Introduction
Cervical cancer is the third most commonly diagnosed
cancer and the fourth leading cause of cancer-related mortalities
in females worldwide. Cervical cancer accounted for 9% (529,800) of
all newly diagnosed cancer cases and 8% (275,100) of all
cancer-related mortalities among females, worldwide in 2008
(1) and >85% of these cases and
mortalities occurred in developing countries (1). According to the American Cancer
Society, the five-year relative survival rate for uterine cervical
cancer diagnosed between 2002 and 2008 in the USA was 69% (2). At present, standard treatment for
cervical carcinoma treatment involves surgery, chemotherapy and
radiotherapy. Combined chemotherapy using cisplatin (DDP) has been
widely approved for the clinical treatment of cervical carcinoma.
Furthermore, a marked decrease in mortality of patients has been
observed with DDP-containing chemotherapy schemes in cervical
carcinoma treatment (3,4). The efficacy of DPP appears to be a
result of its ability to enter cells via multiple pathways and
produce multiple DNA-platinum adducts, which initiate the apoptotic
pathway, resulting in cell death (5). At present, DDP remains a front-line
clinical therapy and constitutes part of the treatment regimen for
patients with various types of cancer. However, drug resistance has
become a major challenge associated with successful DDP treatment
of cervical carcinoma. In addition, the molecular basis for
resistance remains unclear.
DNA-platinum adducts may initiate a cellular
self-defense system, resulting in significant epigenetic and/or
genetic alternations. Previous studies have demonstrated that the
mechanisms underlying DDP resistance are associated with supporting
cell survival, including cell growth-promoting pathways, apoptosis,
developmental pathways, DNA damage repair and endocytosis (6).
In human papillomavirus (HPV) infected cervical
carcinoma, the HPV virus produces E6 and E7 proteins (7) that inactivate cyclin-dependent kinase
inhibitors (CKI), including P16INK4A and retinoblastoma
protein (Rb), or cause the overexpression of cyclin D that releases
active E2F, which induces cell cycle traversal (8). It has been reported that irreversible
proliferation arrest is a drug-responsive program, able to
influence the outcome of cancer chemotherapy (9). Certain CKIs, including P21, P27 and
p53, regulate DDP resistance (8) by
mediating the apoptotic pathway (10,11).
However, the association between the cell cycle and DDP resistance
in HPV-infected cervical carcinoma requires further study. The
tumor suppressor and CKI, P16INK4A, which is usually
overexpressed in cervical carcinoma, is not detectable in cervical
carcinomas, which are predominantly sensitive to DDP (12,13),
and the cyclin D-CDK4,6/p16/phosphorylated RB (pRb)/E2F cascade is
altered in >80% of human tumors (14–17).
However, the association between P16INK4A and DDP
resistance in cervical carcinoma remains unclear.
In the present study, the association between p16
and DDP resistance in cevical carcinoma was investigated. The mRNA
and protein expression levels of P16, Cyclin D1 and pRb in SiHa and
SiHa-DDP cell lines were detected, and p16 knock down of a SiHa-DDP
cell line was performed to investigate the possible mechanism of
DDP chemoresistance, which may lead to the development of novel
treatment strategies for chemoresistant cervical carcinoma.
Materials and methods
Cell culture and transfection of
shRNA
SiHa and SiHa-DPP cells were purchased from
Professor Wang He (West China Second University Hospital, Sichuan,
China). The SiHa cell lines were cultured in Dulbecco’s modified
Eagle medium (DMEM; HyClone, Logan, UT, USA) supplemented with 10%
fetal bovine serum (FBS; Hangzhou Sijiqing Biological Engineering
Materials Co., Ltd., Hangzhou, China) and 50 U/ml penicillin and
streptomycin (all from Beyotime Institute of Biotechnology, Haimen,
China). Human normal oral keratinocytes were cultured in Oral
Keratinocyte Medium (ScienCell Research Laboratories, Carlsbad, CA,
USA) containing 5 ml oral keratinocyte growth supplement and 5 ml
penicillin/streptomycin solution. Transfection was performed when
cells had reached ~80% confluency. P16 shRNA (shP16) and the
control shRNA (0.1 mg; mock) (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA) vectors were transfected with lipofectamine LTX
reagent with PLUS reagent (Invitrogen Life Technologies, Carlsbad,
CA, USA). Following transfection, the cells were isolated using
culture medium. Western blot analysis was then performed to
determine the efficiency of p16 knockdown.
