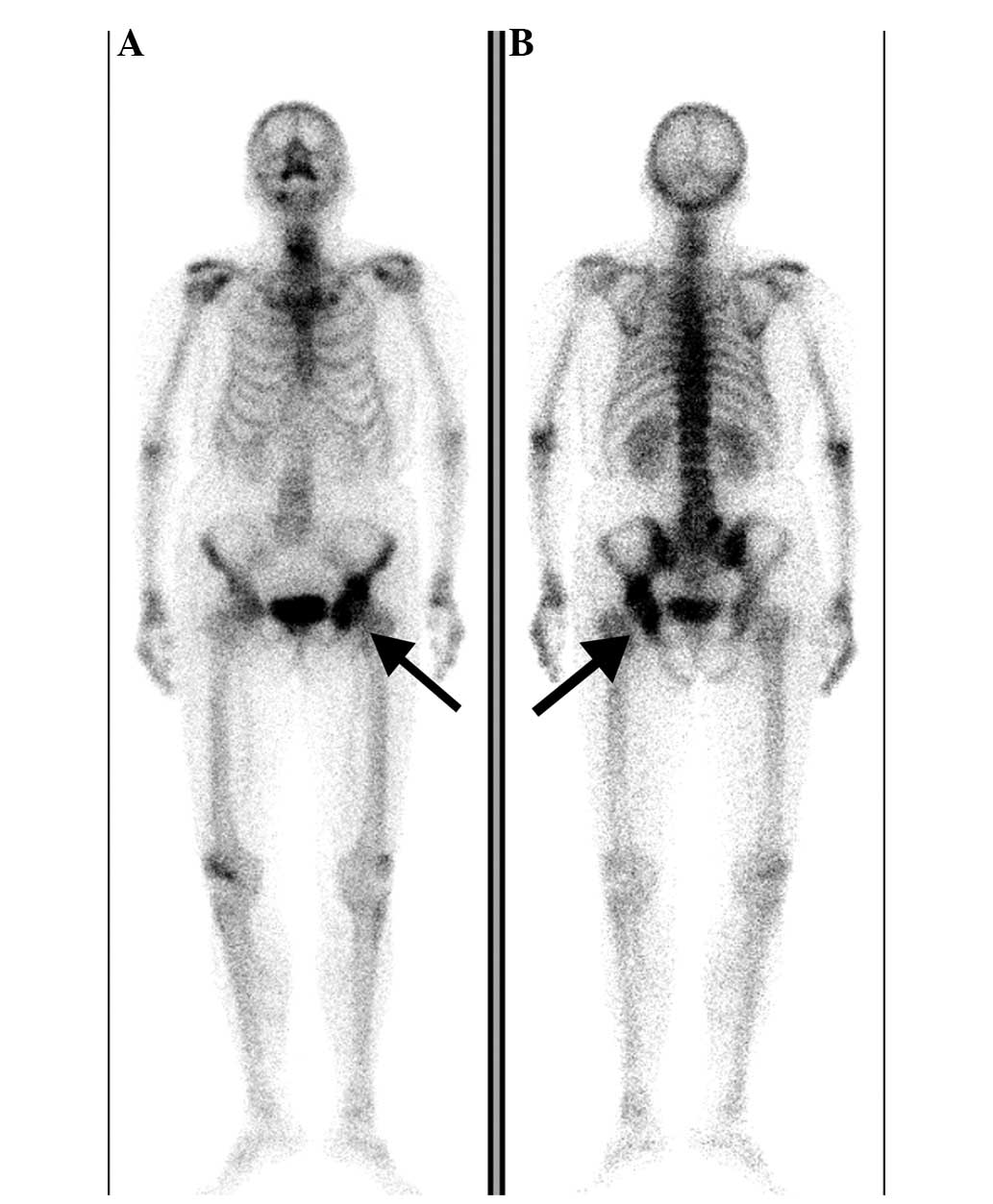
Technetium-99m (99mTc) radionuclide bone
scans were performed to rule out multiple bone lesions, and
increased tracer uptake was shown in the left innominatum,
including the ilium, acetabulum and ischium (Fig. 2).
