Introduction
c-Met is a transmembrane tyrosine kinase receptor
that is phosphorylated and activated upon the binding of its
ligand. The natural ligand for c-Met is hepatocyte growth factor
(HGF)/scatter factor, which is produced by stromal and mesenchymal
cells (1). The phosphorylation of
c-Met activates downstream signaling pathways, which initiate
biological effects in normal and pathological processes (2). Under physiological conditions, the
HGF/c-Met axis is implicated in cell growth and differentiation,
organ development and neovascularization, as well as tissue repair
and regeneration (3). Accumulating
evidence indicates that it is also important in cancer development
(4,5).
A dysfunctional HGF/c-Met axis has been implicated
in the development, invasion and angiogenesis of cancers (6), and an increasing number of studies
have revealed the specific mechanism (7,8).
Overexpression of c-Met occurs via paracrine and autocrine
stimulation of HGF, which in turn stimulates cancer cell
progression. The paracrine signaling pathway is activated by HGF
secretion by stromal cells, whereas the autocrine signaling pathway
is initiated by HGF that is generated from cancer cells. Generally,
the paracrine signaling pathway of HGF is the major cause of c-Met
expression, as not all tumor cells produce HGF (9). Previous studies have also shown that
certain signaling pathways mediate increased cancer progression as
result of the HGF/c-Met axis, typically the phosphoinositide
3-kinase (PI3K) and mitogen-activated protein kinase signaling
pathways (10–13).
HGF induces the disassembly adhesion of epithelial
cancer cells, thereby increasing motility and invasiveness, in a
process termed epithelial-mesenchymal transition (EMT). EMT is
important in cancer metastasis as it promotes the detachment of
cancer cells from the primary tumor areas, leading to invasion of
the vasculature and colonization of distant organs with secondary
tumors (14).
Although EMT induced by HGF has been investigated in
various types of cancer (15,16),
the tumorigenic association between EMT and c-Met, particularly in
prostate cancer, remains unclear. Previous studies have shown that
a high level of c-Met expression is significantly implicated in
prostate cancer aggressiveness and associated with a poor clinical
outcome (17,18). Therefore, it is crucial that an
in-depth understanding of the mechanism by which c-Met signaling
regulates tumorigenic cell processes is gained in order to develop
successful therapeutic strategies. In the present study, the EMT-
and c-Met-negative LNCaP prostate cancer cells (19–22)
were used to demonstrate that the overexpression of c-Met promotes
the progression of prostate cancer via EMT.
Materials and methods
Cell culture
An EMT- and c-Met-negative human prostate cancer
cell line, LNCaP, was cultured in Dulbecco’s modified Eagle’s
medium (DMEM; Gibco-BRL, Carlsbad, CA, USA) supplemented with 10%
fetal bovine serum (FBS; Gibco-BRL), 100 U/ml penicillin and 100
μg/ml streptomycin. Cells were cultured in a 5% CO2
humidified incubator at 37°C.
Cell transfection
The full-length cDNA encoding human c-Met was
amplified and the recombinant plasmid, pcDN- A3.1/c-Met was
constructed (Invitrogen Life Technologies, Carlsbad, CA, USA).
LNCaP cells at 75% confluency were transfected with pcDNA3.1/c-Met
(LNCaP-Met cells) using Lipofectamine 2000 (Invitrogen Life
Technologies) in a six-well plate for 48 h, and transfection with
pcDNA3.1(−) alone was performed for the control group
(LNCaP-pcDNA3.1 cells). Cells were then trypsinized and seeded onto
a 10-cm dish. Stable LNCaP-Met and LNCaP-pcDNA3.1 cells were
subjected to G418 selection (400 mg/ml), and after two weeks
single-cell clones were selected and expanded. After G418
selection, five LNCaP-Met and three LNCaP-pcDNA3.1 cell clones were
selected. Identification of c-Met was performed in all these cell
clones and one clone in each group was selected to be used in the
subsequent study.
Immunofluorescence staining
To identify the c-Met expression, cells were fixed
in 4% paraformaldehyde for 10 min and blocked with goat serum
(Boshide Biotech Co. Ltd., Wuhan, China) for 30 min, then incubated
at 37°C for 1 h with rabbit anti-c-Met primary antibody (1:150;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Following
three washes with phosphate-buffered saline, the cells were
incubated with tetramethylrhodamine isothiocyanate-conjugated
secondary antibody (1:100; Boshide Biotech Co. Ltd.) at 37°C for 1
h and stained with 4′,6-diamidino-2-phenylindole for five min. The
fluorescence staining intensity and intercellular location were
examined using a fluorescence-inverted microscope (Olympus BX51;
Olympus Corporation, Tokyo, Japan).
Western blot analysis
Total cellular proteins were extracted. A 30-μg
protein extract was separated by 12% SDS-PAGE. The separated
proteins were subsequently transferred to nitrocellulose membranes
(Bio-Rad, Hercules, CA, USA), and the membrane was blocked with 5%
non-fat milk in Tris-buffered saline for 1.5 h. The membranes were
incubated for 1.5 h with the primary antibodies, polyclonal rabbit
anti-human C-met, polyclonal rabbit anti-human p-C-met, polyclonal
rabbit anti-human E-cadherin, polyclonal rabbit anti-human ERK and
polyclonal rabbit anti-human AKT, and monoclonal mouse anti-human
vimentin, monoclonal mouse anti-human p-ERK and monoclonal mouse
anti-human p-AKT (Santa Cruz Biotechnology, Inc.) at various
dilutions and hybridized for 1 h with secondary antibodies,
horseradish peroxidase-conjugated goat anti-rabbit and goat
anti-mouse IgG. (Boshide Biotech Co. Ltd.). Imaging was performed
using the electrochemiluminescence detection system (Pierce
Biotechnology, Inc., Rockford, IL, USA) and protein loading
equivalence was assessed by the expression of glyceraldehyde
3-phosphate dehydrogenase.
