Introduction
Glioblastoma multiforme (GBM) is the most frequent,
aggressive and lethal primary malignant tumor of the central
nervous system in adults worldwide. No effective treatment for this
highly aggressive and infiltrative tumor is available, and the
median survival time is <16 months following initial diagnosis,
with a five-year survival rate as low as 4.7% (1,2). High
proportion of mitotically active cells displaying pleomorphic
morphology, pseudopalisading necrosis associated with microvascular
hyperplasia, and infiltrative cell growth towards the parenchyma
are some of the main histopathological features of GBM. Such key
features that are observed in the majority of GBM specimens can be
recapitulated in orthotopic xenograft models by the intracerebral
injection of patient-derived stem-like cells, which are considered
to be responsible for gliomagenesis (3).
A previous genome-wide expression profiling study
identified E2F2 as a hyperexpressed gene in
CD133+ stem-like cells isolated from fresh GBM
specimens. Furthermore, the frequency and levels of E2F2
expression correlated significantly with the malignancy of
astrocytomas, being predominantly hyperexpressed in GBM (4). The E2F2 protein belongs to a large
family of transcription factors regulating cell proliferation, cell
division and cell differentiation. The E2F family has nine members,
which have been divided into two subclasses (activators and
repressors) based on their transcriptional properties and conserved
structural features. E2F2 has a strong transcriptional activation
domain and is able to interact with the tumor suppressor Rb
(5). The Rb/E2F network regulates
the expression of genes involved in cell cycle progression, DNA
replication, checkpoint control, apoptosis, differentiation, DNA
damage repair and development (6).
Despite its well-known positive regulation of cell proliferation,
the contribution of E2F2 to tumorigenesis is not so clear, since it
has been reported to exert either pro-oncogenic or tumor
suppression effects (7).
Since CD133+ GBM cells have originally
been reported to have increased tumor initiating capability in
vivo (3,8), a functional study was carried out to
address the relevance of E2F2 to the tumorigenic properties
of GBM, and its value as a therapeutic target for treatment of this
highly aggressive brain tumor.
Materials and methods
Cell culture
The human glioblastoma cell line U87MG was kindly
provided by Dr Suely K. N. Marie from the Laboratory of Medical
Investigation (LIM15) at the University of São Paulo (São Paulo,
Brazil). Cells were grown in Dulbecco’s modified Eagle’s Medium-low
glucose (DMEM-LG; Invitrogen Life Technologies, Carlsbad, CA, USA)
supplemented with 2 mM L-glutamine, 10% fetal bovine serum (FBS),
100 U/ml penicillin and 100 μg/ml streptomycin (all Life
Technologies, Grand Island, NY, USA), in a humidified atmosphere at
37°C with 5% CO2.
Transient E2F2 silencing
U87MG cells were transfected with Sure Silencing™
shRNA plasmids (Super Array, SABiosciences, Frederick, MD, USA)
designed to specifically knock down the expression of the
E2F2 gene. After 24 h without FBS for synchronization, U87MG
cells were seeded in six-well plates at a density of 105
cells per well and incubated for 24 h. Cells were then transfected
with non-specific DNA (negative, non-specific control; NS) or shRNA
silencing E2F2 (shE2F2) (both SABiosciences, Frederick, MD,
USA), using Lipofectamine™ RNAiMAX (Life Technologies) according to
the manufacturer’s instructions. The total plasmid concentration in
each well was 0.5 μg. Positive control cells were treated with
Lipofectamine RNAiMAX, identically to the other experimental
groups, but received no plasmids. Twenty-four and 72 h after
transfection, the glioblastoma cells displaying neomycin resistance
were selected in medium containing 500 μg/ml G418 (Life
Technologies) and harvested after 96 h of culture for in
vitro and in vivo experiments.
Quantification of gene expression by
quantitative polymerase chain reaction (qPCR)
Total RNA was extracted using an RNeasy®
mini kit (50) (Qiagen GmbH, Hilden, Germany), according to the
manufacturer’s instructions, and quantified by measuring the
absorbance at 260 nm (NanoDrop 2000 spectrophotometer, Thermo
Fisher Scientific, Wilmington, DE, USA). The reverse transcription
(RT) reaction was performed using 1 μg of total RNA with
Superscript™ III Reverse Transcriptase enzyme (Life Technologies).