mRNA expression analysis
Total RNA was isolated using TRIzol Reagent
(Invitrogen Life Technologies) and reverse transcription was
performed using the PrimeScript™ First Strand cDNA synthesis kit
(Takara Biotechnology Co., Ltd., Dalian, China) according to the
manufacturer’s instructions. cDNA was generated from 5 mg of total
RNA, also according to the manufacturer’s instructions. Reverse
transcription (RT) real-time quantitative polymerase chain reaction
(qPCR) was performed to evaluate the expression levels of P16 mRNA
in the SiHa cell lines. Quantitative PCR was performed using Takara
SYBR Premix Ex Taq II (Takara Biotechnology Co., Ltd.) and an ABI
PRISM® 7500 Sequence Detection System (Applied Biosystems, Foster
City, CA, USA). The total reaction volume was 10 μl consisting of 5
μl 2× SYBR premix EX Taq™, 0.5 μl (10 μM) PCR forward primer, 0.5
μl (10 μM) PCR reverse primer, 0.5 μl cDNA and 3.5 μl
dH2O. The sequences of the gene specific primers were as
follows: Forward, 5′-CCTTTGGTTATCGCAAGCTG-3′; and reverse,
5′-CCCTGTAGGACCTTCGGTGA-3′ for P16; forward,
5′-CAAGGGTCATTATGGGTTAGGC-3′ and reverse,
5′-TTAGGTGTAGGGGAGGGGAGA-3′ for pRb; and forward,
5′-AGCCACATCGCTCAGACAC-3′ and reverse, 5′-GCCCAATACGACCAAATCC-3′
for GAPDH.
Protein expression analysis
Proteins were extracted using cell lysis buffer
(Beyotime Institute of Biotechnology) according to the
manufacturer’s instructions. The protein concentration was
quantified using the Enhanced BCA Protein Assay kit (Beyotime
Institute of Biotechnology). For western blot analysis, equal
amounts of total protein (20 μg) were boiled and separated by
SDS-PAGE. Following electrophoresis, the protein was blotted onto a
polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA,
USA) and blocked for 2 h at room temperature. The membranes were
then incubated with human anti-mouse monoclonal p16 antibody
(Sigma-Aldrich, St. Louis, MO, USA), human anti-mouse polyclonal
pRb and β-actin antibodies (Cell Signaling Technology Inc.,
Danvers, MA, USA), human anti-rabbit monoclonal CDK4 antibody (Cell
Signaling Technology, Inc.) overnight at 4°C. The membranes were
then washed with Tris-buffered saline three times prior to
incubation with horseradish peroxidase (HRP)-conjugated goat
anti-rabbit CDK4 and HRP-conjugated goat anti-mouse p16, Rb and
β-actin secondary antibodies (Beyotime Institute of Biotechnology)
and visualized using an ECL substrate (Thermo Fisher Scientific,
Waltham, MA, USA). The membranes were scanned using a myECL Imager
(Thermo Fisher Scientific) and the relative level of protein
expression was analyzed by ImageJ software (imagej.nih.gov/ij/).
3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
Cells proliferation was measured by MTT assay
(Funakoshi Co., Tokyo, Japan) (20)
in 96-well microculture plates (Thermo Fisher Scientific). The
cells were seeded at a density of 1×104 cells/well in
96-well plates in DMEM containing 10% FBS. Three duplicate wells
were set up for each group and the experiment was peformed in
triplicate. After 48 h incubation, 20 μl MTT solution (5 mg/ml) in
phosphate-buffered saline (PBS; Beyotime Institute of
Biotechnology), was added to each well for 4 h. The absorbance of
each well was analyzed using an Infinite F50 microplate reader
(Tecan, Männedorf, Switzerland) at a wavelength of 570 nm.
Proliferation curves were plotted according to the optical
density.
Apoptosis assay
Apoptosis detection was carried out using Annexin V
flow cytometry (Becton Dickinson, San Jose, CA, USA) as previously
described (21). Cells were treated
with 2 mM DDP or left untreated, and adherent cells were then
harvested after 72 h and washed in cold PBS, centrifuged at 200 × g
at 4°C for 5 min and resuspended in Annexin-binding buffer. Annexin
V and propidium iodide (Invitrogen Life Technologies) were added to
the cell resuspension and the stained cells were analyzed by flow
cytometry using the S3 cell sorter (Bio-Rad, Hercules, CA, USA) at
wavelengths of 530 and >575 nm.