Cell proliferation assay
Cell growth was evaluated by MTT assay
(Sigma-Aldrich, St. Louis, MO, USA). A total of 1×104
cells/well were plated into 96-well tissue culture plates in DMEM
containing 10% FBS to a final volume of 0.2 ml. Following
incubation for 24, 48 and 72 h, cells were incubated with 20 μl MTT
to a final concentration of 0.5 mg/ml at 37°C for 4 h. Next, the
medium was removed and the precipitated formazan was dissolved by
adding 200 μl of dimethyl sulfoxide (Sigma-Aldrich). Following
agitation for 10 min, the samples were lysed and the absorbance was
detected at a wavelength of 570 nm using a microplate reader (Model
450 Mioroplate Reader; Bio-Rad).
In vitro transwell invasion assay
Polycarbonate filters (8 μm; Millipore, Billerica,
MA, USA) were coated with 50 μg/cm2 of reconstituted
Matrigel (Sigma-Aldrich). A total of 5×103 cells in 300
μl of serum-free growth medium were then seeded into the upper
chamber. Cells were incubated under normoxic conditions and allowed
to migrate towards the complete growth medium for 24 or 48 h.
Non-invading cells were removed mechanically using cotton swabs.
The inserts were stained with crystal violet and the invasive cells
on the lower surface were counted under a microscope (Olympus BX51;
Olympus Corporation).
Soft agar assay
Cells were resuspended in top agar medium (2 ml;
DMEM containing 0.4% low-melting agarose and 10% FBS), and overlaid
onto bottom agar medium (2 ml; DMEM containing 0.8% low melting
agarose and 10% FBS) in six-well culture plates. After two-three
weeks, colonies >0.1 mm in diameter were scored as positive.
Colony formation efficiency was counted under a light microscope
(Olympus BX51; Olympus Corporation).
In vivo tumorigenicity assay
A total of 30 six-week-old male athymic nude mice,
weighing 30 g (Shanghai Experimental Animal Center, Shanghai,
China) were divided into three groups: LNCaP (control; n=10),
LNCaP-pcDNA3.1 (control; n=10), and LNCaP-Met (test; n=10). The
mice were maintained under specific pathogen-free conditions and
provided with sterile food and water. Cells were harvested, washed
and resuspended in serum-free DMEM at a concentration of
1×107 cells/ml, and injected subcutaneously into the
flank of each mouse. The tumor volume was measured weekly using the
