Introduction
Pancreatic ductal adenocarcinoma behaves
aggressively and is an important cause of cancer-related mortality.
The five-year survival rate following curative surgery has been
reported to be 10–20% (1,2). Malignant pancreatic cancer cells are
characterized by uncontrolled proliferation, an inability to
express the differentiated features of normal duct cells and the
rapid invasion of adjacent tissues (3).
Peroxisome proliferator-activated receptor-γ
(PPAR-γ) is a ligand-activated transcription factor that belongs to
the nuclear hormone receptor super family. Patients who receive
PPAR-γ-activating drugs (used to treat several million patients
with type 2 diabetes mellitus) are at significantly lower risk of
lung cancer (4). The activation of
PPAR-γ in non-small cell lung cancer (NSCLC) has been shown to
inhibit the proliferation of NSCLC cells in vitro and in
xenograft models (5–8). Thiazolidinediones (TZDs), such as
pioglitazone and rosiglitazone, are a novel class of antidiabetic
drugs that attenuate the insulin resistance associated with
obesity, hypertension and impaired glucose tolerance in humans, as
well as in several animal models of non-insulin-dependent diabetes
(9). TZDs have been found to act as
ligands for PPAR-γ, a member of the nuclear receptor superfamily of
ligand-dependent transcription factors predominantly expressed in
adipose tissue (10,11). TZDs have been shown to inhibit the
growth of liposarcoma (12) and
breast (13,14), colon (15–17),
prostatic (18), gastric (19) and pancreatic (20–22)
cancer. The proliferation rates of cultured breast and colon cancer
cells are reduced by treatment with TZDs. The treatment also causes
changes in cell morphology and gene expression that is indicative
of a more differentiated state (16,23,24).
Consequently, PPAR-γ has become a potential molecular target in the
development of anticancer drugs, and TZDs have been submitted for
use in differentiation-mediated therapy in PPAR-γ-expressing
tumors. Despite promising results obtained from in vitro and
in vivo studies of tumor growth following TZD treatment, few
reported studies (24–26) have examined the effect of a PPAR-γ
ligand on the metastatic potential of cancer cells in an animal
model and the underlying molecular mechanisms. The inhibitory
effect of TZD on colon cancer cell metastasis has been previously
demonstrated in an animal model (24) with spontaneous lymph node and
hematological metastases following intra-rectal injection of tumor
cells in the nude mouse rectum. The model is suitable for
assessment of the antimetastatic efficacy of novel agents using
quantitative polymerase chain reaction (PCR) to amplify
cancer-related human β-globin DNA in the target organ (27). PPAR-γ has been shown to be expressed
in human pancreatic cancer, and TZDs have been found to inhibit
pancreatic cell proliferation in vitro (20,28,29).
TZDs inhibit the invasiveness of pancreatic cancer (22) and pioglitazone inhibits pancreatic
cancer growth in vivo (21).
However, the effect of TZDs on the metastatic potential of
pancreatic cancer cells remains unknown.
The present study aimed to investigate the
inhibitory effect of pioglitazone on the proliferation of
pancreatic cancer cell lines. In addition, the study used a rectal
xenograft model to examine whether the oral administration of
pioglitazone prevents tumorigenesis and the spontaneous lymph node
and lung metastases of pancreatic cancer.
Materials and methods
Cell lines
Capan-1 (well-differentiated adenocarcinoma), Aspc-1
(moderately-differentiated adenocarcinoma), BxPC-3
(moderately-differentiated adenocarcinoma), PANC-1
(poorly-differentiated adenocarcinoma) and MIApaCa-2
(undifferentiated carcinoma) human pancreatic cancer cell lines
were obtained from the American Type Culture Collection (Rockville,
MD, USA) and cultured in RPMI 1640 medium (Nissui Pharmaceutical
Co., Ltd., Tokyo, Japan) supplemented with 10% heat-inactivated
fetal bovine serum (JRH Biosciences, Lenexa, KS, USA), and 100 U/ml
penicillin and 100 μg/ml streptomycin, at 37°C in a humidified
atmosphere of 95% air/5% CO2.
Chemicals
Pioglitazone, donated by Takeda Pharmaceutical Co.,
Ltd., (Tokyo, Japan), was dissolved in dimethyl sulfoxide (DMSO)
and then diluted to the appropriate concentrations with culture
medium. The final concentration of DMSO in the medium was ≤0.1%
(v/v).
Cell proliferation assay
The proliferation inhibition of pancreatic cancer
cells treated with pioglitazone was determined by a standard 3-(4,
5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Each
cell line was treated with pioglitazone at various concentrations
(0.01, 0.1, 1, 10 and 100 μM) for 48 h. The percentage inhibition
was determined by comparing the cell density of the drug-treated
cells with that of the untreated control cells.
Reverse transcription (RT)-PCR
Changes in carcinoembryonic antigen (CEA),
interleukin-8 (IL-8) and cyclooxygenase-2 (COX-2) mRNA expression
levels subsequent to pioglitazone treatment in BxPC-3 cells were
evaluated by quantitative RT-PCR. CEA is a known differentiation
marker in pancreatic cancer (30).