Statistical analysis
Statistical significance was determined using SPSS,
version 16.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference. Data
are expressed as the mean ± standard error of the mean.
Results
High P16INK4A expression is
associated with reduced cellular growth and apoptosis in SiHa-DPP
cells
The DDP-sensitive and -resistant cervical carcinoma
cell lines were used to compare DDP responses. MTT assay and flow
cytometry were performed to identify the characteristics of human
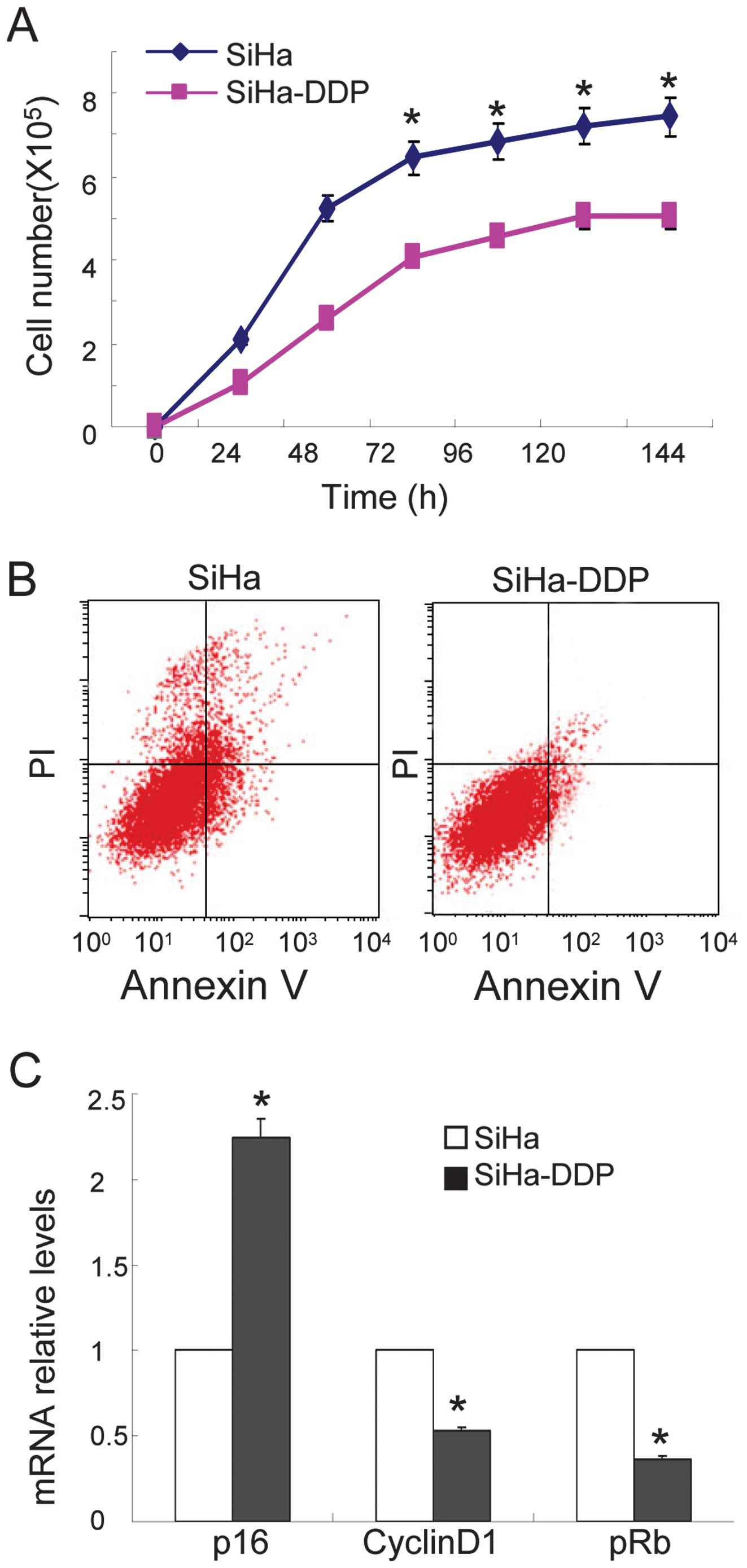
cervical cancer cells (SiHa) and SiHa-DDP cells. As shown in
Fig. 1A, reduced cellular growth
was observed in the SiHa-DDP cells when compared with SiHa cells,
and less apoptosis was observed in SiHa-DDP cells following
treatment with DDP (Fig. 1B). qPCR
demonstrated that the levels of P16INK4A increased
(Fig. 1C) while cyclin D1 and pRb
levels were downregulated in SiHa-DDP cells when compared with SiHa
cells. These results indicate that SiHa cells resistance to DDP was
associated with high P16INK4A expression levels.