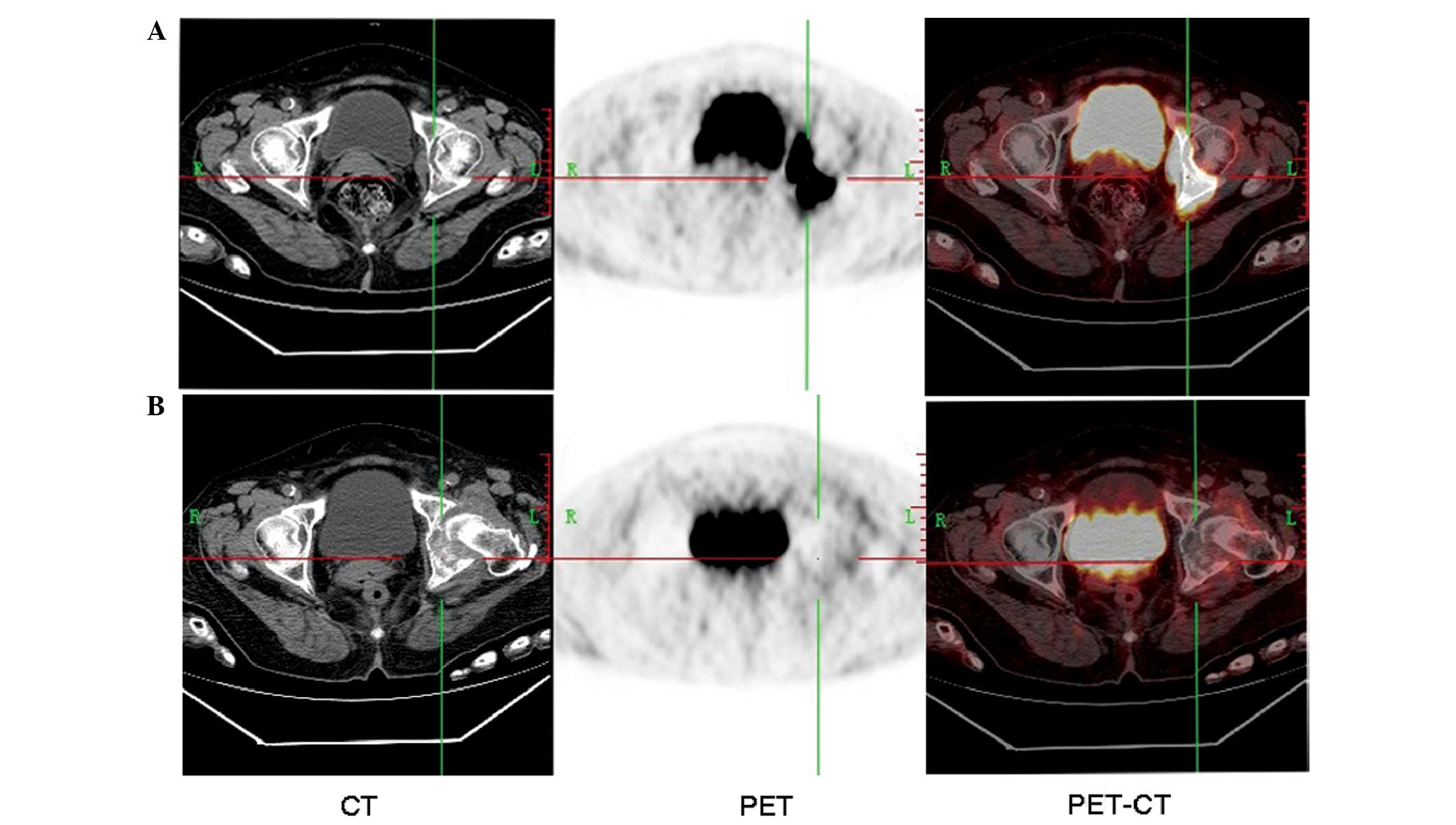
An 18F-fluorodeoxyglucose-positron
emission tomography-computed tomography (FDG-PET-CT) scan was
performed to identify the original site of the tumor. Abnormal
18F-FDG uptake was found in the left innominatum, with a
peak standardized uptake value of 60.7, lytic lesions and
soft-tissue lump formation ~7.8×4.5×8.8 cm in size (Fig. 3A).