following formula: Tumor volume = (length × width2) ×
π/6. The mice were sacrificed after eight weeks. All experiments
performed complied with the Guidelines of Animal Care of Capital
Medical University (Beijing, China).
Statistical analysis
All data are presented as the mean ± standard
deviation. Student’s t-test was performed and P<0.05 was
considered to indicate a statistically significant difference. All
statistical tests were performed using SPSS version 11.0 software
(SPSS, Inc., Chicago, IL, USA).
Results
Overexpression of c-Met in LNCaP-Met
cells
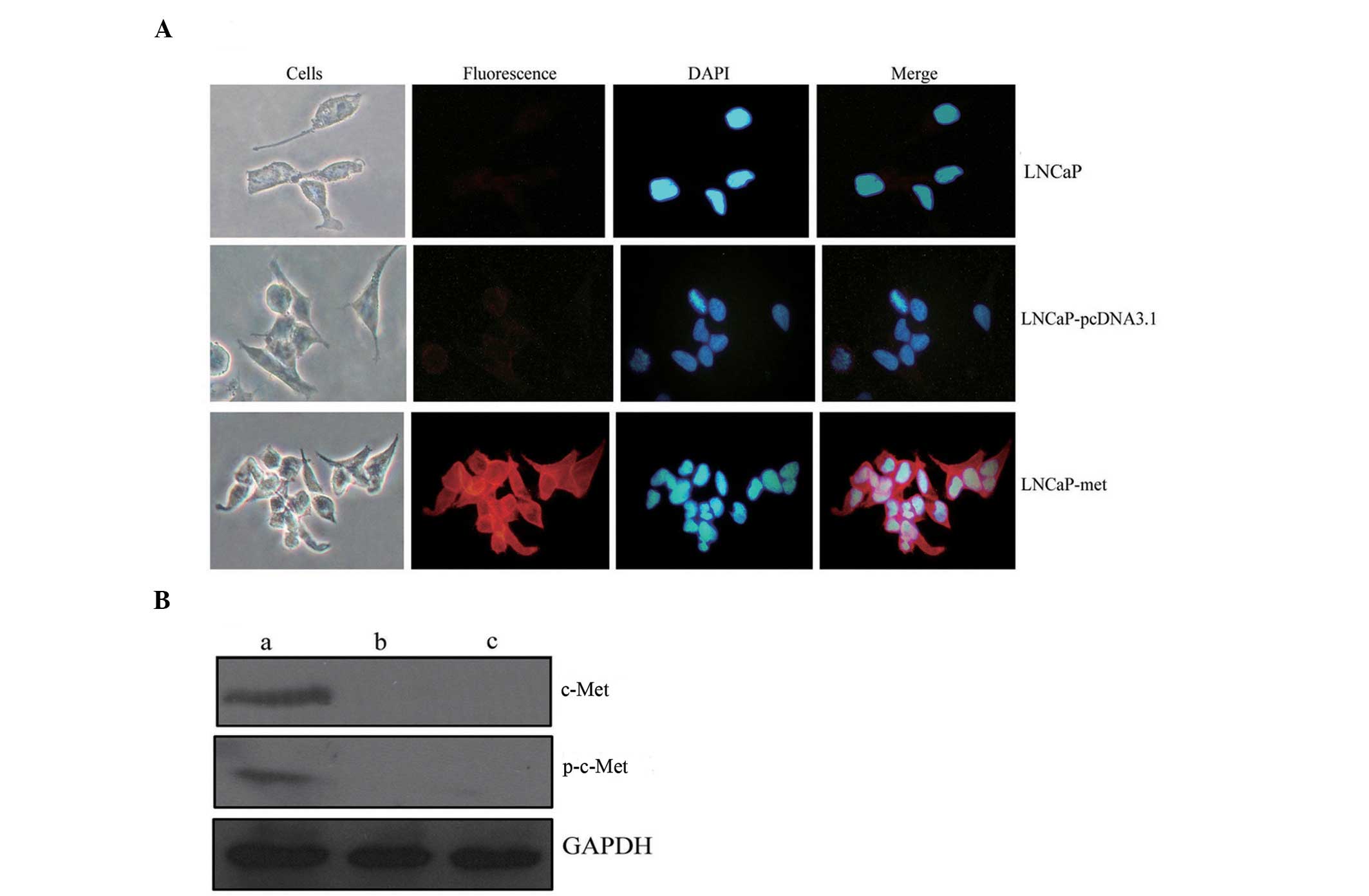
To identify the increased expression of c-Met in the
LNCaP-Met cell line, c-Met was examined using immunofluorescence
staining and western blot analysis. Immunocytochemistry indicated
significant immunofluorescence staining in the membrane of the
LNCaP-Met cells, which, by contrast, was particularly weak in the
membranes of the control LNCaP cells and the LNCaP-pcDNA3.1 cells
(Fig. 1A). Increased expression of
c-Met and phospho-c-Met was observed in the LNCaP-Met cells,
however, no protein was detected in the control LNCaP or the
LNCaP-pcDNA3.1 cells using western blot analysis (Fig. 1B). These results demonstrated that
an LNCaP cell line with stable c-Met overexpression had been
constructed and this was termed the LNCaP-Met cell line.
Effect of c-Met expression on
EMT-associated proteins
The development of EMT is characterized by the loss
of epithelial markers, such as E-cadherin and the gain of
mesenchymal markers, such as vimentin. To investigate changes in
epithelial and mesenchymal markers, western blot analysis was used
to determine the effect of c-Met on E-cadherin and vimentin in
LNCaP-Met cells, compared with the control cells. In LNCaP-Met
cells, the overexpression of c-Met downregulated E-cadherin, but
upregulated vimentin (Fig. 2),
which indicates that the overexpression of c-Met promotes an EMT
phenotype in these cells.
Effect of c-Met expression on the
proliferation, migration and tumorigenicity of LNCaP-Met cells
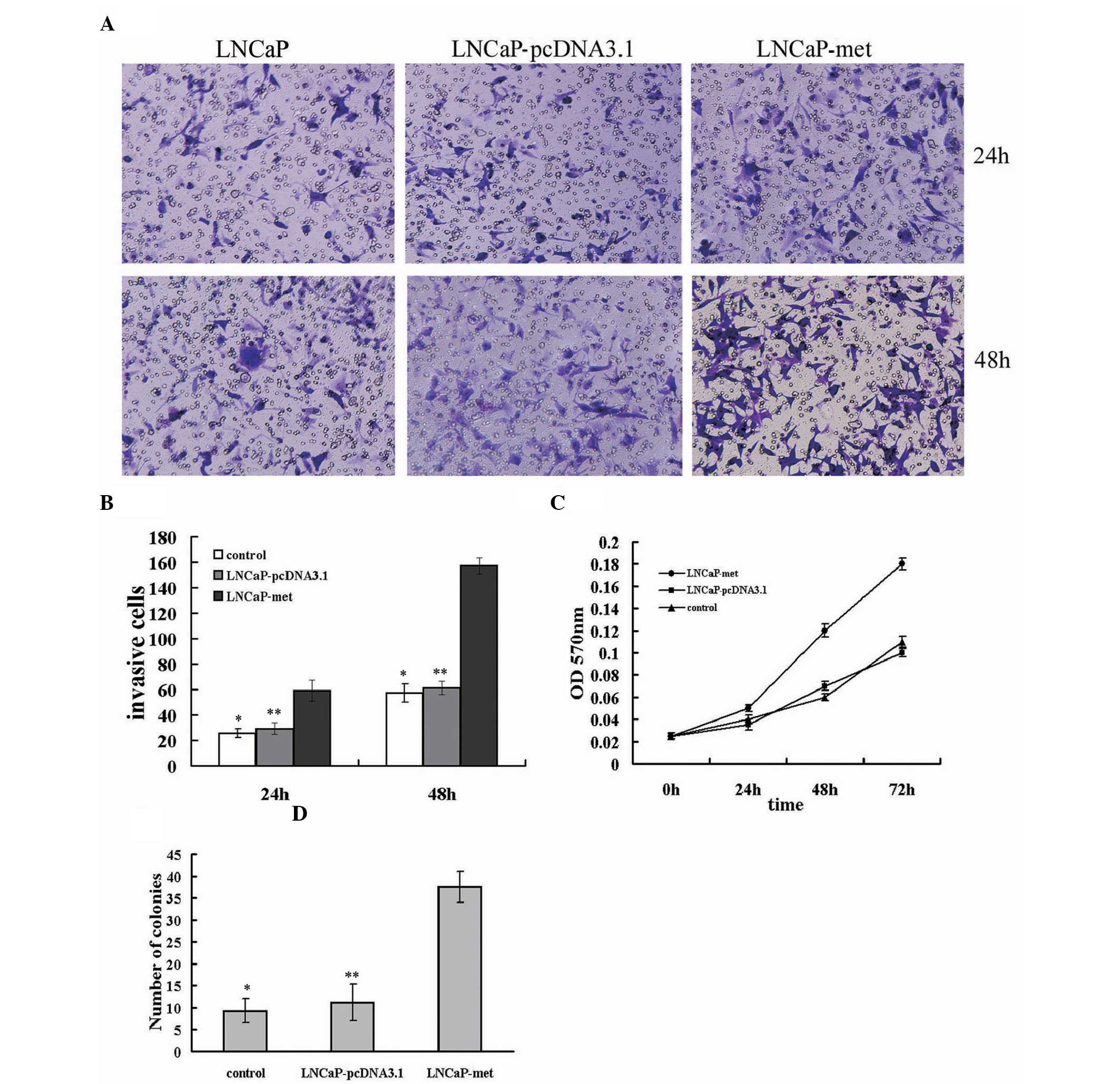
c-Met promotes the migration of cancer cells, which
is critical for metastasis. To determine the effect of increased