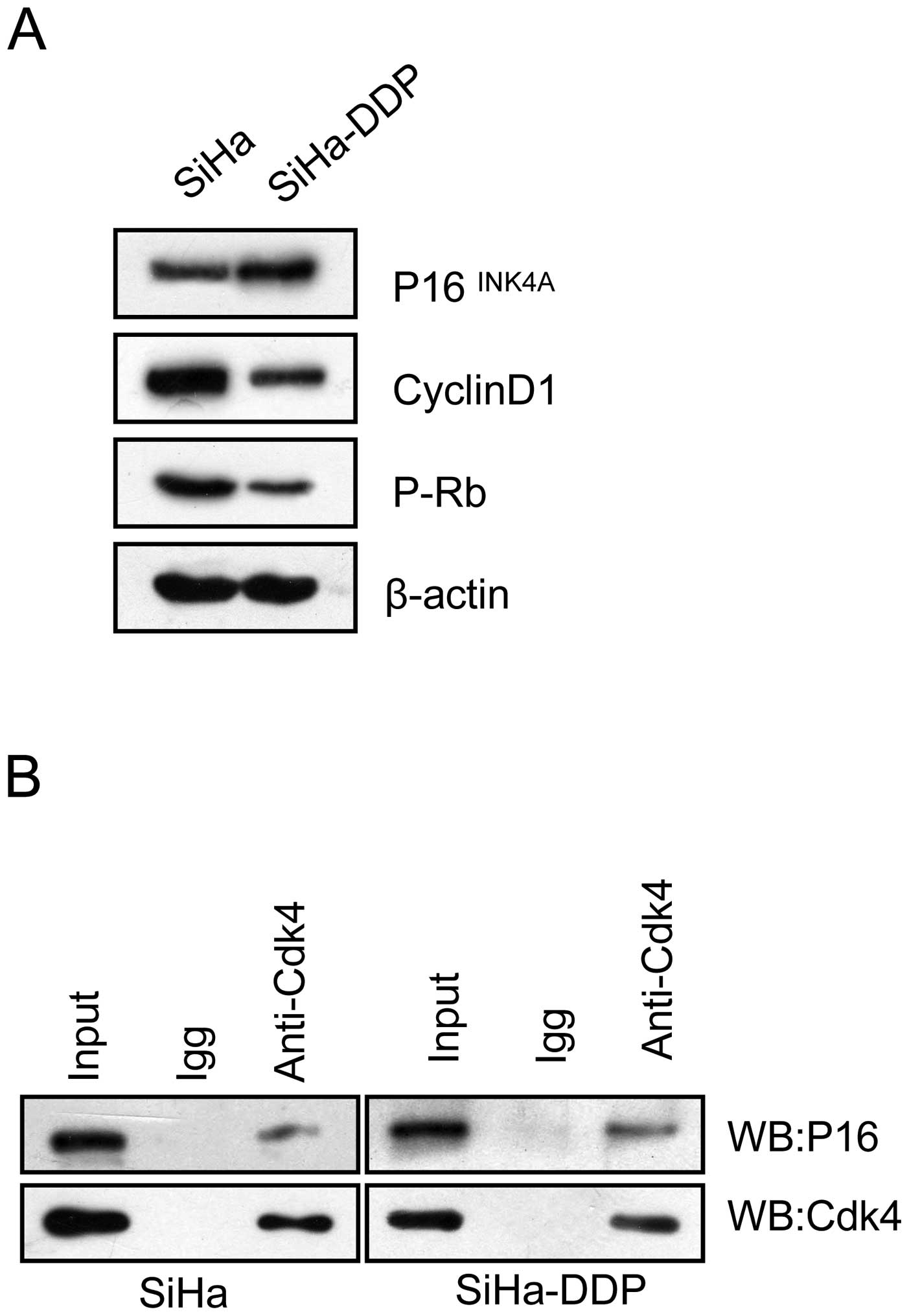
Levels of P16INK4A-CDK4
complex increase in SiHa-DPP cells with pRb inactivation
It has been hypothesized that the cyclin D1-CDK4-pRb
pathway causes DDP resistance via cell cycle arrest (14,15).