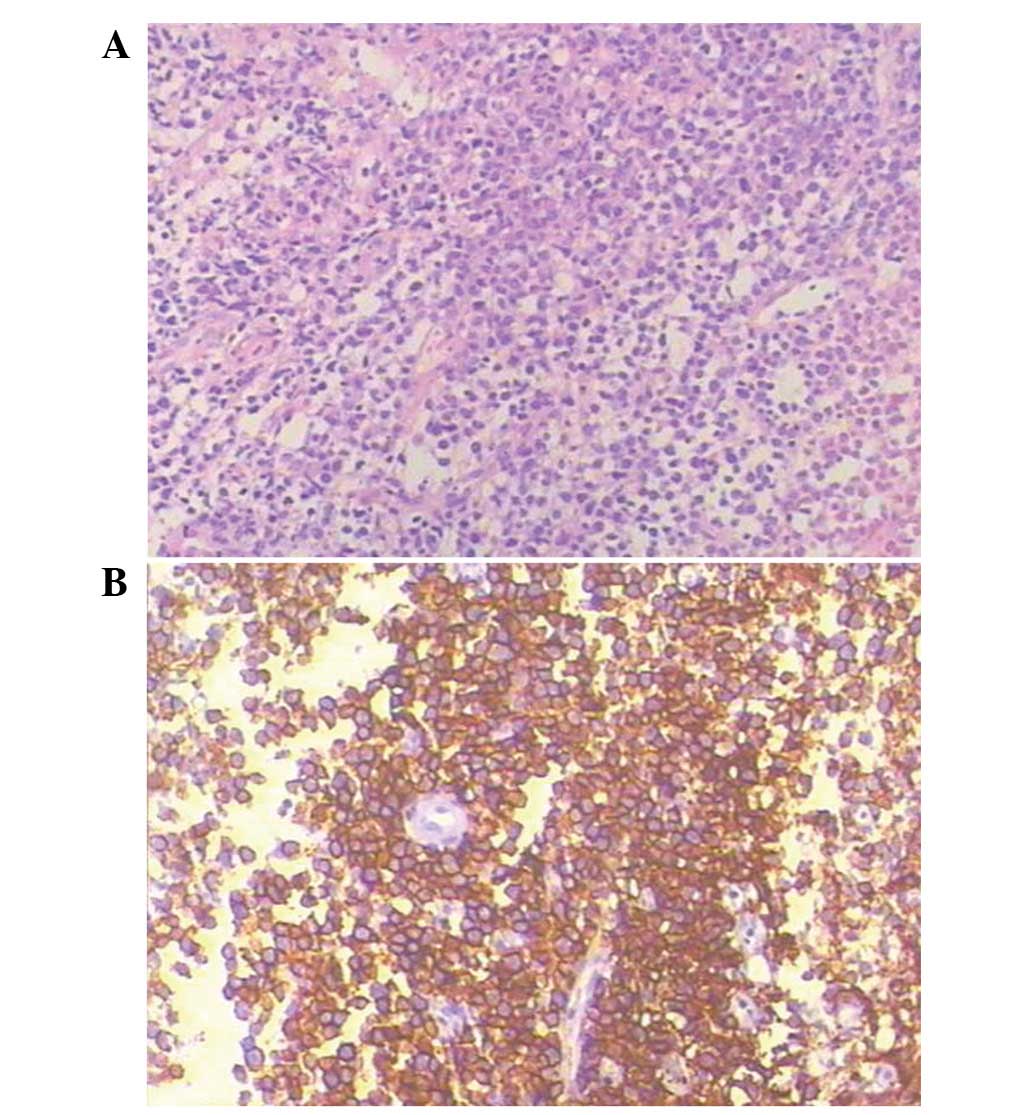
Next, a fine-needle aspiration biopsy of the lesion
was performed and a histopathological examination showed diffuse,
round tumor cells of approximately the same size.
Immunohistochemistry showed that the cell membrane was strongly
positive for cluster of differentiation (CD)20 and negative for
CD79α, CD3, CD78, CD138, cytokeratin (CK) and CK8/18, supporting a
B-cell origin (Fig. 4). Further
examinations, including bone marrow aspiration and biopsy, and CT
scans of the neck, chest and abdomen, were normal.