Real-time RT-PCR was performed in a 7500 Real-time RT-PCR system
(Life Technologies), by the SYBR® GreenER™ incorporation
method (Power SYBR Green PCR Master Mix; Life Technologies). The
cycling conditions were as follows: 95°C for 15 sec, followed by 50
cycles at 60°C for 30 sec, 95°C for 1 h and 55°C for 30 sec. All
primer pairs were designed in different exons using Primer3 Input
version 0.4.0 (http://gmdd.shgmo.org/primer3/?seqid=47), and
synthesized by Promega Corporation (Madison, WI, USA). The primer
sequences were as follows: Forward, 5′-GGACAGGAATGGCCTC-3′ and
reverse, 5′-GTCCTTCGAGGAGCTC-3′ for E2F2; and forward,
5′-GGACAGGAATGGCCTC-3′ and reverse, 5′-GTCCTTCGAGGAGCTC-3′ for
GAPDH.
Cell proliferation assays
U87MG cells were seeded on 96-well plates at an
initial density of 5×104 cells/well, and proliferation
was measured 24, 48 and 72 h after plating by direct counting of
viable cells in a Neubauer chamber with Trypan blue (1:1;
Sigma-Aldrich, St. Louis, MO, USA). The number of viable cells was
also assessed by the 3-(4, 5-dimethylthiazolyl-2)-2,
5-diphenyltetrazolium bromide (MTT) assay, adding 10 μl of MTT to
the cell preparation and incubating for 2 h in a humidified
atmosphere at 37°C with 5% CO2. Cells were lysed in 100
μl of 100% dimethylsulfoxide and absorbance was detected at 550 nm
using a 96-well plate reader (iMark Microplate Absorbance Reader;
Bio-Rad, Hercules, CA, USA). Each sample was run in triplicate.
Anchorage-independent cell growth was also assessed by the soft
agar assay. Briefly, 2 ml of 0.5% agar was added to each well of a
12-well plate. Detached U87MG cells were mixed with an agarose
suspension (0.3% final concentration), and then layered onto the
0.5% agarose underlay. Culture medium was changed every three days,
and the number of cell foci ≥100 mm in diameter was counted after
21 days using the EVOS® XL cell imaging system (Life
Technologies). Each experiment was performed in triplicate.
In vivo tumorigenesis
Subcutaneous xenograft model
U87MG glioblastoma cells (106
cells/mouse) were suspended in DMEM-LG, injected subcutaneously
into the right flank of BALB/c nude mice (male; 4–8 weeks old)
obtained from the University of São Paulo, and allowed to grow for
50 days or until the tumor reached a volume of 2,500 mm3
(tumor weight, 100–200 mg). Animals (n=5 per group) were monitored
daily and tumors were measured with a digital caliper rule twice a
week. Tumor volume was estimated using the formula: Volume = (minor
diameter2 × major diameter)/2.
Orthotopic glioblastoma xenograft
model
Adult BALB/c nude mice (~20 g) were anesthetized by
intraperitoneal administration of ketamine (100 mg/kg)/xylazine (15
mg/kg) (both Syntec Brasil, Cotia, Brazil). Following sedation,
mice were positioned in a stereotaxic frame. The scalp was
sterilized with iodine and 70% ethanol and a median incision of
~1.0 cm was made. The cranial cavity was assessed by a right
frontal hole using an electric mini-drill (Micromotor LB100;
Beltec, Araraquara, Brazil). A total of 106 U87MG cells
(E2F2 knocked down and control) were suspended in 5 μl of DMEM low
glucose without FBS and inoculated with a high-precision
microsyringe (701RN; Hamilton Company, Reno, NV, USA) into the
striatum, 0.9 mm in front of the bregma, 2.5 mm laterally to the
right and 3.0 mm ventrally, at a 0.5 μl/min rate. At the end of
cell injection, the needle was retained in the incision for 5 min
and slowly removed to prevent the cell suspension from flowing
back. The scalp was closed with 2-0 silk suture and the animals
were housed under standard controlled conditions (7:00am to 7:00pm
light/dark cycle; 20–22°C; 45–55% humidity) with food and water ad
libitum. Histological analysis was performed 30 days
post-intracranial implantation of tumor cells. All efforts were
made to minimize animal suffering as proposed by the International
Ethical Guideline for Biomedical Research (CIOMS/OMS, 1985). The
study was approved by the ethics committee for animal research of
the University of São Paulo (CEUA protocol no. 132/2011).