The present study also focused on IL-8 and COX-2 as angiogenic
molecules. The BxPC-3 cells were cultured in medium containing 10
μM pioglitazone. RNA was extracted from the cells using Isogen
systems reagents (Nippon Gene Co., Ltd., Tokyo, Japan). Subsequent
to heat denaturation at 68°C for 15 min with 500 pmol oligo(dT)
primer, 10 μg RNA was reverse-transcribed to first-strand cDNA at
42°C for 60 min in a reverse-transcription solution. This solution
contained 400 units Moloney murine leukemia virus reverse
transcriptase (Invitrogen Japan K. K., Tokyo, Japan), 50 mm
Tris-HCl (pH 8.3), 75 mm KCl, 3 mm MgCl2, 0.01 M DTT,
0.5 mm of each dNTP and 16 units RNasin® (Promega
Corporation, Madison, WI, USA) to obtain a final volume of 100 μl.
Reverse-transcribed cDNA solution corresponding to 100 ng total RNA
was amplified by quantitative PCR using a 5′ nuclease assay and an
ABI Prism 7700 Sequence Detector (TaqMan; PE Biosystems Japan,
Tokyo, Japan). This reaction was conducted in a 50 μl reaction
mixture containing 200 nM forward and reverse primers, 100 nM probe
specific for the targeted cDNA and TaqMan Universal Master mix (PE
Biosystems Japan), which was comprised of Ampli-Taq Gold DNA
polymerase, dNTP, dUTP, AmpErase uracil-N-glycosylase and reaction
buffer. The PCR reaction was conducted using the ABI Prism 7700
Sequence Detector with thermo-cycler conditions as follows: 50°C
for 2 min, 95°C for 5 min, followed by 45 cycles of 95°C for 15 sec
and 60°C for 1 min. The data were examined using Sequence Detection
software (PE Biosystems Japan). The primers and probes for
amplifying CEA, IL-8 and COX-2 mRNA were purchased from PE
Biosystems Japan. Internal standard gene expression levels were
examined using TaqMan GAPDH control reagents (PE Biosystems Japan).
Target gene expression levels were standardized to the internal
standard gene expression levels. The relative expression levels of
CEA, IL-8 and COX-2 mRNA in the pioglitazone-treated BxPC-3 cells
were calculated compared with the levels in the untreated
cells.
Xenograft animal models
Five-week-old nude mice (BALB/cAnNCrj-nu/nu) were
obtained from Charles River Japan, Inc. (Kanagawa, Japan).
Following at least one week of observation, the mice were 6–7 weeks
old when the experiments were conducted. All mice were housed in
the Laboratory for Animal Experiments, Research Institute, Kanazawa
University School of Medicine (Kanazawa, Japan), under laminar
airflow conditions. Housing was temperature-controlled with a
12-h/12-h light/dark cycle. For inoculation, log-phase BxPC-3 cells
were harvested with trypsin EDTA, washed three times with RPMI and
resuspended in RPMI at 1×107 cells/ml density. The mice
were anesthetized with ether and placed in a supine position. A
7-mm incision was made in the anterior end of the anorectal wall in
the anorectal region to prevent colonic obstruction due to rectal
tumor progression. The BxPC-3 tumor cells suspended in RPMI
(1×106 cells/0.1 ml/mouse) were slowly injected
submucosally into the posterior wall with a 27-gauge needle. The
mice received solute (DMSO) or pioglitazone (20 mg/kg/day) orally
by gavage. Treatment was initiated seven days after tumor cell
inoculation and was continued five times per week for five weeks.
To evaluate the antitumor and antimetastatic effects of
pioglitazone, the mice were sacrificed by anesthesia with
pentobarbital sodium six weeks after tumor cell inoculation. The
antitumor effect of pioglitazone in the primary site was evaluated
by measuring the weight of the peri-anorectal tumor. To evaluate
metastases, the lungs and the lymph nodes surrounding the iliac
artery and abdominal aorta were excised. DNA from each specimen was
extracted by the standard proteinase K digestion-phenol chloroform
extraction method, as previously described (31). The human β-globin-related sequence
in 1 μg of each extracted DNA was amplified with primers and a
probe specific for human the β-globin gene using the ABI Prism 7700
Sequence Detector (TaqMan). The concentrations of the reaction
components, with the exception of template DNA and the
thermo-cycler conditions, were the same as those for the
aforementioned cDNA amplification. The number of metastasized tumor
cells in the excised whole organ was calculated from the standard
curve of a serial dilution series of BxPC-3 DNA, following
quantitative PCR, as previously described (27). The primer and probe sequences for
human β-globin gene amplification were as follows: Forward,
CACTGACTCTCTCTGCTATTGGTC and reverse, AGGAGTGGACAGATCCCCAAA; TaqMan
probe, 6FAM5′-CTACCCTTGGACCCAGAGGTTCTTTGAGTC-3′TAMRA. The study
design was approved by the local ethics committee for animal
experiments at the Takara-machi Campus of Kanazawa University.