The patient was diagnosed with PLB of the left
innominatum, classified as stage I–E according to the Ann Arbor
system (5). The patient was treated
with 50 Gy radiation therapy in 25 fractions over five weeks,
followed by six cycles of CHOP regimen (1,000 mg cyclophosphamide,
80 mg epirubicine and 2 mg vincristine on day one, and 100 mg
prednisone on days one to five, every three weeks). Following the
third cycle of CHOP, the patient experience abnormal pain in the
left hip. The FDG-PET-CT examination was repeated, but no abnormal
FDG uptake was observed (Fig. 3B).
However, the neck of the left femur was broken, with displacement
of the distal section. At present, the patient is being regularly
followed up and has remained disease-free since the last treatment,
with the exception of the broken femoral neck.
Discussion
The cause of PLB is not well-known now, however,
viral infection, immunodeficiency, organ transplantation, Paget’s
disease of the bone and inherited factors have been identified as
possible causes in the process; although this has only been found
in retrospective studies (7). The
majority of PLB patients are >45 years of age and there is a
slight male preponderance, with a male to female ratio of 1.2:1.8
(5). Involvement of any region of
the skeleton is possible, however, a trend exists in favor of the
long bones with persistent bone marrow (3). The most commonly affected site is the
femur, which accounts for ~50%, with tumor cell infiltration along
the shaft of the bone longitudinally. The pelvis is the secdonary
affected site with a proportion of ~20%, while other sites include
the spine, ribs, mandible, scapula and proximal phalanx of the
thumb (8). PLB differs from
secondary lymphoma of the bone, where the axial bones are the most
common sites of presentation. Furthermore, certain clinical
characteristics of PLB in the Asian population differ from those in
Western populations, with the pelvis being the most commonly
involved site (52%) (9–11). Patients with PLB commonly present
with local bone pain, soft-tissue swelling, a mass or pathological
fracture, or hypercalcemic crisis (12). ‘B’ symptoms are rare and are only
observed in stage IV patients (13).
The staging of PLB varies with the diagnostic
criteria as this changes over time. According to the Ann Arbor
system (3), currently the most
widely accepted staging system, PLB is divided into the following
four stages: i) Stage I, single lesion in the bone with or without
soft-tissue infiltration; ii) stage II, more than two lesions
beside one side of the diaphragm, or a single lesion in the bone
with soft-tissue infiltration; iii) stage III, lesions beside two
sides of the diaphragm; and iv) stage IV, infiltration of the
central or peripheral nervous system, or bone marrow, as determined
by staging biopsy at different times.
According to the Ann Arbor system, when a full
staging evaluation is performed, the majority of patients exhibit
stage IE or IIE disease (7,10). A study by Heyning et al
(11) classified 46% stage I, 16%
stage II and 16% stage IV PLB patients and 20% with an unknown
stage. Stage IV disease was exclusively caused by the presence of
multiple bone lesions (11). In a
prospective study that included 28 PLB cases, the Ann Arbor stage
distribution was 54% for stage I–II and 46% for stage III–IV
(14). Other studies have also
obtained similar results, with disease stages IE or IIE
constituting the majority of PLB cases (13,15,16).
Non-Hodgkin’s lymphoma (NHL) forms the majority of
PLB, with the most common subtype being of B-cell origin (4,17)
whereas primary Hodgkin’s lymphoma of the bone is extremely rare
(7). The B phenotype constitutes
78–100% of PLB (9,14,15),
and among these, diffuse large BCL (DLBCL) represents 54–92%
(10,13,14,16).
The frequency of T-cell subtypes is relatively high (24%) in the
Chinese population compared with that in the Western population
(9).
According to the Kiel classification (7), 45–78% of primary NHL of the bone are
centroblastic and multilobulated (10,15,18).
BCL-6 was positive in 30% of cases and strong p53 protein
expression was observed in 11 out of 20 (55%) cases. A clonal
B-cell process by immunoglobulin heavy gene rearrangement was also
found in the majority of cases (13/18; 72%) (18). Another study has also demonstrated
that p53 and Bcl-2 may be involved in the pathogenesis of PLB
(19).
The literature has defined PLB in numerous different
ways. Certain studies have only included patients with Ann Arbor
stage I and II disease in the diagnosis of PLB (19,20),
others have included patients with stage IV disease and yet more
have included patients with involvement of the lymph nodes
(3,21–23).
