Introduction
Pheochromocytomas and paragangliomas are
neuroendocrine tumors that arise from the chromaffin cells of the
adrenal medulla and extra-adrenal paraganglia, respectively.
Pheochromocytomas are occasionally termed intra-adrenal
paragangliomas (1). The annual
incidence rate of paraganglioma is ~1.5 cases per million
individuals worldwide, and the peak incidence occurs between the
ages of 30 and 50 years (2).
Paragangliomas may be derived from the parasympathetic or
sympathetic ganglia. The majority of paragangliomas derived from
the parasympathetic ganglia occur in the head and neck, while the
majority of paragangliomas derived from the sympathetic ganglia
occur in the abdomen, adjacent to the adrenal glands (1). However, paragangliomas may occur in
any region where the sympathetic or parasympathetic ganglia exist.
Despite the majority of paragangliomas being sporadic, a genetic
association has been identified in <50% of cases due to the
evolution in genetic testing (3).
In total, ~25% of paragangliomas are malignant and metastasis may
arise ≥20 years following the first presentation (4). Due to the difficulty of confirming
malignancy, all paragangliomas should be considered malignant. The
only definitive sign of malignancy is distant metastasis to organs
such as the bone, liver and lymph nodes (2). Due to the majority of paragangliomas
are chemo- and radioresistant, surgical excision of the tumor is
the primary treatment (2,5,6). The
five-year survival rate of patients with malignant paragangliomas
is ~60% (6). The present study
reports a case of a jejunal mass resulting in an obstruction that
was treated laparoscopically and diagnosed as malignant
paraganglioma. The patient possessed a history of left nephrectomy
due to malignant pheochromocytoma that had invaded into the left
kidney eight months prior to presentation. Written informed consent
was obtained from the patient.
Case report
The present study reports the case of a 70-year-old
male that presented to the Emergency Department of Bezmialem Vakif