Histological analysis
Brain samples were frozen in cold isopentane
solution (Sigma-Aldrich) at −25°C, and then sectioned at 20 μm on a
cryostat. Coronal histological sections of the tumor xenograft and
surrounding brain area were mounted on silanized microscope slides
(StarFrost®, Knittel-Gläser, Braunschweig, Germany), and
stained with hematoxylin and eosin. Microscope images were captured
by an ExwaveHAD Color video digital camera (Sony Corporation,
Tokyo, Japan) attached to a Nikon Eclipse E600 microscope (Nikon,
Corporation, Tokyo, Japan), using the WinAVI Video Capture software
(WinAVI, Eden Prairie, MN, USA).
Statistical Analysis
All experiments were performed in triplicate and
three independent experiments were performed. Data were analyzed by
one way analysis of variance with Bonferroni as the post hoc test,
using GraphPad Prism 3.0 (GraphPad Software, San Diego, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference. Data are presented as the mean ± standard
deviation.
Results
E2F2 knockdown inhibits glioblastoma cell
proliferation in vitro
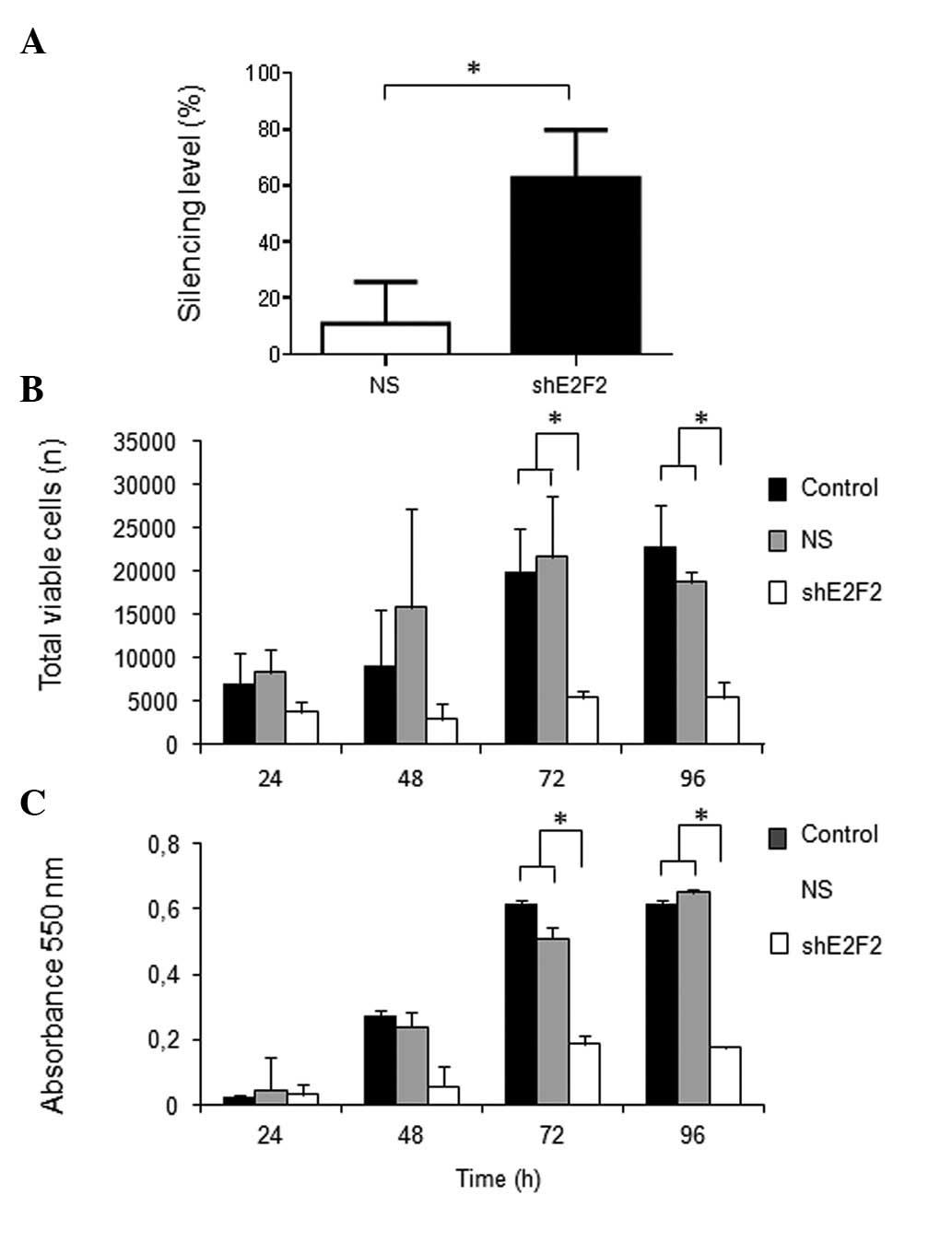
Specific knockdown of E2F2 expression was
previously confirmed in U87MG cells, reaching a silencing level of
~60% after 96 h of transfection with shRNA. Under standard growth
conditions in vitro, the total number of viable tumor cells