c-Met expression on LNCaP-Met cell migration, cells migrating to
the bottom of the insert were counted at 24 and 48 h. As shown in
Fig. 3A and B, the migratory
capacity was significantly increased, in a time-dependent manner,
in LNCaP-Met cells when compared with LNCaP and LNCaP-pcDNA3.1
cells at 24 and 48 h (P<0.05).
To examine the effect of c-Met on LNCaP cell
proliferation, the growth rate of the c-Met-transfected, control
and parental LNCaP cells was determined. The LNCaP-Met cells
exhibited a significantly higher growth rate than the
LNCaP-pcDNA3.1 and LNCaP cells (P<0.05;Fig. 3C). However, no significant
differences in cell growth rate were identified between the
LNCaP-pcDNA3.1 and LNCaP cells (P>0.05; Fig. 3C).
The increased expression of c-Met resulted in a
significant increase in the number of LNCaP-Met cell colonies
formed when compared with the number of colonies formed in the
parental LNCaP or LNCaP-pcDNA3.1 cells (P<0.05; Fig. 3D). By contrast, no significant
differences were identified between the colony numbers of the LNCaP
and LNCaP-pcDNA3.1 cells (P>0.05; Fig. 3D). Overall, these data indicate that
EMT induced by c-Met overexpression stimulates LNCaP cell
migration, proliferation and tumorigenicity.
Effect of c-Met on tumorigenicity in
vivo
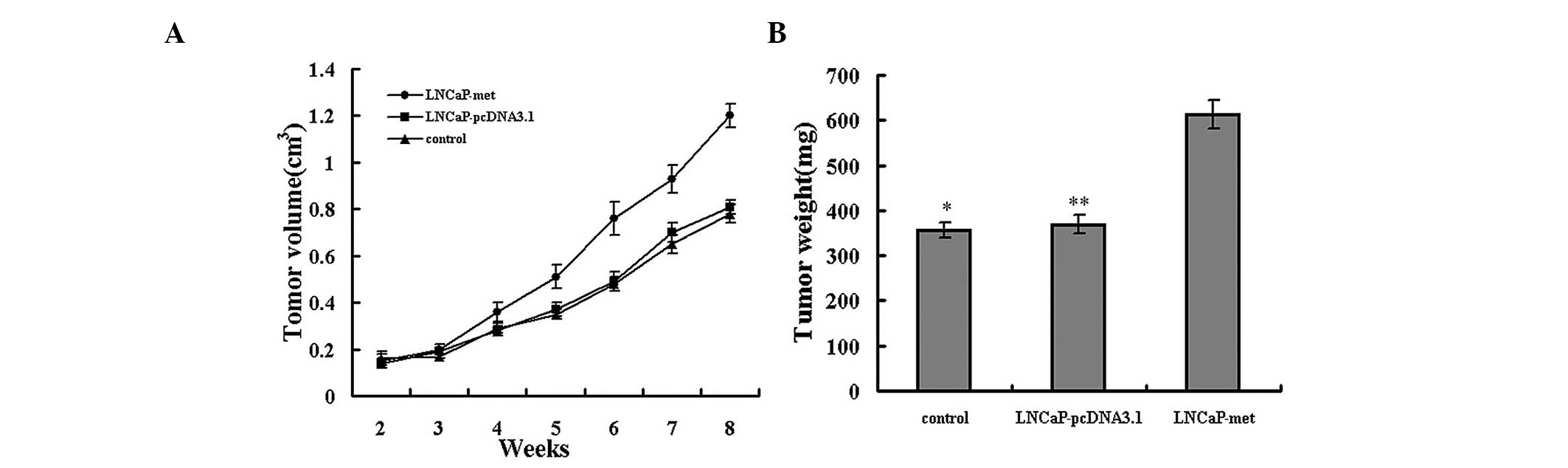
To corroborate the observation that the upregulation
of c-Met increased the invasive capacity of prostate cancer cells,
the growth rates and metastatic behavior were analyzed in
vivo. Tumor xenografts were established via subcutaneous
injection into athymic nude mice. Subcutaneous tumors developed in
five mice from the LNCaP-Met cells group, however, only grew in two
mice from the LNCaP and LNCaP-pcDNA3.1 cell groups. Metastasis was
not observed in any of the groups. The mean tumor volumes on days
14, 21, 28, 35, 42, 49 and 56 are shown in Fig. 4A. The volume of LNCaP and
LNCaP-pcDNA3.1 cell tumors were significantly smaller than the
LNCaP-Met cell tumors. On day 56 following implantation, the
average weights of the LNCaP and LNCaP-pcDNA3.1 cell tumors were
significantly lower than those of the LNCaP-Met cell tumors
(Fig. 4B). These results indicate
that EMT induction by overexpression of c-Met accelerates the
growth of tumors in vivo.