University Hospital (Istanbul, Turkey) complaining of abdominal
pain, a lack of defecation for one week and vomiting of two-day
duration. The medical history of the patient included left
nephrectomy due to malignant pheochromocytoma that had invaded into
the left kidney eight months prior to presentation. A coronary
artery bypass graft operation two years previously and
acetylsalicylic acid use were also reported in the medical history.
The vital signs were stable, without hypertension and tachycardia.
Physical examination revealed abdominal tenderness in the
epigastric region, with mild abdominal distention. No abdominal
guarding was present, but rebound tenderness occurred. The physical
examination also identified hyperkinetic bowel sounds and empty
rectal ampulla upon digital examination. Sternotomy and left
subcostal incision scars were clearly visible on the patient. A
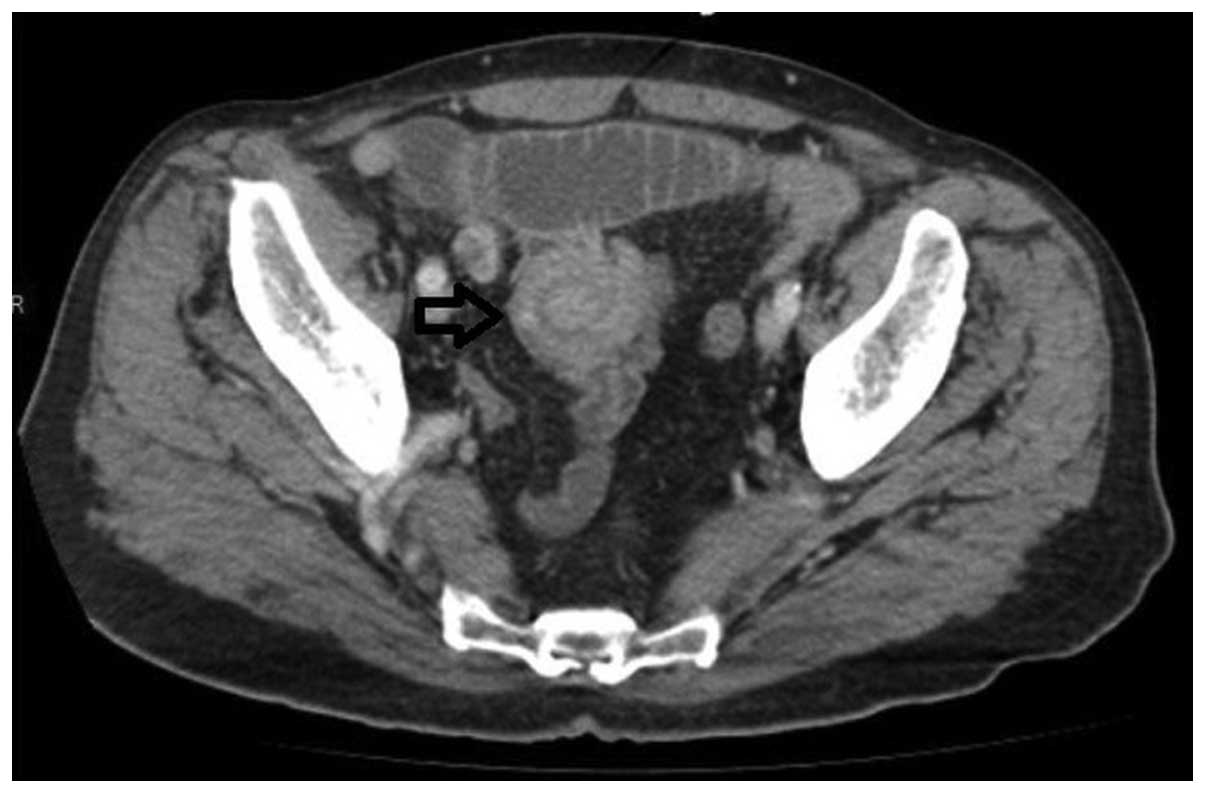
contrast-enhanced abdominal computed tomography (CT) scan was