was significantly lower after 48, 72 and 96 h of E2F2
knockdown compared with that in the NS control group (P=0.0044,
P=0.0007 and P=0.0035, respectively). Similar results were observed
by the MTT assay, based on the activity of mitochondrial succinate
dehydrogenase, which indicated significantly lower numbers of
viable tumor cells after 72 h and 96 h of E2F2 knockdown,
compared with the controls (P<0.0001 for the two time points)
(Fig. 1). The absorbance levels
acquired from cells subjected to E2F2 knockdown were
virtually unchanged over the time course examined (24–96 h),
suggesting inhibition of cell proliferation.
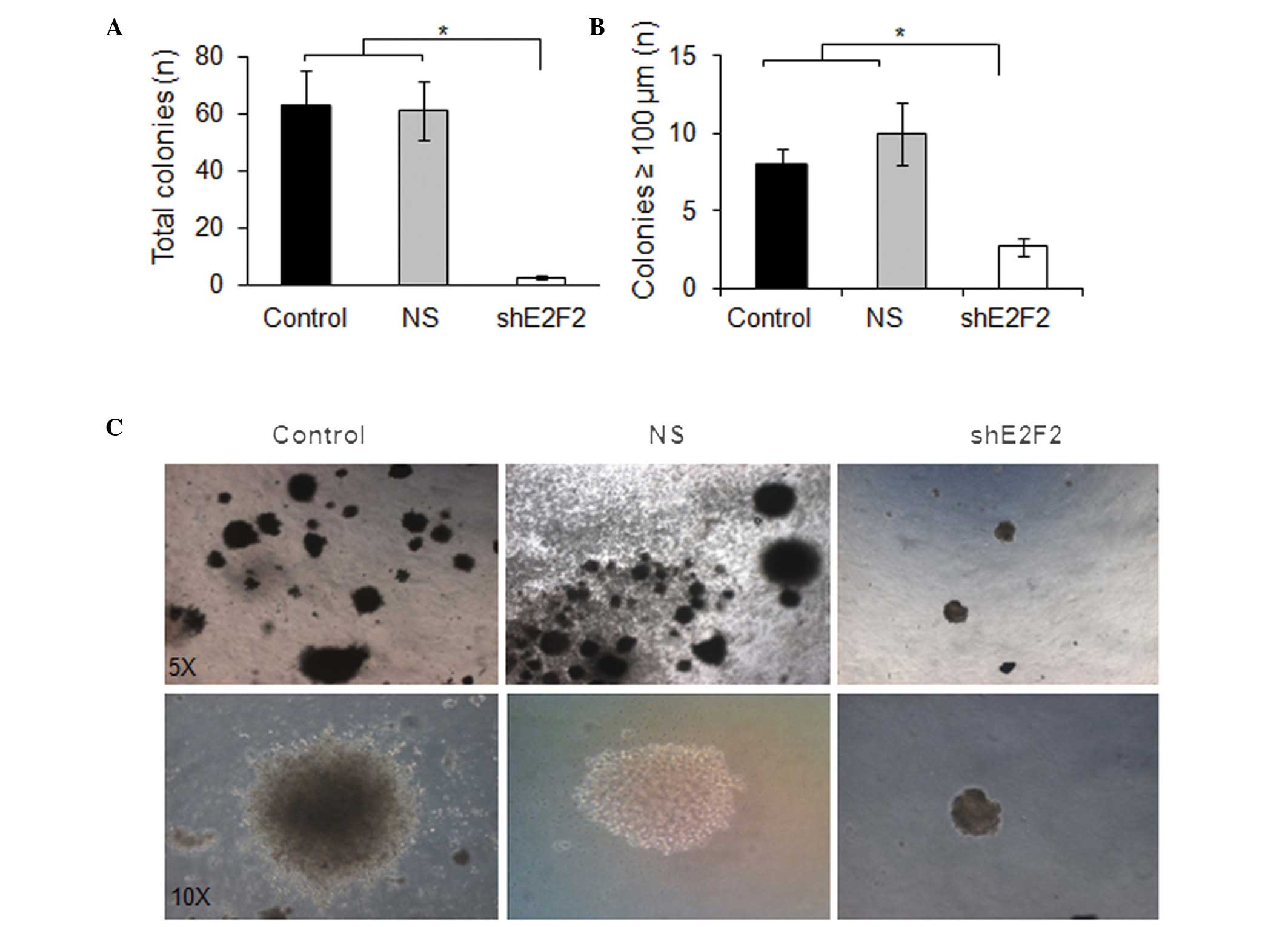
Anchorage-independent cell growth is a valuable
indicator of tumorigenic capability, since it is associated with
neoplastic transformation and metastatic potential. In agreement
with the previous cell viability experiments, the efficiency of
U87MG cells to generate tumor cell colonies by
anchorage-independent growth in a semi-solid medium was
significantly reduced by knocking down E2F2. Both the total
amount of colonies (≥100 μm) and the average size of the colonies
were significantly lower when assaying U87MG cells subjected to
E2F2 knockdown, compared with those of the control cells
(P=0.0081 and P=0.0076, respectively) (Fig. 2).
E2F2 knockdown inhibits gliomagenesis in
xenograft models