At present, the following diagnostic criteria of PLB is widely
accepted and includes the following conditions (6): i) Primary site of tumor origin in the
bone marrow, with no other site indicating the existence of the
lesion on physical or imagining examination; ii) no identification
of lymphoma at any other site six months after the diagnosis of
PLB; iii) the diagnosis must be confirmed by pathology and
immunohistochemistry; and iv) malignant lymphomas, with the
exception of PLB and secondary lymphoma of the bone, must be
excluded.
A common complaint of patients with PLB is pain in
the bones. However, as non-steroidal anti-inflammatory drugs may
partly relieve these symptoms, PLB patients can be referred to
rheumatologists and misdiagnosed with rheumatic diseases (24). Chronic myelitis, metastatic tumor of
the bone and other primary bone tumors, such as osteosarcoma, must
be excluded prior to determining the diagnosis.
This current case study presents a review of the
radiological imaging of skeletal lymphoma with conventional
radiographs, scintigraphic studies, computed tomography, MRI and
FDG-PET-CT (25).
At the time of the initial radiograph, the results
of plain X-rays are usually normal. A solitary lytic lesion near
the end of a long bone, with a permeative or moth-eaten pattern of
destruction, and a periosteal reaction can be observed in
aggressive types, and this appearance is similar to that of
metastatic lymphomatous involvement of the bone, osteosarcomas and
Ewing’s sarcoma (26).
Radionuclide bone scans (99mTc
radionuclide imaging) show increased tracer uptake in 98% of
patients, with markedly increased activity in 64% of patients
(26), which is usually
non-specific. However, bone scintigraphy of
99mTc-methylene diphosphonate is a valuable tool in the
staging of PLB. It detects multifocal involvement, which alters the
prognosis and possible treatment (27).
CT is excellent in delineating cortical destruction,
however, the features are usually non-specific. Within months of
successful treatment, CT shows bone remodeling with a persistent
architecture that is similar to that of Paget’s disease of the bone
(28). The diagnosis of PLB can be
indicated by CT and MRI, particularly when upon the observation of
a large soft-tissue mass and abnormal marrow attenuation or signal
intensity without extensive cortical destruction.
Compared with Ewing’s sarcoma or osteosarcoma, PLB
shows significantly less frequent cortical abnormality, complete
penetration, focal destruction and complete destruction on MRI
(29,30). On T1-weighted MRI, the signal
intensities in the lesion range between isointense and hypointense
relative to the muscle, while on T2-weighted MRI, the signal
intensities are varied and do not appear to just reflect the
histological findings of intralesional vascularity or fibrosis
(31). Following successful
treatment, a rapidly decreasing tumor volume can be observed, with
complete disappearance of the soft tissue component. Minor bone
marrow signal abnormalities that have no clinical relevance may
persist for up to two years (28).
PLB is usually shown as a hypermetabolic lesion on
FDG-PET. PET-CT is a sensitive tool for accurately determining a
response to therapy, particularly a complete response (CR). In
cases of CR, PET scanning following treatment shows no
hypermetabolic lesions, with a rapid decline in FDG uptake, in
contrast to MRI or plain X-ray, which show a persistent bone lesion
following a partial response (32).
Newly developed lesions with rapid increase of FDG uptake, found by
PET during the follow-up period in patients with a CR, are
determined to be recurrence (33).
There is no standard therapy or guideline for PLB,
as all previous literature studies have been retrospective. Therapy
in general is multimodal and includes surgery, radiotherapy,
chemotherapy and rituximab.
Although the likelihood of local control following
the treatment of stage IE PLB is extremely high with radiotherapy
alone, radiation alone in limited-stage disease has a poor
five-year overall survival (OS) rate of ~45%, even when patients
with apparently limited-stage disease have been carefully selected
for treatment (27). Radiotherapy
alone has not been found to improve survival, and 10-year survival
has been shown to decrease in stage III patients. Therefore,
radiation alone should only be used in patients with spinal cord
compression (34), and more
effective systemic regimens are required (35). It has been reported that the
survival time is longer for patients treated with a combination of
chemotherapy and radiotherapy than those treated with radiotherapy
alone (16). Other studies have
also found that chemotherapy combined with radiotherapy is superior
to chemotherapy or radiotherapy alone, with five-year survival
rates of 58–95 versus 70–78%, respectively (34,35).