performed. The CT scan revealed the jejunojejunal invagination of a
contrast enhancing mass 20×9 mm in size located at the distal
jejunum. The jejunum was dilated, resulting in a proximal diameter
≤3.5 cm. The CT findings featured a hypervascular target sign
resembling a neuroendocrine tumor (Fig.
1).
Informed consent was obtained from the patient prior
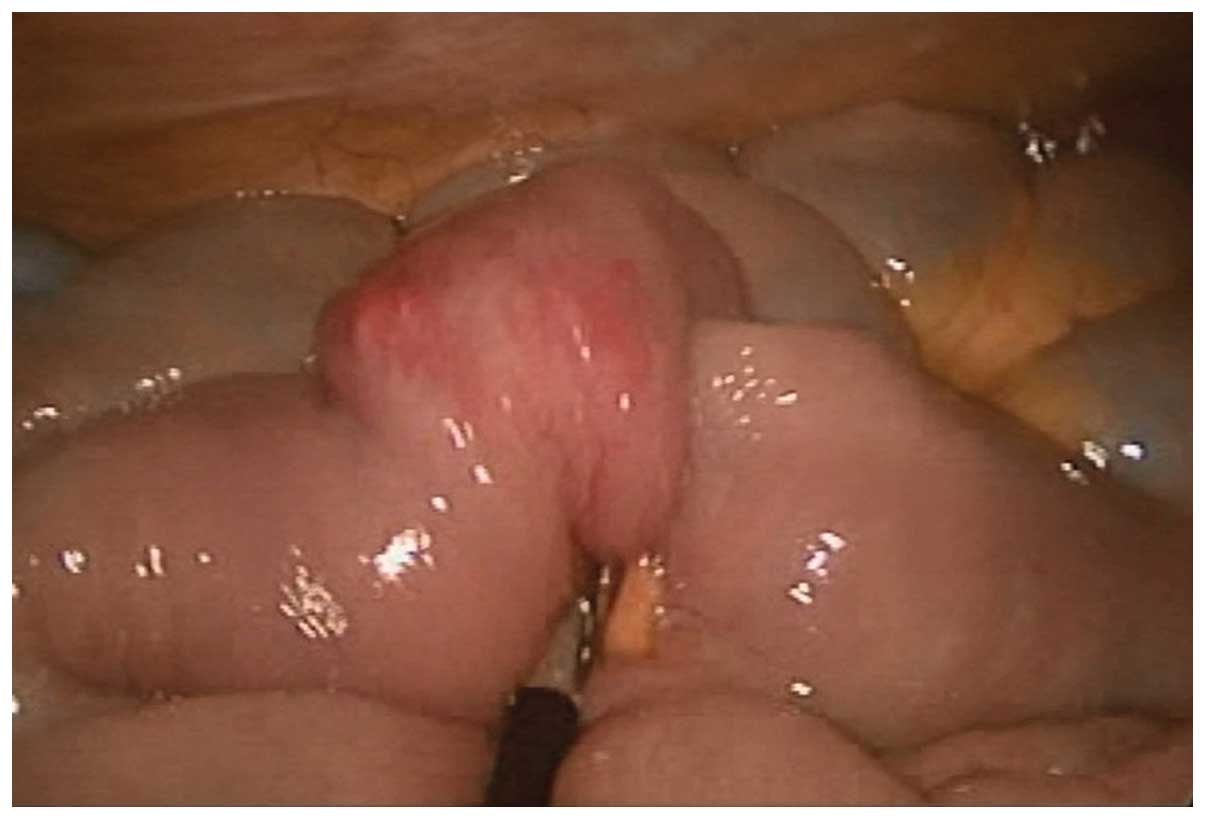
to undergoing surgery. In the present patient, a laparoscopic
approach was preferred. The invaginated jejunal segment was
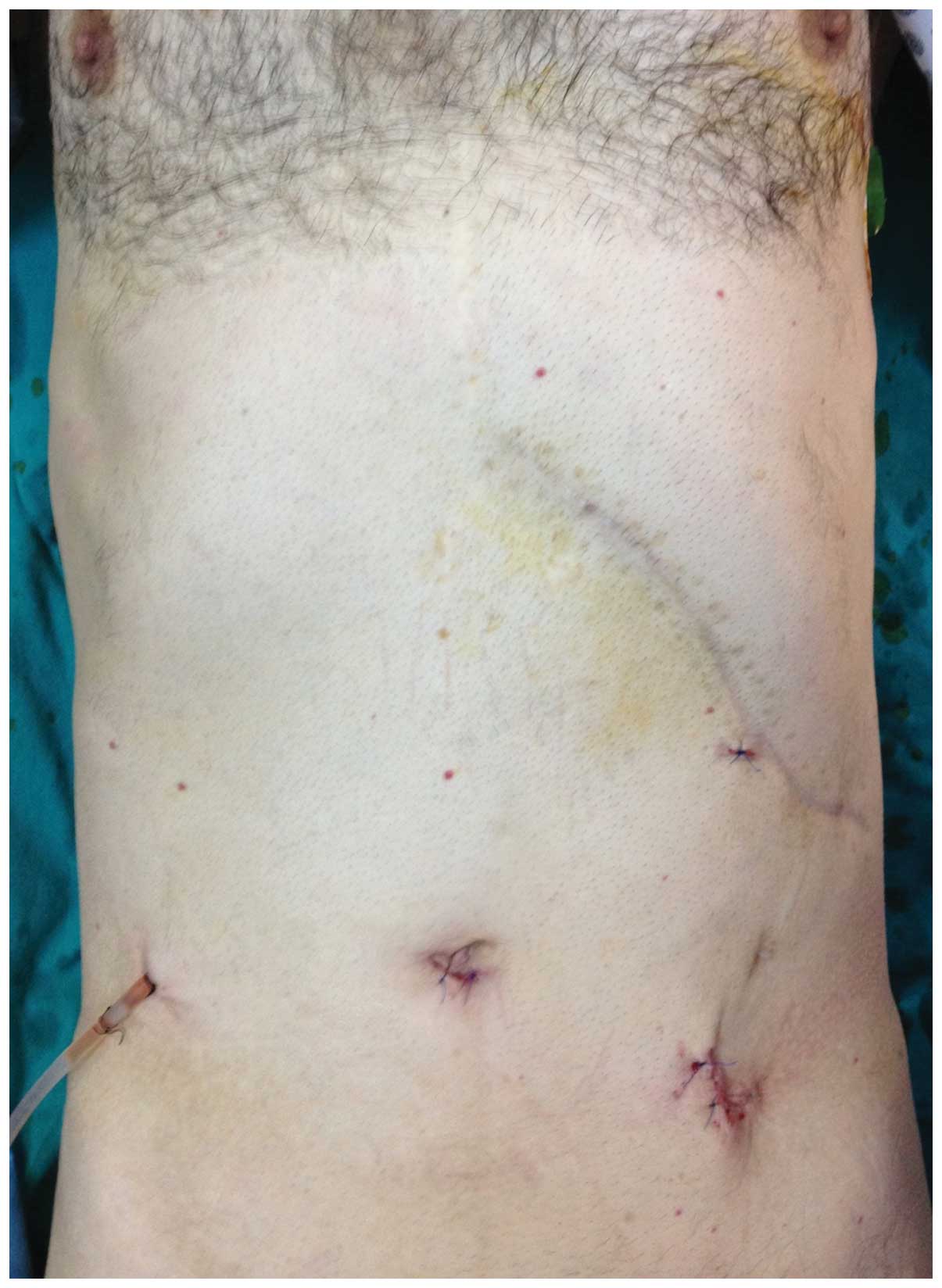
identified at 120 cm distal to the ligament of Treitz (Fig. 2). The segmentary resection was
performed laparoscopically to provide a minimally invasive approach
and a soft suction drainage tube was inserted into the abdominal
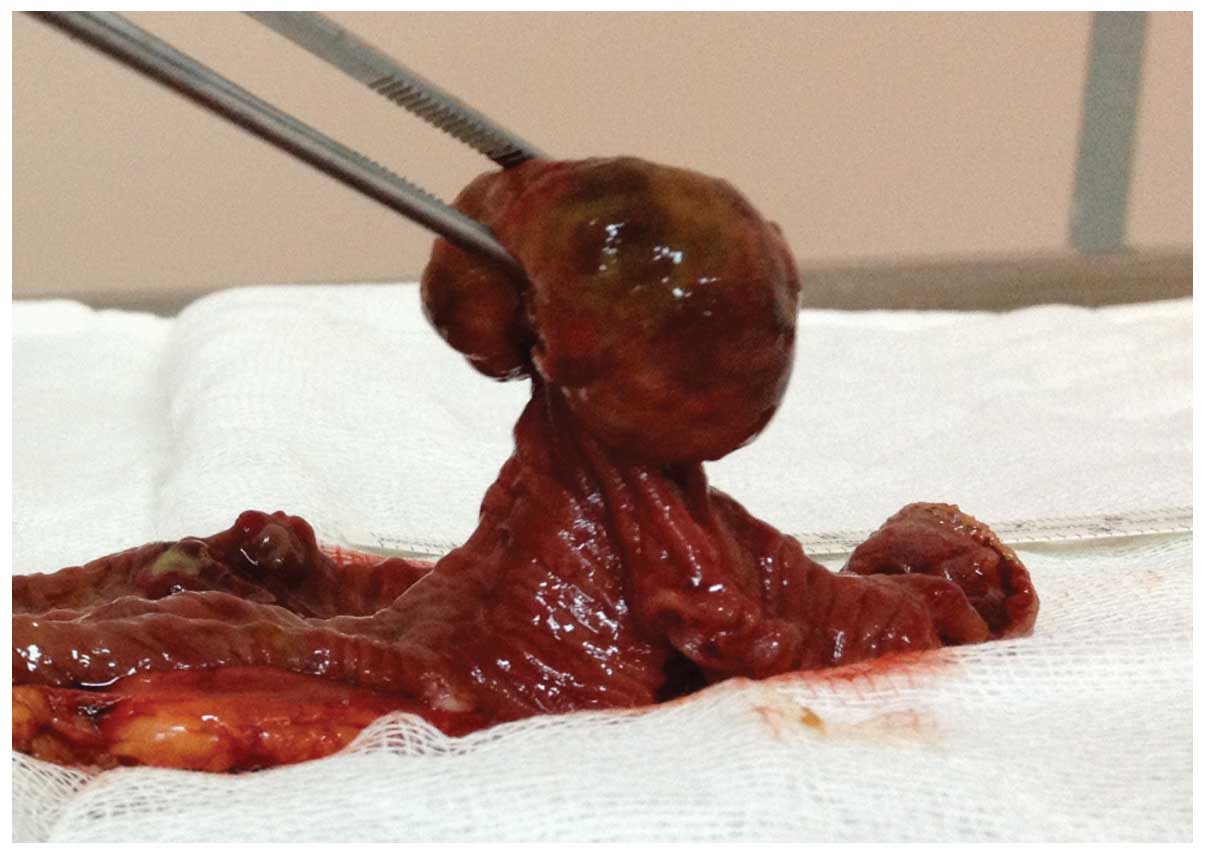
cavity (Fig. 3). A polypoid mass
3.5×3.5×2 cm in size was identified subsequent to specimen
dissection (Fig. 4).
Pathological examination of the mass revealed a
malignant mesenchymal neuroendocrine tumor with vascular invasion.