Statistical analysis
The Mann-Whitney U test was used for statistical
analysis of the antitumor and antimetastatic effect of pioglitazone
in the xenograft model. Student’s t-test was used for statistical
analysis of the effect of pioglitazone on cancer cell proliferation
and the alteration of mRNA expression in vitro. P<0.05 was
considered to indicate a statistically significant difference.
Results
Proliferation inhibition effect of
pioglitazone in vitro
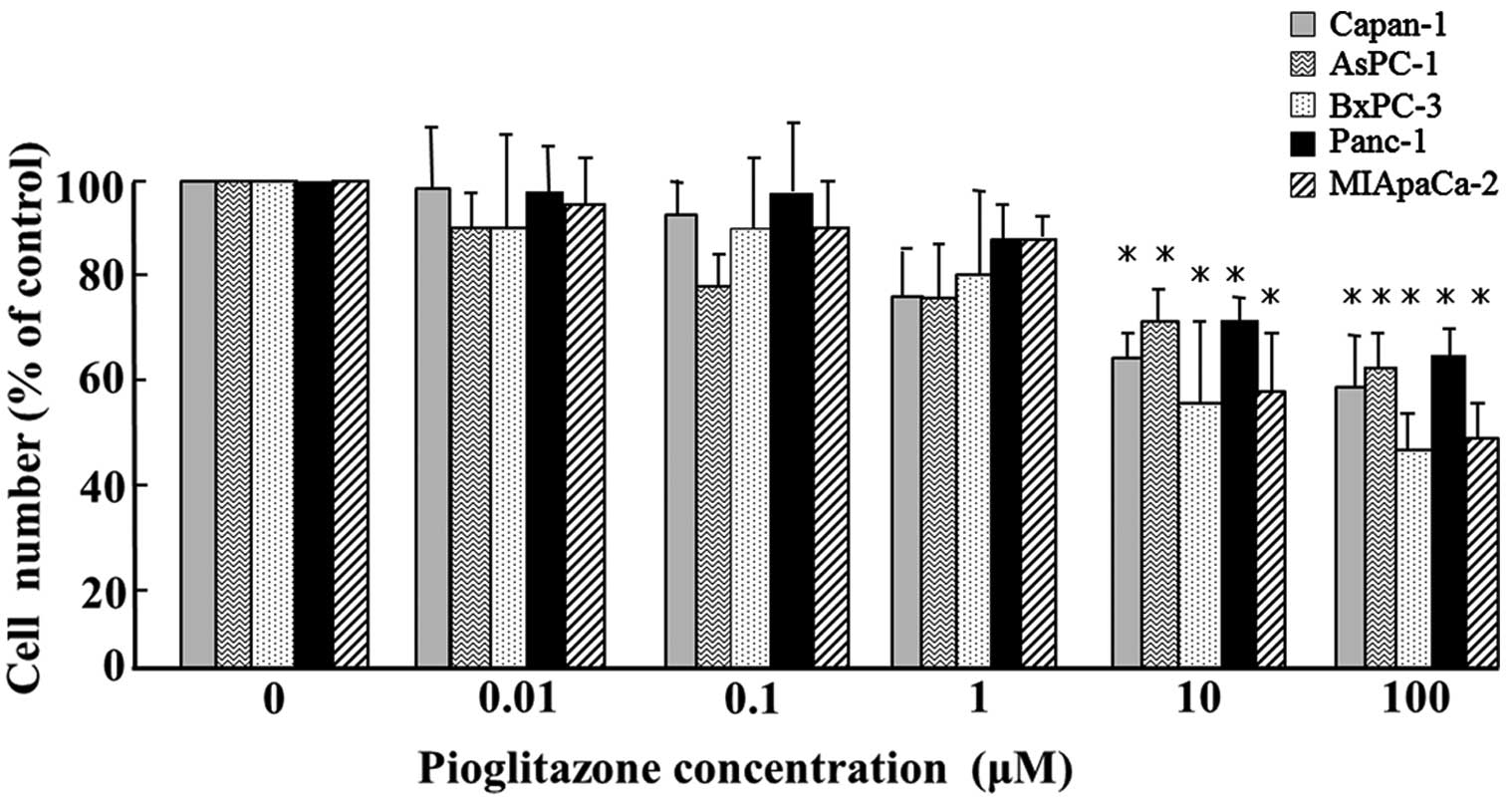
Pioglitazone significantly inhibited the
proliferation of all five pancreatic cancer cell lines (Capan-1,
Aspc-1, BxPC-3, PANC-1 and MIApaCa-2) in vitro at
concentrations >10 μM (P<0.05; Fig. 1).