PLB is sensitive to chemotherapy, however,
Adriamycin- or anthracycline-based regimens have been confirmed to
be successful in achieving excellent long-term, disease-free
survival, particularly when followed by involved-field radiotherapy
(13).
Progression-free survival (PFS) and OS times in
patients with CD20-positive BCL have been markedly improved by
adding rituximab to CHOP chemotherapy (37), however, the superiority of rituximab
in PLB of DLBCL is controversial (27). A previous study showed marked
improvements in the three-year PFS rate for PLB patients following
the introduction of rituximab (88%) compared with those treated
earlier without rituximab (52%) (17), and also provided evidence of
improved survival with combined systemic therapy using rituximab
and combination chemotherapy with CHOP (17). However, a retrospective analysis of
patients with PLB demonstrated that the addition of rituximab to
chemotherapy resulted in a non-significant trend toward a superior
OS rate (38).
As surgery has not been found to improve OS or PFS,
surgery is only indicated for prophylactic fixation of impending
fractures or the treatment of pathological fractures or spinal cord
compression.
PLB has an improved prognosis compared with other
bone malignant tumors, such as osteosarcoma or secondary lymphoma
of the bone (14). A younger age
has also been identified as an independent predictor of survival
(3). Heyning et al (3) found that patients who were >60
years old at the time of presentation exhibited poorer OS (76 vs.
37%) and a smaller progression-free period (58 vs. 28%) (14). Disease stage has also been found to
have a significant effect on five-year OS. Patients with localized
disease have statistically improved survival times compared with
patients with systemic disease, and survival rates have been
recorded as 90% for stage I and 41% for stage IV (10).
The international prognostic index (IPI) is a
prognostic factor for PLB. A significant difference has been
identified in the OS of patients low and low-intermediate versus
high-intermediate IPI scores (P=0.0035), regardless of stage
(27). In addition, younger
patients with good IPI scores have a favorable prognosis (18).
Heyning et al (3) also found poorer survival times in
patients with the immunoblastic subtype compared with the
centroblastic mono/polymorphic or centroblastic multilobulated
subtypes (P=0.015). This was also confirmed in a study by Lewis
et al (4), in which
statistically improved survival times were observed in patients
with DLBCL with multilobulated nuclei (15).
A statistically significant difference has been
identified in OS favoring the use of combined chemotherapy (with or
without rituximab) and radiation compared with either modality
alone (P=0.02) (27). Furthermore,
the addition of rituximab has been found to result in a
non-significant trend towards improved OS (P=0.11).
PLB is a distinct clinicopathological entity with a
relatively homogeneous morphology and clinical behavior, and is
usually of B-cell type. PET-CT is of great importance in evaluating
CR, and patients with PLB treated with combined modality therapy
have been found to exhibit a superior outcome compared with those
treated by single modality therapy. In addition, younger patients
with good IPI scores and localized disease have a favorable
prognosis. The present PLB patient is a well documented case, who
underwent full evaluation, received proper treatment, and had a
good prognosis. This case demonstrates that PLB has an improved
prognosis compared with primary lymphoma of other sites and that
combined therapy may further improve outcome. However, future
prospective studies must be performed in order to gain an improved
understanding of the disease.
References
|
1
|
Singh T, Satheesh CT, Lakshmaiah KC,
Suresh TM, Babu GK, Lokanatha D, Jacob LA and Halkud R: Primary
bone lymphoma: a report of two cases and review of the literature.
J Cancer Res Ther. 6:296–298. 2010.
|
|
2
|
Oberling C: The reticulosarcomas and the
reticulotheliomas of Ewing’s osterosarcoma. Bull Assoc Fr Etude
Cancer (Paris). 17:259–296. 1928.(In French).
|
|
3
|
Heyning FH, Hogendoorn PC, Kramer MH,
Hermans J, Kluin-Nelemans JC, Noordijk EM and Kluin PM: Primary
non-Hodgkin’s lymphoma of bone: a clinicopathological investigation
of 60 cases. Leukemia. 13:2094–2098. 1999.
|
|
4
|
Lewis VO, Primus G, Anastasi J, Doherty D,
Montag AG, Peabody TD and Simon MA: Oncologic outcomes of primary
lymphoma of bone in adults. Clin Orthop Relat Res. 415:90–97.