Mitosis was identified at a level of three mitoses per 10
high-power fields and the Ki-67 index was 40%. The surgical margins
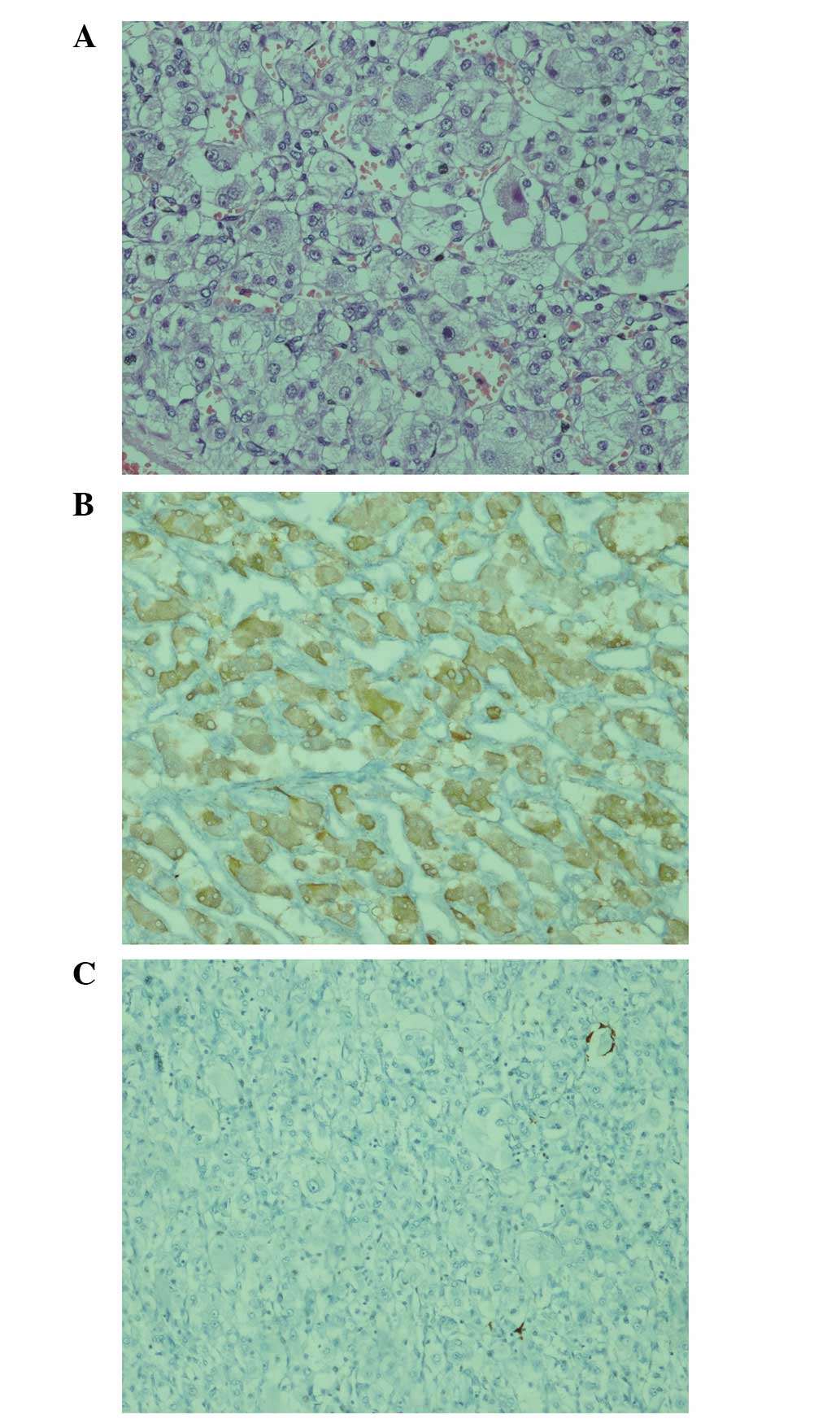
were tumor-free. Immunohistochemistry (IHC) revealed islets of
ganglion-like cells that possessed large eosinophilic cytoplasms
and were located in the submucosa under the epithelium. These cells
were positive for the expression of neuron-specific enolase and did
not express S-100 (Fig. 5). The
final pathological diagnosis was malignant paraganglioma. No
post-operative early complications occurred and the patient was
discharged on post-operative day five. It was decided to perform a
follow-up without the administration of adjuvant therapy at the
oncological council of Bezmialem Vakif University (Istanbul,
Turkey). Currently, the patient remains alive without recurrence
six months subsequent to the start of the follow-up.
Discussion
Pheochromocytomas are neuroendocrine tumors that
arise from the chromaffin cells of the adrenal medulla. The
neuroendocrine tumors that arise from the extra-adrenal paraganglia
are termed paraganglioma. Pheochromocytomas are occasionally termed
as intra-adrenal paraganglia (1).