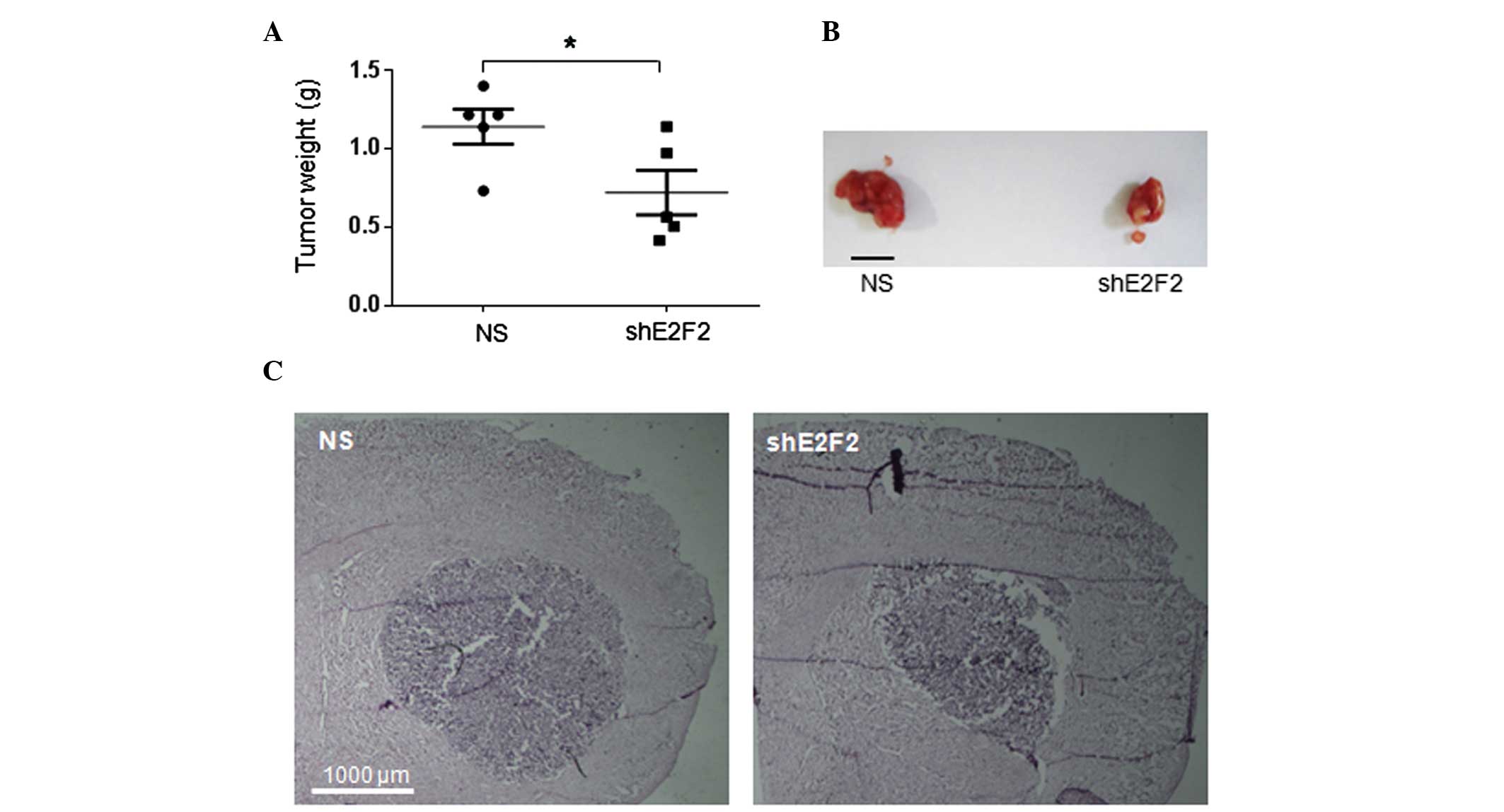
In order to test whether E2F2 knockdown would
affect in vivo tumorigenesis, two xenograft models of human
GBM were employed. Tumors derived from the subcutaneous injection
of U87MG cells in nude mice were measurable ~30 days following
injection, reaching volumes usually higher than 1,000
mm3 in the subsequent 20 days of in vivo growth.
In a period of 50 days post-cell injection, although transient, the
E2F2 knockdown in U87MG cells inhibited tumor development. Tumors
generated from U87MG cells with E2F2 knockdown were smaller
and significantly lighter than tumors resulting from control cells
(P=0.04) (Fig. 3A). Mice bearing
orthotopic U87MG tumors also revealed differences in brain tumor
development due to E2F2 knockdown. In agreement with the
previous subcutaneous xenograft model, brain tumors derived from
the stereotaxic intracerebral injection of U87MG cells with
E2F2 knockdown were somewhat smaller than tumors derived
from control cells, 30 days following injection in nude mice
(Fig. 3B).
Discussion
Despite the conserved functions in cell cycle
regulation, development and tissue maintenance, E2F transcription
factors may affect tumorigenic processes in different ways due to
the fact that each member displays individual mechanisms of action
and may control the expression of other family members through a
complex feedback regulation (9).
The function of E2F2 is less characterized relative to other
members of the E2F family, and its involvement in tumorigenesis
remains a matter of debate, since evidence of both tumor
suppression and pro-oncogenic activities have been reported
(5).
It has been shown in mice that deficiency in E2F2
caused by gene targeting (E2F2−/−) significantly
increased the population of self-reactive peripheral T cells,
causing symptoms similar to severe autoimmunity. Such increment in
self-reactive T cells was demonstrated to be due to increased cell
proliferation rates without evidence of differential resistance to
apoptosis (10). More recently,
however, overexpression of E2F2 was reported to induce
p53-mediated apoptosis of mouse retina neurons lacking Rb
and p107, independent of other activating E2Fs (11). In a conditional bitransgenic mouse
model of Myc-induced T-cell lymphomagenesis, Opavsky et al
(12) demonstrated that
inactivation of E2F2 (either E2F2+/− or
E2F2−/−), but not of E2F1 or E2F3,
significantly accelerated tumor onset and progression, indicating
an haploinsufficient tumor suppressor function for E2F2 in T
cells. Similar results were obtained with MMTV-Myc transgenic mice,
in which E2F2 knockout delayed latency and reduced the
incidence of Myc-driven mammary tumors (13).