Effect of c-Met expression on
extracellular signal-regulated kinase (ERK) and AKT signaling
pathways
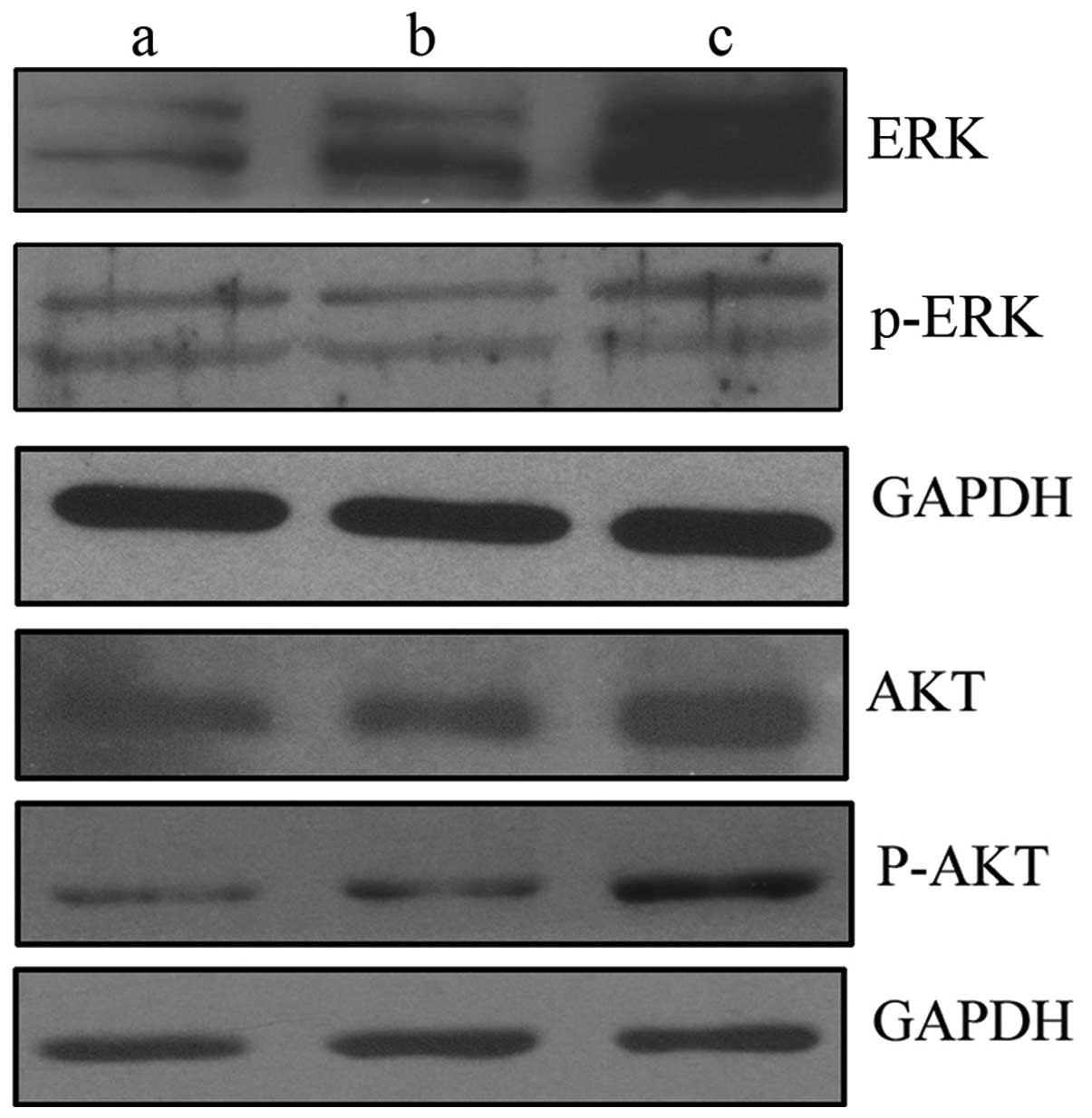
To investigate the mechanisms involved in
c-Met-induced EMT development, the levels of ERK and AKT signaling
pathway components were determined by western blot analysis. ERK
and AKT are two typical signaling pathways of c-Met. The results
indicated that the increased expression of c-Met promotes EMT by
upregulating the levels of ERK, phospho-ERK, AKT and phospho-AKT in
LNCaP-Met cells (Fig. 5).