Changes in molecular markers following
pioglitazone treatment
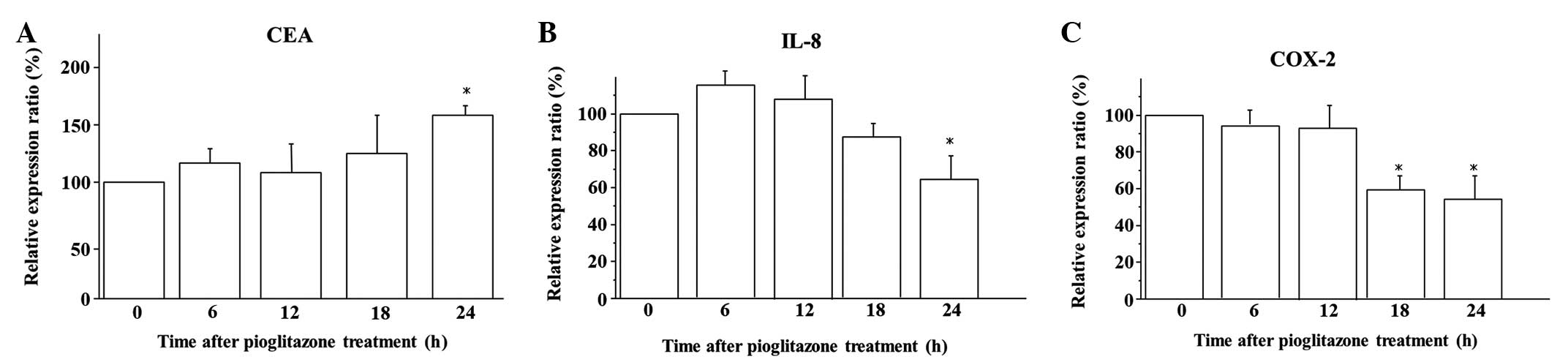
The kinetics of mRNA expression subsequent to
pioglitazone treatment in the BxPC-3 cells was analyzed by
quantitative RT-PCR. Exposure for 24 h to 10 μM pioglitazone
induced significant overexpression of CEA mRNA compared with that
observed in the untreated cells (P<0.05; Fig. 2A). Furthermore, 10 μM pioglitazone
significantly suppressed IL-8 mRNA expression after 24 h of
exposure (P<0.05; Fig. 2B) and
COX-2 mRNA expression after 18 h of exposure (P<0.05; Fig. 2C).
Antitumor and antimetastatic effects of
pioglitazone in the xenograft model
Macroscopically, the BxpPC-3 xenograft produced a
locally aggressive rectal tumor, and subsequently, lymph node
metastases surrounding the abdominal aorta appeared six weeks after
tumor cell inoculation. Pioglitazone macroscopically inhibited
xenograft growth and abdominal lymph node metastasis. The antitumor
activity of pioglitazone in the xenograft was examined by comparing
the wet weight of each xenograft, and pioglitazone was found to
significantly inhibit BxPC-3 xenograft growth by 82.6% (P=0.046;
Table I). The antimetastatic
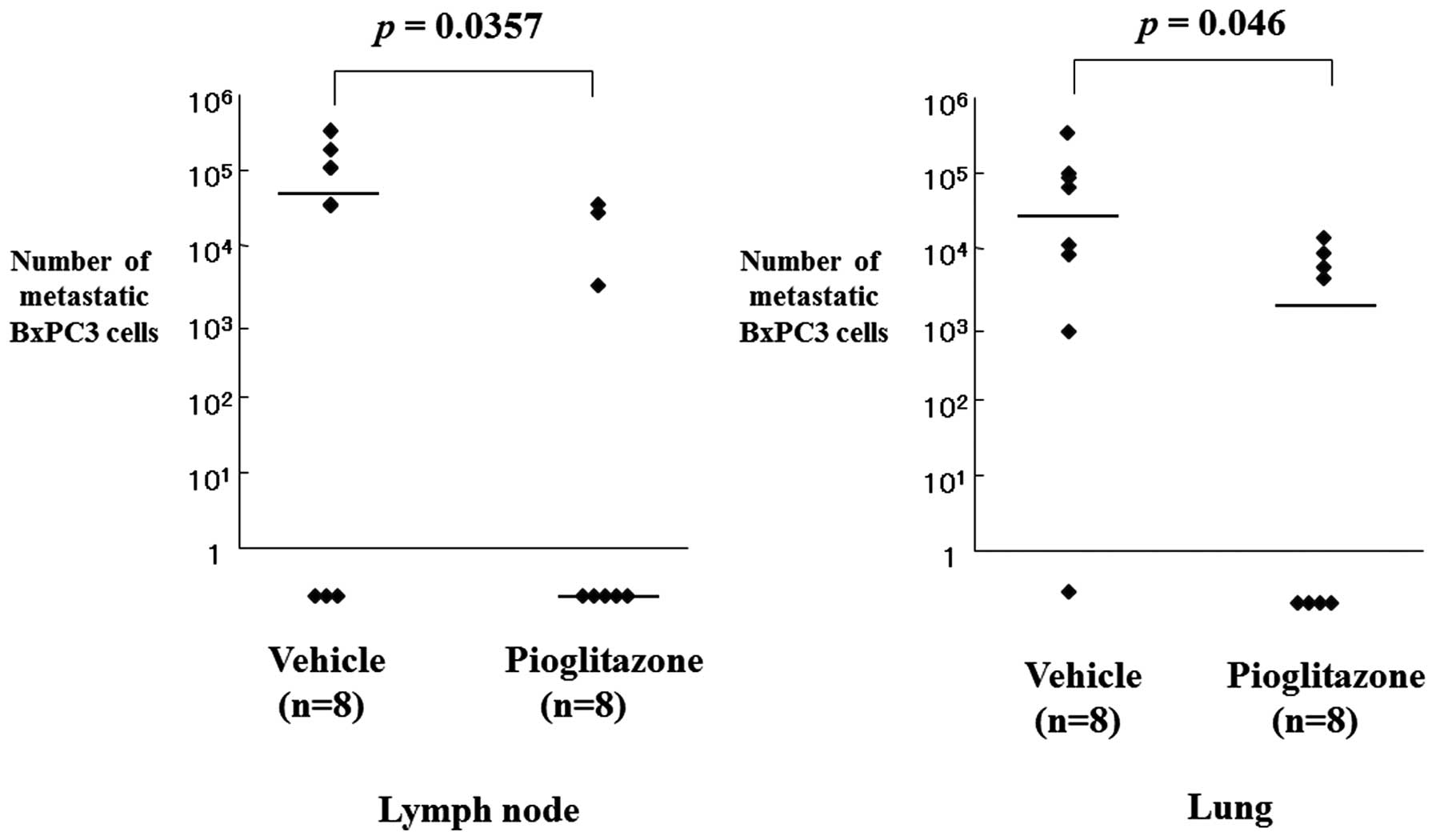
activity of pioglitazone was examined by amplifying the human
β-globin-related sequence in the lymph nodes and the lungs of
rectal xenograft mice. Quantification of cancer metastasis by
calculating the number of metastasized tumor cells using the
quantitatively-amplified β-globin gene revealed that pioglitazone
significantly inhibited lymph node and lung metastasis (P=0.035 and
P=0.046, respectively; Fig. 3;
Table II).
 | Table IEffect of pioglitazone on BxPC-3
xenografts (n=8). |
Table I
Effect of pioglitazone on BxPC-3
xenografts (n=8).
| Median tumor weight
(range), g | |
|---|
|
| |
|---|
| Tissue | Vehicle | Pioglitazone | P-value |
|---|
| Rectal xenograft | 1.67 (1.23–2.18) | 1.38 (0.67–1.74) | 0.046 |
| Table IIAntimetastatic effect of pioglitazone
in the BxPC-3 rectal xenografts. |
Table II
Antimetastatic effect of pioglitazone
in the BxPC-3 rectal xenografts.
| Median number of
metastatic BxPC3 cells (range)a | |
|---|
|
| |
|---|
| Target organ | Vehicle (n=8) | Pioglitazone
(n=8) | P-value |
|---|
| Lymph node | 7.07×104
(0–3.29×105) | 0
(0–3.57×104) | 0.035 |
| Lung | 4.92×104
(0–4.65×105) | 2.45×103
(0–1.73×105) | 0.046 |
Discussion
In the present study, a ligand of PPAR-γ
pioglitazone inhibited pancreatic cancer cell proliferation in