By contrast, in neuroblastomas, E2F2 was shown to
positively regulate MYCN transcription and thought to be
required for full activity of MYCN expression in aggressive
neuroblastomas usually associated with poor prognosis (14). Stable overexpression of E2F2
in fibroblasts indeed revealed a strong oncogenic capacity for this
E2F member (15). Transgenic mice
have also supported a pro-oncogenic role for E2F2. In an Eμ-pp-E2F2
mouse model, overexpression of E2F2 induced mild hyperplasia
of the thymus in young mice and subsequent development of thymomas
(16). Notably, overexpression of
E2F2 was predominantly found in cortical thymic epithelial
cells, which are highly proliferative cells involved in T-cell
development and regeneration capacity of the thymus (17). In E2F2 knockout mice, loss of
this transcription factor resulted in cell cycle arrest in
hematopoietic progenitors (18) and
increased DNA double-strand breaks in erythroblasts (19).
Regarding human cancers, in addition to the
abovementioned study in neuroblastomas, E2F2 has been shown
to be under control of the AP-1 transcription factor in breast
cancer cells, where it positively regulates cell proliferation
(20). Accordingly, high
E2F2 expression was recently reported to be associated with
poor survival of breast cancer patients (21). In prostate cancer cells, E2F2
expression was reported to be inhibited by let-7a (22) and miR-31 (23) microRNAs, resulting in suppression of
tumorigenesis in a nude mice ectopic xenograft model.
In GBM, E2F2 was identified as one of the
hyper-expressed genes in CD133+ tumor cells, compared
with their counterparts, and its expression correlated with
malignancy grade (4). Such
subcellular population of GBM had been reported to have neural stem
cells characteristics and enhanced in vivo tumor initiation
capability (8). Studies also
isolated and characterized CD133+ stem-like cells in
different GBM cell lines, including U87MG (24), establishing a useful experimental
model to study cancer stem cell biology.
The effects of E2F2 knockdown in U87MG cells
verified in the present study by in vitro and in vivo
models of tumorigenesis are consistent with a pro-tumorigenic
activity of E2F2 in GBM. In agreement with this notion, a recent
study demonstrated that overexpression of the microRNA miR-125b
inhibits proliferation of CD133+ GBM cells in
vitro, by targeting E2F2 transcripts, and that such
effect on in vitro proliferation is rescued by
overexpression of E2F2 (25). Overall, these concordant findings
suggest that E2F2 is an important transcription factor regulating
the tumor-initiating capability of human GBM cells. Inhibitors of
E2F2 expression may therefore be considered as candidates
for drug development to locally treat GBM, a highly malignant and
devastating tumor of the central nervous system.
Acknowledgements
This study was supported by grants from the National
Institute of Science and Technology-Stem Cells in Human Genetic
Diseases, the National Council for Scientific and Technological
Development (CNPq), the Coordination for the Improvement of Higher
Education Personnel (CAPES), and the São Paulo Research Foundation
(FAPESP). Dr Adriana M. Nakahata and Dr Daniela E. Suzuki were
recipients of fellowships from CAPES and CNPq, respectively. Miss
Carolina O. Rodini and Miss Mayara L. Fiuza received fellowships
from FAPESP.
References
|
1
|
Karak AK, Singh R, Tandon PN and Sarkar C:
A comparative survival evaluation and assessment of
interclassification concordance in adult supratentorial astrocytic
tumors. Pathol Oncol Res. 6:46–52. 2000.
|
|
2
|
Dolecek TA, Propp JM, Stroup NE and
Kruchko C: CBTRUS statistical report: primary brain and central
nervous system tumors diagnosed in the United States in 2005–2009.
Neuro Oncol. 14(Suppl 5): v1–v49. 2012.
|
|
3
|
Dimova DK and Dyson NJ: The E2F
transcriptional network: old acquaintances with new faces.
Oncogene. 24:2810–2826. 2005.
|
|
4
|
Bracken AP, Ciro M, Cocito A and Helin K:
E2F target genes: unraveling the biology. Trends Biochem Sci.
29:409–417. 2004.
|
|
5
|
DeGregori J: The genetics of the E2F
family of transcription factors: shared functions and unique roles.