Discussion
EMT is a process during which cancer cells lose
their epithelial phenotype and acquire a mesenchymal phenotype,
thus promoting the loosening of intercellular adhesions, detachment
from the tumor mass and invasion of neighboring tissue, blood or
lymph vessels, leading to the development of secondary tumors.
Previous studies have indicated that EMT is implicated in cancer
metastasis and invasion (23,25).
A number of factors, which induce EMT have been
identified, including transforming growth factor-β, HGF, epidermal
growth factor, fibroblast growth factor, platelet-derived growth
factor, insulin-like growth factor, microRNA, hypoxia as well as
transcription factors (25–27). Among these factors, EMT induction by
the HGF/c-Met axis is essential in certain types of cancer
(28–31), however, to the best of our
knowledge, the role of c-Met in the progression of prostate cancer
has not yet been reported.
In the present study, the effect of c-Met on the
invasiveness of human prostate cancer was examined in LNCaP cells
in vitro and in vivo. The results clearly
demonstrated that by inducing EMT, c-Met overexpression enhances
the invasion, migration and proliferation capability of LNCaP
cells.
c-Met stable expression cell lines were constructed
in c-Met- and HGF-negative LNCaP prostate cancer cells by cell
transfection. Western blot analysis revealed that the expression of
phospho-c-Met, as well as c-Met, was enhanced, which indicates that
c-Met may be activated in an HGF-independent manner. A similar
study demonstrated that c-Met may be activated via an
HGF-independent signaling pathway, following transfection, in lung
cancer (32). In addition, c-Met
and HGF may be transactivated by the mutant of epidermal growth
factor receptor (EGFR), also termed EGFRvIII (33). Furthermore, recent studies have
demonstrated that c-Met activation occurs in the absence of ligand
binding. Integrin activation, plexins, CD44, certain G
protein-coupled receptors and other receptor tyrosine kinases have
all been implicated in c-Met activation without a requirement for
HGF binding (34).
Furthermore, activation of c-Met was found to
activate the downstream signaling pathways PI3K and ERK, which
resulted in the downregulation of E-cadherin and upregulation of
vimentin. E-cadherin downregulation is regarded as a characteristic
change of EMT (35). As
intercellular adhesions are critical for the maintenance of the
epithelial phenotype, the downregulation of E-cadherin (an
essential component for adherent junctions) results in abnormal
differentiation and the loss of cell polarity, which ultimately
facilitates EMT (36). In addition,
in vitro and in vivo studies demonstrated that the
LNCaP-Met cell lines exhibited the greatest proliferative,
migratory and tumorigenic potential, indicating a potential
association between c-Met-mediated signaling pathways, EMT and
prostate cancer aggressiveness. These results are consistent with a
study showing that the acquisition of EMT characteristics is
associated with the upregulation of c-Met mRNA and increased
responsiveness to HGF in breast cancer (37).
To investigate the mechanism of EMT induction
involving c-Met, the PI3K and ERK signaling pathways were examined.
The two signaling pathways are considered to be typical
c-Met-mediated signaling pathways (38) as well as EMT signaling pathways
(39). Activation of the PI3K
signaling pathway is associated with cell motility, whereas the ERK
signaling pathway regulates cell proliferation and differentiation
(40). The results of the present
study revealed that AKT, ERK, phospho-AKT and phospho-ERK
expression increased, indicating a molecular connection between EMT
and c-Met. However, the mechanism at the transcriptional level
requires further investigation.
Abnormal c-Met activation may occur in certain
cancer types due to gene amplification, mutation or transactivation
(41). However, c-Met
overexpression as a result of upregulation at the transcriptional
level is predominant in the majority of human malignancies
(42). In the present study, it was
found that the overexpression of c-Met signaling and the subsequent
induction of EMT is potentially a common phenomenon in prostate
cancer cells. Furthermore, EMT increased prostate cancer cell
invasiveness in vitro and in vivo. All of these
results demonstrated the potential association between c-Met, EMT
and invasiveness in prostate cancer. Similarly, in other solid
human tumors, various studies have shown that c-Met-mediated
signaling activation drives EMT and cancer cell migration, invasion
and metastasis (43–45).
In conclusion, the present study demonstrates that
EMT induced by c-Met expression is involved in prostate cancer
metastasis. Through the process of EMT, LNCaP prostate cancer cells
acquire an increased proliferative, migrative and tumorigenic
capability. Thus far, the role of c-Met in the progression and
metastasis of numerous cancers has been described and a number of
c-Met inhibitors have been investigated in clinical trials
(46–48). Therefore, in addition to the
transitional prognostic and predictive value, c-Met may present a
promising therapeutic target in the fight against prostate
cancer.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 30700968 and
81341066).
References
|
1
|
Jeffers M, Rong S and Vande Woude GF:
Enhanced tumorigenicity and invasion-metastasis by hepatocyte
growth factor/scatter factor-met signaling in human cells
concomitant with induction of the urokinase proteolysis network.