vitro and in vivo. Pioglitazone induced pancreatic
cancer differentiation with CEA overexpression, and inhibited
angiogenic factor IL-8 and COX-2 mRNA expression. Furthermore,
pioglitazone prevented spontaneous pancreatic cancer lymph node and
lung metastases in a xenograft model.
High PPAR-γ expression levels have been previously
identified in human pancreatic cancer cells, and TZD treatment has
been shown to inhibit cellular proliferation and induce cellular
differentiation (20). In the
present study, pioglitazone induced the upregulation of CEA mRNA
expression in the BxPC-3 cells. These results indicate that
pioglitazone also increased pancreatic cancer cell differentiation,
as CEA expression has been previously observed to be associated
with the degree of differentiation in pancreatic cancer (30). PPAR-γ activation may also result in
the differentiation of pancreatic cancer cells themselves. PPAR-γ
ligands inhibit the cellular proliferation of pancreatic cancer, a
process comparable with the terminal differentiation induced by
cessation of cell proliferation and the accumulation of cells in
the G1 phase of the cell cycle (20). The detailed mechanism of
proliferation inhibition, however, is considered to differ from
cell to cell, with the exception of that caused by PPAR-γ.
In the present study, treatment with pioglitazone
was demonstrated to inhibit lymph node and lung metastasis, as well
as tumor growth in a rectal xenograft model. Several previous
studies concerning the metastasis inhibition effect of TZDs have
been published (24,25). However, the underlying mechanism of
metastasis inhibition by TZDs remains unclear. The data from the
present study revealed that pioglitazone suppressed IL-8 and COX
mRNA expression in vitro. IL-8 is commonly overexpressed in
surgical specimens of pancreatic cancer tumor tissue (32,33),
and expression levels correlate with metastatic potential and tumor
growth (34,35). IL-8 promotes the growth of
pancreatic tumors as a primary mediator of angiogenesis (32,36),
and IL-8 expression levels have been observed to correlate with
angiogenesis, tumorigenicity and metastasis in numerous xenograft
and orthotopic in vivo tumor models, including pancreatic
cancer models (34). COX-2 is
almost undetectable in the majority of tissues under normal
physiological conditions (37),
although it is a markedly inducible molecule that is involved in
proliferation and the inflammatory response (38,39).