Therefore, the influence of P16 on the cyclin D1-CDK4-pRb pathway
in SiHa-DPP cells was analyzed. Co-immunoprecipitation was
performed to evaluate the interaction between P16INK4A
and CDK4 in SiHa-DDP cells and SiHa cells. The results showed that
P16 was coprecipitated with CDK4 in both cell lines (Fig. 2A), and the interaction in SiHa-DDP
was enhanced when compared with SiHa cells, indicating that the
P16-CDK4 complex existed and may be involved in DDP-resistance. To
investigate whether the enhanced levels of the P16-CDK4 complex
contributes to cell cycle arrest, western blot analysis was
performed to detect changes in pRb activity. As shown in Fig. 2B, pRb levels decreased concurrently
with the upregulation of P16INK4A protein levels in
SiHa-DDP cells, indicating that P16 may compete with cyclin D1 to
interact with CDK4, thus inhibiting pRb activation associated with
DDP-resistance in SiHa-DDP cells.
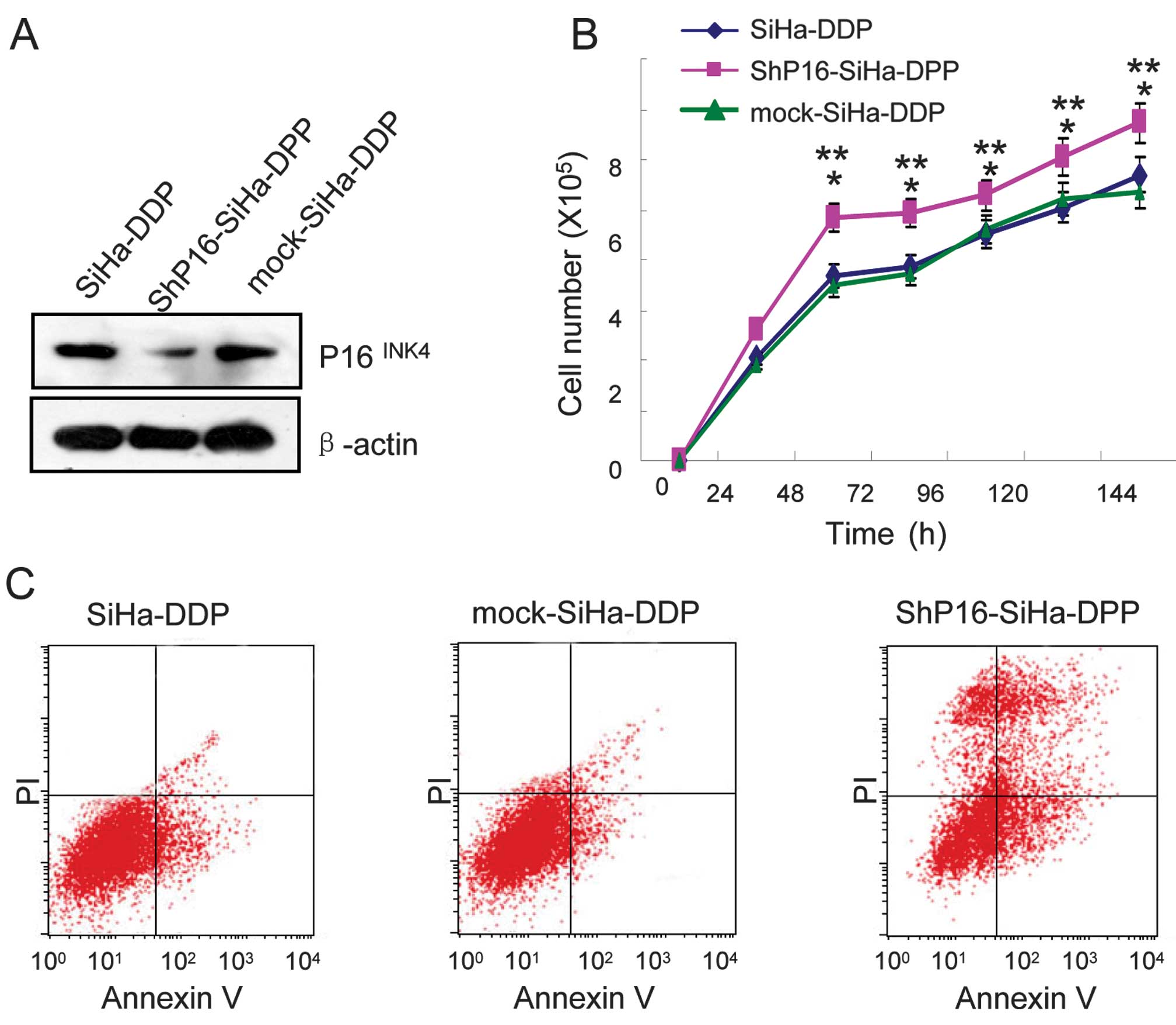
Knockdown of P16INK4A induces
growth and apoptosis in cells treated with DDP
To investigate whether P16INK4A regulates
cellular growth and apoptosis in SiHa-DPP cells and contributes to
DDP-resistance, shRNA and mock of P16INK4A were
transfected into SiHa-DPP cells. Western blot analysis was used to
evaluate the protein levels of P16INK4A. It was
indicated that P16INK4A in shRNA-transfected cells was
markedly downregulated when compared with mock-transfected cells
(Fig. 3A). To investigate the
proliferative effects in shP16-transfected cells, cellular growth
was monitored for 144 h. The shP16-transfected SiHa-DPP cells