Mol Cell Biol. 16:1115–1125. 1996.
|
|
2
|
Ponzetto C, Bardelli A, Zhen Z, et al: A
multifunctional docking site mediates signaling and transformation
by the hepatocyte growth factor/scatter factor receptor family.
Cell. 77:261–271. 1994.
|
|
3
|
Birchmeier C and Gherardi E: Developmental
roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends
Cell Biol. 8:404–410. 1998.
|
|
4
|
Bhardwaj V, Cascone T, Cortez MA, et al:
Modulation of c-Met signaling and cellular sensitivity to
radiation: potential implications for therapy. Cancer.
119:1768–1775. 2013.
|
|
5
|
Gibney GT, Aziz SA, Camp RL, et al: c-Met
is a prognostic marker and potential therapeutic target in clear
cell renal cell carcinoma. Ann Oncol. 24:343–349. 2013.
|
|
6
|
Knudsen BS and Vande Woude G: Showering
c-Met dependent cancers with drugs. Curr Opin Genet Dev. 18:87–96.
2008.
|
|
7
|
De Bacco F, Luraghi P, Medico E, et al:
Induction of MET by ionizing radiation and its role in
radioresistance and invasive growth of cancer. J Natl Cancer Inst.
103:645–661. 2011.
|
|
8
|
Qian LW, Mizumoto K, Inadome N, et al:
Radiation stimulates HGF receptor/c-Met expression that leads to
amplifying cellular response to HGF stimulation via upregulated
receptor tyrosine phosphorylation and MAP kinase activity in
pancreatic cancer cells. Int J Cancer. 104:542–549. 2003.
|
|
9
|
Graveel CR, Tolbert D and Vande Woude GF:
MET: a critical player in tumorigenesis and therapeutic target.
Cold Spring Harb Perspect Biol. 5:a0092092013.
|
|
10
|
Luraghi P, Schelter F, Krüger A and
Boccacciol C: The MET oncogene as a therapeutical target in cancer
invasive growth. Front Pharmacol. 3:1642012.
|
|
11
|
Feng Y, Thiagarajan PS and Ma PC: MET
signaling: novel targeted inhibition and its clinical development
in lung cancer. J Thorac Oncol. 7:459–467. 2012.
|
|
12
|
Li B, Torossian A, Sun Y, Du R, Dicker AP
and Lu B: Higher levels of c-Met expression and phosphorylation
identify cell lines with increased sensitivity to AMG-458, a novel
selective c-Met inhibitor with radiosensitizing effects. Int J
Radiat Oncol Biol Phys. 84:e525–e531. 2012.
|
|
13
|
Seiden-Long I, Navab R, Shih W, et al:
Gab1 but not Grb2 mediates tumor progression in Met overexpressing
colorectal cancer cells. Carcinogenesis. 29:647–655. 2008.
|
|
14
|
Chung JY, Davis JA, Price BD, et al:
Competitive enhancement of HGF-induced epithelial scattering by
accessory growth factors. Exp Cell Res. 317:307–318. 2011.
|
|
15
|
Ogunwobi OO and Liu C: Hepatocyte growth
factor upregulation promotes carcinogenesis and
epithelial-mesenchymal transition in hepatocellular carcinoma via
Akt and COX-2 pathway. Clin Exp Metastasis. 28:721–731. 2011.
|
|
16
|
Previdi S, Maroni P, Matteucci E, Broggini
M, Bendinelli P and Desiderio MA: Interaction between human-breast
cancer metastasis and bone microenvironment through activated
hepatocyte growth factor/Met and β-catenin/Wnt pathways. Eur J
Cancer. 46:1679–1691. 2010.
|
|
17
|
Pisters LL, Troncoso P, Zhau HE, Li W, von
Eschenbach AC and Chung LW: c-met proto-oncogene expression in
benign and malignant human prostate tissues. J Urol. 154:293–298.
1995.
|
|
18
|
Cecchi F, Rabe DC and Bottaro DP:
Targeting the HGF/Met signaling pathway in cancer. Eur J Cancer.
46:1260–1270. 2010.
|
|
19
|
Luo Y, He DL and Ning L: Expression of
‘epithelial-mesenchymal transition’ associated proteins in prostate
cancer cell lines with different metastatic potentials and its
significance. Zhonghua Nan Ke Xue. 12:696–700. 2006.(In
Chinese).
|
|
20
|
McKeithen D, Graham T, Chung LW and
Odero-Marah V: Snail transcription factor regulates neuroendocrine
differentiation in LNCaP prostate cancer cells. Prostate.
70:982–992. 2010.
|
|
21
|
Wang Y, Yue D, Li K, Liu YL, Ren CS and
Wang P: The role of TRPC6 in HGF-induced cell proliferation of
human prostate cancer DU145 and PC3 cells. Asian J Androl.
12:841–852. 2010.
|
|
22
|
Tate A, Isotani S, Bradley MJ, Sikes RA,
Davis R, Chung LW and Edlund M: Met-independent hepatocyte growth
factor-mediated regulation of cell adhesion in human prostate
cancer cells. BMC Cancer. 6:1972006.
|
|
23
|
Książkiewicz M, Markiewicz A and Zaczek
AJ: Epithelial-mesenchymal transition: a hallmark in metastasis
formation linking circulating tumor cells and cancer stem cells.