COX-2 and COX-2-derived prostaglandins have been observed to
mediate tumor growth and metastasis in animal models by inducing
the formation of blood vessels (40). The selective COX-2 inhibitor
celecoxib has been previously reported to inhibit lung and lymph
node metastasis in a colon cancer xenograft, and COX-2 inhibition
by celecoxib has been observed to decrease angiogenesis, vascular
endothelial growth factor expression levels and prostaglandin
E2 production (41).
Pioglitazone also suppresses COX-2 expression and inhibits the
metastasis of colon cancer cells in vivo (26), and it may inhibit xenograft
angiogenesis through the inhibition of IL-8 and COX-2 mRNA
expression. In conclusion, pioglitazone may inhibit metastasis by
the induction of cellular differentiation and a stable cell
phenotype, and by inhibition of tumor cell dissociation from the
primary tumor through antiangiogenic effects.
TZD has also been demonstrated to inhibit pancreatic
cancer cell invasiveness, a process that affects gelatinolytic and
fibrinolytic activity with a mechanism independent of PPAR-γ
activation, which involves matrix metalloproteinase 2 and
plasminogen activator inhibitor 1 expression (22). There may be several unknown
mechanisms of pioglitazone-mediated metastasis inhibition. To
clarify the precise mechanism of metastasis inhibition, further
studies are required.
Type 2 diabetes is the most common form of diabetes
and is associated with a higher risk of cancer (42). The association between diabetes and
the risk of pancreatic cancer has been investigated for a long
time. In addition, various preclinical and observational studies
have revealed that antidiabetic medications may affect the risk of
developing pancreatic cancer, although a meta-analysis of these
studies did not reveal a protective or harmful association between
antidiabetic therapies and the risk of pancreatic cancer in
patients with diabetes (43).
Recently, attention has focused on the associated between
pioglitazone and an increased risk of bladder cancer. Laboratory
animals have been shown to develop bladder tumors subsequent to the
administration of experimental drugs with dual PPAR-α and PPAR-γ
activity (44). Muraglitazar, a
dual human PPAR-α/γ agonist, has been found to induce a
dose-related increased incidence in transitional cell papilloma and
carcinoma of the urinary bladder in male rats (45). These experimental data indicate that
TZDs may be a risk factor for bladder cancer in patients with
diabetes, but findings from clinical and epidemiological studies
are inconsistent (46,47). A recent meta-analysis revealed that
pioglitazone treatment appears to be associated with a
significantly increased risk of bladder cancer in patients with
diabetes (48). A current concern
is whether the use of pioglitazone is associated with the risk of
cancer. However, the association between TZD treatment and the risk
of cancer remains controversial. The mechanisms underlying the
pro-tumor potential of pioglitazone for bladder cancer are not yet
fully understood.
As shown in the present study, pioglitazone may be
useful to prevent pancreatic cancer metastases, particularly in
cases of unresectable advanced disease in diabetic patients.
However, careful attention is required with regard to the
prophylactic use of pioglitazone following curative treatment due
to the potential to induce another malignant tumor, such as bladder
cancer. To confirm the usefulness of pioglitazone in pancreatic
cancer treatment, clinical trials are warranted.
References
|
1
|
Matsuno S, Egawa S, Fukuyama S, et al:
Pancreatic Cancer Registry in Japan: 20 years of experience.
Pancreas. 28:219–230. 2004.
|
|
2
|
Schmidt CM, Powell ES, Yiannoutsos CT, et
al: Pancreaticoduodenectomy: a 20-year experience in 516 patients.
Arch Surg. 139:718–727. 2004.
|
|
3
|
Höhne MW, Halatsch ME, Kahl GF and Weinel
RJ: Frequent loss of expression of the potential tumor suppressor
gene DCC in ductal pancreatic adenocarcinoma. Cancer Res.
52:2616–2619. 1992.
|
|
4
|
Govindarajan R, Ratnasinghe L, Simmons DL,
et al: Thiazolidinediones and the risk of lung, prostate, and colon
cancer in patients with diabetes. J Clin Oncol. 25:1476–1481.
2007.
|
|
5
|
Choudhary R, Li H, Winn RA, et al:
Peroxisome proliferator-activated receptor-gamma inhibits
transformed growth of non-small cell lung cancer cells through
selective suppression of Snail. Neoplasia. 12:224–234. 2010.
|
|
6
|
Keshamouni VG, Reddy RC, Arenberg DA, et
al: Peroxisome proliferator-activated receptor-gamma activation
inhibits tumor progression in non-small-cell lung cancer. Oncogene.