2003.
|
|
5
|
Jawad MU, Schneiderbauer MM, Min ES,
Cheung MC, Koniaris LG and Scully SP: Primary lymphoma of bone in
adult patients. Cancer. 116:871–879. 2010.
|
|
6
|
Mulligan ME, McRae GA and Murphey MD:
Imaging features of primary lymphoma of bone. AJR Am J Roentgenol.
173:1691–1697. 1999.
|
|
7
|
Song YG, Hahn JS, Choi YH, Yeom JS, Yang
WI, Seo CO and Kim JM: A case of primary bone lymphoma associated
with acquired immunodeficiency syndrome. Yonsei Med J. 39:383–389.
1998.
|
|
8
|
Bao J: Young males with primary lymphoma
of bone presenting with musculoskeletal pain are prone to be
misdiagnosed as ankylosing spondylitis: a case report. Rheumatol
Int. 32:263–264. 2012.
|
|
9
|
ter Braak BP, Guit GL and Bloem JL: Case
111: Soft-tissue lymphoma. Radiology. 243:293–296. 2007.
|
|
10
|
O’Neill J, Finlay K, Jurriaans E and
Friedman L: Radiological manifestations of skeletal lymphoma. Curr
Probl Diagn Radiol. 38:228–236. 2009.
|
|
11
|
Heyning FH, Hogendoorn PC, Kramer MH,
Holland CT, Dreef E and Jansen PM: Primary lymphoma of bone:
extranodal lymphoma with favourable survival independent of
germinal centre, post-germinal centre or indeterminate phenotype. J
Clin Pathol. 62:820–824. 2009.
|
|
12
|
Catlett JP, Williams SA, O’Connor SC,
Krishnan J and Malkovska V: Primary lymphoma of bone: an
institutional experience. Leuk Lymphoma. 49:2125–2132. 2008.
|
|
13
|
O’Connor AR, Birchall JD, O’Connor SR and
Bessell E: The value of 99mTc-MDP bone scintigraphy in staging
primary lymphoma of bone. Nucl Med Commun. 28:529–531. 2007.
|
|
14
|
Park YH, Choi SJ, Ryoo BY and Kim HT: PET
imaging with F-18 fluorodeoxyglucose for primary lymphoma of bone.
Clin Nucl Med. 30:131–134. 2005.
|
|
15
|
Kitsoulis P, Vlychou M, Papoudou-Bai A,
Karatzias G, Charchanti A, Agnantis NJ and Bai M: Primary lymphomas
of bone. Anticancer Res. 26:325–337. 2006.
|
|
16
|
Park YH, Kim S, Choi SJ, Ryoo BY, Yang SH,
Cheon GJ, Choi CW, Lim SM, Yoo JY and Lee SS: Clinical impact of
whole-body FDG-PET for evaluation of response and therapeutic
decision-making of primary lymphoma of bone. Ann Oncol.
16:1401–1402. 2005.
|
|
17
|
Yuste AL, Segura A, López-Tendero P,
Gironés R, Montalar J and Gómez-Codina J: Primary lymphoma of bone:
a clinico-pathological review and analysis of prognostic factors.
Leuk Lymphoma. 45:853–855. 2004.
|
|
18
|
Stein ME, Kuten A, Gez E, Rosenblatt KE,
Drumea K, Ben-Shachar M, Zidan J, Haim N and Epelbaum R: Primary
lymphoma of bone - a retrospective study. Experience at the
Northern Israel Oncology Center (1979–2000). Oncology. 64:322–327.
2003.
|
|
19
|
Amara H, Elomri H, Cherni N, Tlili K,
Mrad-Dali K, Sriha B, Kraiem C and Nabli S: Primary lymphoma of
bone: imaging features. J Radiol. 83:55–58. 2002.(In French).
|
|
20
|
Huebner-Chan D, Fernandes B, Yang G and
Lim MS: An immunophenotypic and molecular study of primary large
B-cell lymphoma of bone. Mod Pathol. 14:1000–1007. 2001.
|
|
21
|
Marshall DT, Amdur RJ, Scarborough MT,
Mendenhall NP and Virkus WW: Stage IE primary non-Hodgkin’s
lymphoma of bone. Clin Orthop Relat Res. 405:216–222. 2002.