Pathobiology. 79:195–208. 2012.
|
|
24
|
Voutsadakis IA: The ubiquitin-proteasome
system and signal transduction pathways regulating Epithelial
Mesenchymal transition of cancer. J Biomed Sci. 19:672012.
|
|
25
|
Matsuoka J, Yashiro M, Doi Y, et al:
Hypoxia stimulates the EMT of gastric cancer cells through
autocrine TGFβ signaling. PLoS One. 8:e623102013.
|
|
26
|
Luo Y, He DL, Ning L, et al:
Over-expression of hypoxia-inducible factor-1α increases the
invasive potency of LNCaP cells in vitro. BJU Int. 98:1315–1319.
2006.
|
|
27
|
Lamouille S, Subramanyam D, Blelloch R and
Derynck R: Regulation of epithelial-mesenchymal and
mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell
Biol. 25:200–207. 2013.
|
|
28
|
Davidson B, Tropé B and Reich R:
Epithelial-mesenchymal transition in ovarian carcinoma. Front
Oncol. 2:332012.
|
|
29
|
Hamada S, Satoh K, Masamune A and
Shimosegawa T: Regulators of epithelial mesenchymal transition in
pancreatic cancer. Front Physiol. 3:2542012.
|
|
30
|
Talbot LJ, Bhattacharya SD and Kuo PC:
Epithelial-mesenchymal transition, the tumor microenvironment, and
metastatic behavior of epithelial malignancies. Int J Biochem Mol
Biol. 3:117–136. 2012.
|
|
31
|
Leopold PL, Vincent J and Wang H: A
comparison of epithelial-to-mesenchymal transition and
re-epithelialization. Semin Cancer Biol. 22:471–483. 2012.
|
|
32
|
Navab R, Liu J, Seiden-Long I, et al:
Co-overexpression of Met and hepatocyte growth factor promotes
systemic metastasis in NCI-H460 non-small cell lung carcinoma
cells. Neoplasia. 11:1292–1300. 2009.
|
|
33
|
Garnett J, Chumbalkar V, Vaillant B, et
al: Regulation of HGF expression by ΔEGFR-mediated c-Met activation
in glioblastoma cells. Neoplasia. 15:73–84. 2013.
|
|
34
|
Varkaris A, Gaur S, Parikh NU, et al:
Ligand-independent activation of MET through IGF-1/IGF-1R
signaling. Int J Cancer. 133:1536–1546. 2013.
|
|
35
|
Nurwidya F, Takahashi F, Murakami A and
Takahashi K: Epithelial mesenchymal transition in drug resistance
and metastasis of lung cancer. Cancer Res Treat. 44:151–156.
2012.
|
|
36
|
Shimada S, Mimata A, Sekine M, et al:
Synergistic tumour suppressor activity of E-cadherin and p53 in a
conditional mouse model for metastatic diffuse-type gastric cancer.
Gut. 61:344–353. 2012.
|
|
37
|
Jahn SC, Law ME, Corsino PE, et al: An in
vivo model of epithelial to mesenchymal transition reveals a
mitogenic switch. Cancer Lett. 326:183–190. 2012.
|
|
38
|
Martin TA, Mason MD and Jiang WG:
Hepatocyte growth factor signaling in cancer metastasis. Curr
Signal Transduct Ther. 6:180–190. 2011.
|
|
39
|
Thiery JP, Acloque H, Huang RY and Nieto
A: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009.
|
|
40
|
Gentile A, Trusolino L and Comoglio PM:
The Met tyrosine kinase receptor in development and cancer. Cancer
Metastasis Rev. 27:85–94. 2008.
|
|
41
|
Birchmeier C, Birchmeier W, Gherardi E and
Vande Woude GF: Met, metastasis, motility and more. Nat Rev Mol
Cell Biol. 4:915–925. 2003.
|
|
42
|
Benvenuti S and Comoglio PM: The MET
receptor tyrosine kinase in invasion and metastasis. J Cell
Physiol. 213:316–325. 2007.
|
|
43
|
Torres KE, Zhu QS, Bill K, et al:
Activated MET is a molecular prognosticator and potential
therapeutic target for malignant peripheral nerve sheath tumors.
Clin Cancer Res. 17:3943–3955. 2011.
|
|
44
|
Lengyel E, Prechtel D, Resau JH, et al:
C-Met overexpression in node-positive breast cancer identifies
patients with poor clinical outcome independent of Her2/neu. Int J
Cancer. 113:678–682. 2005.
|
|
45
|
Zhou AX, Toylu A, Nallapalli RK, et al:
Filamin a mediates HGF/c-MET signaling in tumor cell migration. Int
J Cancer. 128:839–846. 2011.
|
|
46
|
Inagaki Y, Qi F, Gao J, et al: Effect of
c-Met inhibitor SU11274 on hepatocellular carcinoma cell growth.
Biosci Trends. 5:52–56. 2011.
|
|
47
|
Falchook GS, Fu S, Amin HM, et al: Phase I
dose-escalation study of the oral selective C-Met inhibitor EMD
1204831 in patients with advanced solid tumours. Eur J Cancer.
47:S1582011.
|
|
48
|
Yap TA, Olmos D, Brunetto AT, et al: Phase
I trial of a selective c-MET inhibitor ARQ 197 incorporating proof
of mechanism pharmacodynamic studies. J Clin Oncol. 29:1271–1279.
2011.
|