Biochim Biophys Acta. 1602:131–150. 2002.
|
|
6
|
Okamoto OK, Oba-Shinjo SM, Lopes L and
Nagahashi Marie SK: Expression of HOXC9 and E2F2 are up-regulated
in CD133(+) cells isolated from human astrocytomas and associate
with transformation of human astrocytes. Biochim Biophys Acta.
1769:437–442. 2007.
|
|
7
|
Galli R, Binda E, Orfanelli U, et al:
Isolation and characterization of tumorigenic, stem-like neural
precursors from human glioblastoma. Cancer Res. 64:7011–7021.
2004.
|
|
8
|
Singh SK, Hawkins C, Clarke ID, et al:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004.
|
|
9
|
Chen HZ, Tsai SY and Leone G: Emerging
roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev
Cancer. 9:785–797. 2009.
|
|
10
|
Murga M, Fernández-Capetillo O, Field SJ,
et al: Mutation of E2F2 in mice causes enhanced T lymphocyte
proliferation, leading to the development of autoimmunity.
Immunity. 15:959–970. 2001.
|
|
11
|
Chen D, Chen Y, Forrest D and Bremner R:
E2f2 induces cone photoreceptor apoptosis independent of E2f1 and
E2f3. Cell Death Differ. 20:931–940. 2013.
|
|
12
|
Opavsky R, Tsai SY, Guimond M, et al:
Specific tumor suppressor function for E2F2 in Myc-induced T cell
lymphomagenesis. Proc Natl Acad Sci USA. 104:15400–15405. 2007.
|
|
13
|
Fujiwara K, Yuwanita I, Hollern DP and
Andrechek ER: Prediction and genetic demonstration of a role for
activator E2Fs in Myc-induced tumors. Cancer Res. 71:1924–1932.
2011.
|
|
14
|
Strieder V and Lutz W: E2F proteins
regulate MYCN expression in neuroblastomas. J Biol Chem.
278:2983–2989. 2003.
|
|
15
|
Chen C and Wells AD: Comparative analysis
of E2F family member oncogenic activity. PLoS One. 2:e9122007.
|
|
16
|
Scheijen B, Bronk M, van der Meer T, De
Jong D and Bernards R: High incidence of thymic epithelial tumors
in E2F2 transgenic mice. J Biol Chem. 279:10476–10483. 2004.
|
|
17
|
Rode I and Boehm T: Regenerative capacity
of adult cortical thymic epithelial cells. Proc Natl Acad Sci USA.
109:3463–3468. 2012.
|
|
18
|
Li FX, Zhu JW, Hogan CJ and DeGregori J:
Defective gene expression, S phase progression, and maturation
during hematopoiesis in E2F1/E2F2 mutant mice. Mol Cell Biol.
23:3607–3622. 2003.
|
|
19
|
Dirlam A, Spike BT and Macleod KF:
Deregulated E2f-2 underlies cell cycle and maturation defects in
retinoblastoma null erythroblasts. Mol Cell Biol. 27:8713–8728.
2007.
|
|
20
|
Shen Q, Uray IP, Li Y, Krisko TI, Strecker
TE, Kim HT and Brown PH: The AP-1 transcription factor regulates
breast cancer cell growth via cyclins and E2F factors. Oncogene.
27:366–377. 2008.
|
|
21
|
Nguyen-Vu T, Vedin LL, Liu K, et al: Liver
X receptor ligands disrupt breast cancer cell proliferation through
an E2F-mediated mechanism. Breast Cancer Res. 15:R512013.
|
|
22
|
Dong Q, Meng P, Wang T, et al: MicroRNA
let-7a inhibits proliferation of human prostate cancer cells in
vitro and in vivo by targeting E2F2 and CCND2. PLoS One.
5:e101472010.
|
|
23
|
Lin PC, Chiu YL, Banerjee S, et al:
Epigenetic repression of miR-31 disrupts androgen receptor
homeostasis and contributes to prostate cancer progression. Cancer
Res. 73:1232–1244. 2013.
|
|
24
|
Yu SC, Ping YF, Yi L, et al: Isolation and
characterization of cancer stem cells from a human glioblastoma
cell line U87. Cancer Letters. 265:124–134. 2008.
|
|
25
|
Wu N, Xiao L, Zhao X, et al: miR-125b
regulates the proliferation of glioblastoma stem cells by targeting
E2F2. FEBS Lett. 586:3831–3839. 2012.
|