23:100–108. 2004.
|
|
7
|
Reddy RC, Srirangam A, Reddy K, et al:
Chemotherapeutic drugs induce PPAR-gamma expression and show
sequence-specific synergy with PPAR-gamma ligands in inhibition of
non-small cell lung cancer. Neoplasia. 10:597–603. 2008.
|
|
8
|
Lyon CM, Klinge DM, Do KC, et al:
Rosiglitazone prevents the progression of preinvasive lung cancer
in a murine model. Carcinogenesis. 30:2095–2099. 2009.
|
|
9
|
Olefsky JM: Treatment of insulin
resistance with peroxisome proliferator-activated receptor gamma
agonists. J Clin Inv. 106:467–472. 2000.
|
|
10
|
Braissant O, Foufelle F, Scotto C, Dauça M
and Wahli W: Differential expression of peroxisome
proliferator-activated receptors (PPARs): tissue distribution of
PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology.
137:354–366. 1996.
|
|
11
|
Kliewer SA, Forman BM, Blumberg B, et al:
Differential expression and activation of a family of murine
peroxisome proliferator-activated receptors. Proc Natl Acad Sci
USA. 91:7355–7359. 1994.
|
|
12
|
Tontonoz P, Singer S, Forman BM, et al:
Terminal differentiation of human liposarcoma cells induced by
ligands for peroxisome proliferator-activated receptor gamma and
the retinoid X receptor. Proc Natl Acad Sci USA. 94:237–241.
1997.
|
|
13
|
Mueller E, Sarraf P, Tontonoz P, et al:
Terminal differentiation of human breast cancer through PPAR gamma.
Mol Cell. 1:465–470. 1998.
|
|
14
|
Elstner E, Müller C, Koshizuka K, et al:
Ligands for peroxisome proliferator-activated receptorgamma and
retinoic acid receptor inhibit growth and induce apoptosis of human
breast cancer cells in vitro and in BNX mice. Proc Natl Acad Sci
USA. 95:8806–8811. 1998.
|
|
15
|
Sarraf P, Mueller E, Jones D, et al:
Differentiation and reversal of malignant changes in colon cancer
through PPARgamma. Nat Med. 4:1046–1052. 1998.
|
|
16
|
Brockman JA, Gupta RA and Dubois RN:
Activation of PPARgamma leads to inhibition of
anchorage-independent growth of human colorectal cancer cells.
Gastroenterology. 115:1049–1055. 1998.
|
|
17
|
Kitamura S, Miyazaki Y, Shinomura Y, et
al: Peroxisome proliferator-activated receptor gamma induces growth
arrest and differentiation markers of human colon cancer cells. Jpn
J Cancer Res. 90:75–80. 1999.
|
|
18
|
Kubota T, Koshizuka K, Williamson EA, et
al: Ligand for peroxisome proliferator-activated receptor gamma
(troglitazone) has potent antitumor effect against human prostate
cancer both in vitro and in vivo. Cancer Res. 58:3344–3352.
1998.
|
|
19
|
Takahashi N, Okumura T, Motomura W, et al:
Activation of PPARgamma inhibits cell growth and induces apoptosis
in human gastric cancer cells. FEBS Lett. 455:135–139. 1999.
|
|
20
|
Elnemr A, Ohta T, Iwata K, et al:
PPARgamma ligand (thiazolidinedione) induces growth arrest and
differentiation markers of human pancreatic cancer cells. Int J
Oncol. 17:1157–1164. 2000.
|
|
21
|
Itami A, Watanabe G, Shimada Y, et al:
Ligands for peroxisome proliferator-activated receptor gamma
inhibit growth of pancreatic cancers both in vitro and in vivo. Int
J Cancer. 94:370–376. 2001.
|
|
22
|
Galli A, Ceni E, Crabb DW, et al:
Antidiabetic thiazolidinediones inhibit invasiveness of pancreatic
cancer cells via PPARgamma independent mechanisms. Gut.
53:1688–1697. 2004.
|
|
23
|
Pignatelli M, Cortés-Canteli M, Lai C,
Santos A and Perez-Castillo A: The peroxisome
proliferator-activated receptor gamma is an inhibitor of ErbBs
activity in human breast cancer cells. J Cell Sci. 114:4117–4126.
2001.
|
|
24
|
Yoshizumi T, Ohta T, Ninomiya I, et al:
Thiazolidinedione, a peroxisome proliferator-activated
receptor-gamma ligand, inhibits growth and metastasis of HT-29
human colon cancer cells through differentiation-promoting effects.
Int J Oncol. 25:631–639. 2004.
|
|
25
|
Panigrahy D, Singer S, Shen LQ, et al:
PPARgamma ligands inhibit primary tumor growth and metastasis by
inhibiting angiogenesis. J Clin Invest. 110:923–932. 2002.
|
|
26
|
Takano S, Kubota T, Nishibori H, et al:
Pioglitazone, a ligand for peroxisome proliferator-activated
receptor-gamma acts as an inhibitor of colon cancer liver
metastasis. Anticancer Res. 28:3593–3599. 2008.
|
|
27
|
Ninomiya I, Terada I, Yoshizumi T, et al:
Anti-metastatic effect of capecitabine on human colon cancer
xenografts in nude mouse rectum. Int J Cancer. 112:135–142.