exhibited a significant increase in cellular growth when compared
with mock-transfected cells (P=0.05; Fig. 3B). Therefore, the effect of DDP
treatment on the sensitivity of P16INK4A knockdown cells
to DNA damage was assessed. Flow cytometry revealed that
shP16-transfected cells were more sensitive to DDP-induced
apoptosis when compared with mock-transfected cells (Fig. 3C).
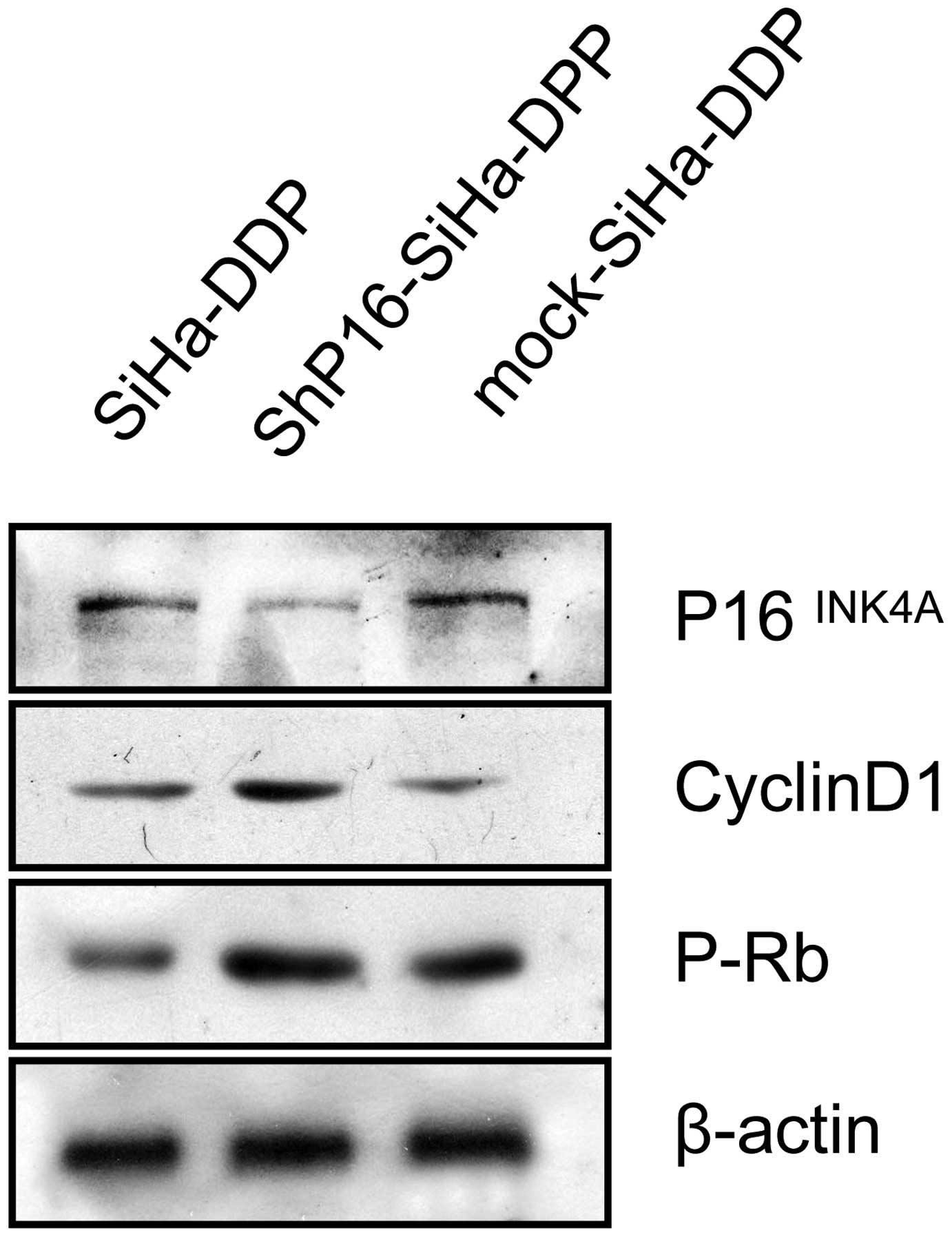
P16INK4A inactivates pRb in
SiHa-DDP cells
To identify the mechanism by which downregulated
P16INK4A influences proliferation, which contributes to
DDP resistance, the protein expression of pRb and cyclin D1 in
SiHa, mock-transfected SiHa-DDP cells and shP16-transfected
SiHa-DDP cells was investigated. pRb and cyclin D1 were
significantly increased in shP16-transfected SiHa-DDP cells
(Fig. 4).
Discussion
Rapidly proliferating cancer cells may be
successfully killed by numerous chemotherapeutic drugs; however,
several healthy proliferating cells are also damaged in the
process, including hair follicle cells, intestinal cells and
hematopoietic precursors. This nonselective killing of rapidly
proliferating healthy cells often causes adverse effects in cancer
patients. Several studies have indicated that CKIs may be used to
protect normal cells from the toxicity caused by chemotherapy
(8).
The cyclin D-CDK4,6/p16/pRb/E2F cascade has been
found to be altered in a number of tumors (18). During the G1 phase of the cell
cycle, Rb is inactivated by sequential phosphorylation events,