|
|
22
|
Brousse C, Baumelou E and Morel P: Primary
lymphoma of bone: a prospective study of 28 cases. Joint Bone
Spine. 67:446–451. 2000.
|
|
23
|
Evron E, Goland S, Klepfish A, Malnick SD,
Sokolowski N and Sthoeger ZM: Primary multifocal lymphoma of bone
presenting as hypercalcemic crisis: report of a rare manifestation
of extranodal lymphoma. Leuk Lymphoma. 34:197–200. 1999.
|
|
24
|
Haussler MD, Fenstermacher MJ, Johnston DA
and Harle TS: MRI of primary lymphoma of bone: cortical disorder as
a criterion for differential diagnosis. J Magn Reson Imaging.
9:93–100. 1999.
|
|
25
|
White LM, Schweitzer ME, Khalili K,
Howarth DJ, Wunder JS and Bell RS: MR imaging of primary lymphoma
of bone: variability of T2-weighted signal intensity. AJR Am J
Roentgenol. 170:1243–1247. 1998.
|
|
26
|
Niitsu N, Nakayama M and Umeda M: A
clinical study of primary lymphoma of bone. Rinsho Ketsueki.
39:432–435. 1998.(In Japanese).
|
|
27
|
Ramadan KM, Shenkier T, Sehn LH, Gascoyne
RD and Connors JM: A clinicopathological retrospective study of 131
patients with primary bone lymphoma: a population-based study of
successively treated cohorts from the British Columbia Cancer
Agency. Ann Oncol. 18:129–135. 2007.
|
|
28
|
Power DG, McVey GP, Korpanty G, et al:
Primary bone lymphoma: single institution case series. Ir J Med
Sci. 177:247–251. 2008.
|
|
29
|
Freeman C, Berg JW and Cutler SJ:
Occurrence and prognosis of extranodal lymphomas. Cancer.
29:252–260. 1972.
|
|
30
|
Rudders RA, Ross ME and DeLellis RA:
Primary extranodal lymphoma: response to treatment and factors
influencing prognosis. Cancer. 42:406–416. 1978.
|
|
31
|
Barbieri E, Cammelli S, Mauro F, et al:
Primary non-Hodgkin’s lymphoma of the bone: treatment and analysis
of prognostic factors for Stage I and Stage II. Int J Radiat Oncol
Biol Phys. 59:760–764. 2004.
|
|
32
|
Fidias P, Spiro I, Sobczak ML, et al:
Long-term results of combined modality therapy in primary bone
lymphomas. Int J Radiat Oncol Biol Phys. 45:1213–1218. 1999.
|
|
33
|
Ferreri AJ, Reni M, Ceresoli GL and Villa
E: Therapeutic management with adriamycin-containing chemotherapy
and radiotherapy of monostotic and polyostotic primary
non-Hodgkin’s lymphoma of bone in adults. Cancer Invest.
16:554–561. 1998.
|
|
34
|
Shoji H and Miller TR: Primary reticulum
cell sarcoma of bone. Significance of clinical features upon the
prognosis. Cancer. 28:1234–1244. 1971.
|
|
35
|
Glotzbecker MP, Kersun LS, Choi JK, Wills
BP, Schaffer AA and Dormans JP: Primary non-Hodgkin’s lymphoma of
bone in children. J Bone Joint Surg Am. 88:583–594. 2006.
|
|
36
|
Durr HR, Müller PE, Hiller E, Maier M,
Baur A, Jansson V and Refior HJ: Malignant lymphoma of bone. Arch
Orthop Trauma Surg. 122:10–16. 2002.
|
|
37
|
Beal K, Allen L and Yahalom J: Primary
bone lymphoma: treatment results and prognostic factors with
long-term follow-up of 82 patients. Cancer. 106:2652–2656.
2006.
|
|
38
|
Sehn LH, Donaldson J, Chhanabhai M,
Fitzgerald C, Gill K, Klasa R, MacPherson N, O’Reilly S, Spinelli
JJ, Sutherland J, et al: Introduction of combined CHOP plus
rituximab therapy dramatically improved outcome of diffuse large
B-cell lymphoma in British Columbia. J Clin Oncol. 23:5027–5033.
2005.
|