2004.
|
|
28
|
Motomura W, Okumura T, Takahashi N, Obara
T and Kohgo Y: Activation of Peroxisome Proliferator-activated
Receptor gamma by Troglitazone Inhibits Cell Growth through the
Increase of p27Kip1 in Human Pancreatic Carcinoma Cells.
Cancer Res. 60:5558–5564. 2000.
|
|
29
|
Kawa S, Nikaido T, Unno H, et al: Growth
inhibition and differentiation of pancreatic cancer cell lines by
PPAR gamma ligand troglitazone. Pancreas. 24:1–7. 2002.
|
|
30
|
Allum WH, Stokes HJ, Macdonald F and
Fielding JW: Demonstration of carcinoembryonic antigen (CEA)
expression in normal, chronically inflamed, and malignant
pancreatic tissue by immunohistochemistry. J Clin Pathol.
39:610–614. 1986.
|
|
31
|
Blin N and Stafford DW: A general method
for isolation of high molecular weight DNA from eukaryotes. Nucleic
Acids Res. 3:2303–2308. 1976.
|
|
32
|
Xie K: Interleukin-8 and human cancer
biology. Cytokine Growth Factor Rev. 12:375–391. 2001.
|
|
33
|
Kuwada Y, Sasaki T, Morinaka K, et al:
Potential involvement of IL-8 and its receptors in the invasiveness
of pancreatic cancer cells. Int J Oncol. 22:765–771. 2003.
|
|
34
|
Shi Q, Abbruzzese JL, Huang S, et al:
Constitutive and inducible interleukin 8 expression by hypoxia and
acidosis renders human pancreatic cancer cells more tumorigenic and
metastatic. Clin Cancer Res. 5:3711–3721. 1999.
|
|
35
|
Le X, Shi Q, Wang B, et al: Molecular
regulation of constitutive expression of interleukin-8 in human
pancreatic adenocarcinoma. J Interferon Cytokine Res. 20:935–946.
2000.
|
|
36
|
Koch AE, Polverini PJ, Kunkel SL, et al:
Interleukin-8 as a macrophage-derived mediator of angiogenesis.
Science. 258:1798–1801. 1992.
|
|
37
|
Wolfe MM, Lichtenstein DR and Singh G:
Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N
Engl J Med. 340:1888–1899. 1999.
|
|
38
|
Masferrer JL, Seibert K, Zweifel B and
Needleman P: Endogenous glucocorticoids regulate an inducible
cyclooxygenase enzyme. Proc Natl Acad Sci USA. 89:3917–3921.
1992.
|
|
39
|
Kujubu DA, Fletcher BS, Varnum BC, Lim RW
and Herschman HR: TIS10, a phorbol ester tumor promoter-inducible
mRNA from Swiss 3T3 cells, encodes a novel prostaglandin
synthase/cyclooxygenase homologue. J Biol Chem. 266:12866–12872.
1991.
|
|
40
|
Masferrer JL, Leahy KM, Koki AT, et al:
Antiangiogenic and antitumor activities of cyclooxygenase-2
inhibitors. Cancer Res. 60:1306–1311. 2000.
|
|
41
|
Ninomiya I, Nagai N, Oyama K, et al:
Antitumor and anti-metastatic effects of cyclooxygenase-2
inhibition by celecoxib on human colorectal carcinoma xenografts in
nude mouse rectum. Oncol Rep. 28:777–784. 2012.
|
|
42
|
Seshasai SR, Kaptoge S, Thompson A, et al:
Emerging Risk Factors Collaboration: Diabetes mellitus, fasting
glucose, and risk of cause-specific death. N Engl J Med.
364:829–841. 2011.
|
|
43
|
Singh S, Singh PP, Singh AG, et al:
Anti-diabetic medications and risk of pancreatic cancer in patients
with diabetes mellitus: a systematic review and meta-analysis. Am J
Gastroenterol. 108:510–519. 2013.
|
|
44
|
Cohen SM: Effects of PPARgamma and
combined agonists on the urinary tract of rats and other species.
Toxicol Sci. 87:322–327. 2005.
|
|
45
|
Tannehill-Gregg SH, Sanderson TP, Minnema
D, et al: Rodent carcinogenicity profile of the antidiabetic dual
PPAR alpha and gamma agonist muraglitazar. Toxicol Sci. 98:258–270.
2007.
|
|
46
|
Lewis JD, Ferrara A, Peng T, et al: Risk
of bladder cancer among diabetic patients treated with
pioglitazone: interim report of a longitudinal cohort study.
Diabetes Care. 34:916–922. 2011.
|
|
47
|
Ferrara A, Lewis JD, Quesenberry CP Jr, et
al: Cohort study of pioglitazone and cancer incidence in patients
with diabetes. Diabetes Care. 34:923–929. 2011.
|
|
48
|
Zhu Z, Shen Z, Lu Y, Zhong S and Xu C:
Increased risk of bladder cancer with pioglitazone therapy in
patients with diabetes: A meta-analysis. Diabetes Res Clin Pract.
98:159–163. 2012.
|