which are mediated by various CKIs, including P16INK4A,
which leads to the release of the E2F transcription factors and
promotion of the cell cycle. It appears that the threshold level of
the E2F1 protein, as well as the cell type, determine the function
of the gene (17). DDP-resistant
cells exhibited less and delayed growth when compared with
non-resistant (siHa) cells (13). A
reasonable explanation may be that the depressed growth of siHa-DDP
cells was attributable to an elevated cellular p16 level, which
caused proliferation arrest.
In the present study, SiHa human cervical carcinoma
cells, which are known to be infected with HPV, were used (8). The results showed high
P16INK4A expression and its enhanced interaction with
CDK4 in cervical carcinoma DDP-resistance cells (SiHa-DPP), which
may cause irreversible proliferation arrest. Knockdown of
P16INK4A induced cells exiting the G1 phase into the S
phase and promoted their proliferation, which enhanced DDP
chemosensitivity as DDP targets cells in the S phase. Previous
studies have revealed that the induction of P16INK4A,
p21Waf1 or p27Kip1 lead to significant
resistance to DDP-mediated cytotoxicity in the human cervical
cancer cells, but not in the SiHa cells (19). This indicated that high expression
of p16INK4A in SiHa-DDP cells was required for DDP
resistance, but did not cause it, implying that additional factors
coordinated with P16INK4A, leading to DDP resistance in
HPV-infected cervical carcinoma cells. Therefore, future studies
are required to confirm the identity of such factors.