The incidence of paraganglioma is estimated as 1.5 cases per
million individuals each year, and the peak incidence occurs at
30–50 years (2). Paragangliomas can
be derived from either parasympathetic or sympathetic ganglia.
Paragangliomas derived from parasympathetic ganglia are mostly
located in the head and neck, whereas the majority of sympathetic
ganglia-derived paragangliomas are located in the abdomen, adjacent
to the adrenals (1). However,
paragangliomas can be found in any region that the sympathetic or
parasympathetic ganglia exist. The present study reports the case
of a patient exhibiting paraganglioma that arose from the jejunum
of the small intestine, which is an extremely rarely reported
location in the literature. Despite the majority of paragangliomas
being sporadic, a genetic association has been identified in up to
one-half of cases due to the evolution in genetic testing (3). In total, ~25% of paragangliomas are
malignant and metastasis may arise≥20 years after the first
presentation, which indicates the importance of long-term follow-up
(4). In the present case,
paraganglioma arose eight months subsequent to the first appearance
of a tumor in the left adrenal gland, which invaded into the left
kidney. The majority of paragangliomas are chemo- and
radioresistant (2). Thus, surgical
excision of the tumor is the primary treatment (5,6).
For the surgical excision of paragangliomas, either
an open or laparoscopic approach can be performed. In a case of
suspected malignancy, the open approach is recommended by certain
literature in order to protect the capsule of the tumor and avoid
seeding, therefore preventing recurrence (7). However, experienced surgeons are able
to perform the laparoscopic approach safely even in emergency
settings (8, 9). In the present study, the laparoscopic
approach was preferred due to advantages that included a low rate
of occurrence of intraperitoneal adhesions, a decrease in the
severity of post-operative pain, decreased occurrence of surgical
site infections, an earlier recovery and a shorter hospital stay
compared with the open approach (10). The five-year survival rate of
patients with malignant paragangliomas is ~60% (6). Recurrence is observed within 10 years
of the first surgical procedure in 16% of pheochromocytoma or
paraganglioma cases (11).
Therefore, a long-term follow-up is required.
References
|
1
|
Ilias I and Pacak K: A clinical overview
of pheochromocytomas/paragangliomas and carcinoid tumors. Nucl Med
Biol. 35(Suppl 1): S27–S34. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moslemi MK, Abolhasani M and Vafaeimanesh
J: Malignant abdominal paraganglioma presenting as a giant
intra-peritoneal mass. Int J Surg Case Rep. 3:537–540. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Young WF Jr: Paragangliomas: clinical
overview. Ann N Y Acad Sci. 1073:21–29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fishbein L and Nathanson KL:
Pheochromocytoma and paraganglioma: understanding the complexities
of the genetic background. Cancer Genet. 205:1–11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matro J, Giubellino A and Pacak K: Current
and future therapeutic approaches for metastatic pheochromocytoma
and paraganglioma: focus on SDHB tumors. Horm Metab Res.
45:147–153. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jimenez C, Rohren E, Habra MA, Rich T,
Jimenez P, Ayala-Ramirez M and Baudin E: Current and future
treatments for malignant pheochromocytoma and sympathetic
paraganglioma. Curr Oncol Rep. 15:356–371. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Adjallé R, Plouin PF, Pacak K and Lehnert
H: Treatment of malignant pheochromocytoma. Horm Metab Res.
41:687–696. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Al-Mulhim AS, Nasser MA, Abdullah MM, Ali
AM and Kaman L: Emergency laparoscopy for acute abdominal
conditions: a prospective study. J Laparoendosc Adv Surg Tech A.
18:599–602. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Karamanakos SN, Sdralis E, Panagiotopoulos
S and Kehagias I: Laparoscopy in the emergency setting: a
retrospective review of 540 patients with acute abdominal pain.
Surg Laparosc Endosc Percutan Tech. 20:119–124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liauw JJ and Cheah WK: Laparoscopic
management of acute small bowel obstruction. Asian J Surg.
28:185–188. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Amar L, Servais A, Gimenez-Roqueplo AP,
Zinzindohoue F, Chatellier G and Plouin PF: Year of diagnosis,
features at presentation and risk of recurrence in patients with
pheochromocytoma or secreting paraganglioma. J Clin Endocrinol
Metab. 90:2110–2116. 2005. View Article : Google Scholar : PubMed/NCBI
|