CKI-mediated resistance to chemotherapy may be a
useful approach to protect normal cells from chemotherapy-induced
toxicity in patients with pRb pathway-impaired cancer. Our results
provided the first evidence that P16INK4A regulated DDP
resistance in cervical carcinoma SiHa cells, which may lead to the
development of novel strategies for the treatment of chemoresistant
cervical carcinoma.
Acknowledgements
The authors would like to thank all members of the
Department of Obstetrics and Gynecology, The Third Xiangya Hospital
of Central South University (Changsha, China).
References
|
1
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kondagunta GV, Sheinfeld J, Mazumdar M, et
al: Relapse-free and overall survival in patients with pathologic
stage II nonseminomatous germ cell cancer treated with etoposide
and cisplatin adjuvant chemotherapy. J Clin Oncol. 22:464–467.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Einhorn LH: Role of the urologist in
metastatic testicular cancer. J Clin Oncol. 25:1024–1025. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Siddik ZH: Cisplatin: mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2004. View Article : Google Scholar
|
|
6
|
Shen DW, Pouliot LM, Hall MD and Gottesman
MM: Cisplatin resistance: a cellular self-defense mechanism
resulting from multiple epigenetic and genetic changes. Pharmacol
Rev. 64:706–721. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
del Moral-Hernández O, López-Urrutia E,
Bonilla-Moreno R, Martínez-Salazar M, Arechaga-Ocampo E, Berumen J
and Villegas-Sepúlveda N: The HPV-16 E7 oncoprotein is expressed
mainly from the unspliced E6/E7 transcript in cervical carcinoma
C33-A cells. Arch Virol. 155:1959–1970. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schmidt M and Fan Z: Protection against
chemotherapy-induced cytotoxicity by cyclin-dependent kinase
inhibitors (CKI) in CKI-responsive cells compared with
CKI-unresponsive cells. Oncogene. 20:6164–6171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rebbaa A: Targeting senescence pathways to
reverse drug resistance in cancer. Cancer Lett. 219:1–13. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Koster R, di Pietro A, Timmer-Bosscha H,
Gibcus JH, van den Berg A, Suurmeijer AJ and Bischoff R:
Cytoplasmic p21 expression levels determine cisplatin resistance in
human testicular cancer. J Clin Invest. 120:3594–3605. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mandic R, Schamberger CJ and Müller JF:
Reduced cisplatin sensitivity of head and neck squamous cell
carcinoma cell lines correlates with mutations affecting the
COOH-terminal nuclear localization signal of p53. Clin Cancer Res.
11:6845–6852. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sari Aslani F, Safaei A, Pourjabali M and
Momtahan M: Evaluation of Ki67, p16 and CK17 markers in
differentiating cervical intraepithelial neoplasia and benign
lesions. Iran J Med Sci. 38:15–21. 2013.PubMed/NCBI
|
|
13
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nevins JR: The Rb/E2F pathway and cancer.
Hum Mol Genet. 10:699–703. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kaye FJ: RB and cyclin dependent kinase
pathways: defining a distinctionbetween RB and p16 loss in lung
cancer. Oncogene. 21:6908–6914. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Giacinti C and Giordano A: RB and cell
cycle progression. Oncogene. 25:5220–5227. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Crosby ME and Almasan A: Opposing roles of
E2Fs in cell proliferation and death. Cancer Biol Ther.
3:1208–1211. 2004. View Article : Google Scholar
|
|
18
|
Pei XH and Xiong Y: Biochemical and
cellular mechanisms of mammalian CDK inhibitors: a few unresolved
issues. Oncogene. 24:2787–2795. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen XL, Wang H, Zhang XM, et al:
Establishment of a cisplatin-resistant human cervical cancer cell
line. Sichuan Da Zue Zue Bao Yixue Ban. 43:151–155. 2012.(In
Chinese).
|
|
20
|
Uchida F, Uzawa K and Kasamatsu A:
Overexpression of CDCA2 in human squamous cell carcinoma:
correlation with prevention of G1 phase arrest and apoptosis. PLoS
One. 8:e563812013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Al-Khalaf HH, Colak D and Al-Saif M: p16
(INK4a) positively regulates cyclin D1 and E2F1 through negative
control of AUF1. PLoS One. 6:e211112013. View Article : Google Scholar
|