Introduction
Ovarian cancer is one of the most lethal malignant
gynecological cancers worldwide (1).
Compared with other cancers in women, ovarian carcinoma confers a
relatively high risk of recurrence. Although there is a high
initial response rate to standard surgery and chemotherapy, 30–40%
of patients relapse within one year (2,3).
Therefore, the prediction of patients at a high risk of recurrence
may provide novel therapeutic avenues to improve their outcomes.
Although common clinicopathological parameters, such as stage and
histological grade, and several biomarkers were proposed for
recurrence prediction, these factors demonstrated insufficient
sensitivity and specificity (4).
Thus, there is an urgent requirement to identify novel markers or
models to increase the power of recurrence prediction for patients
with ovarian carcinoma.
The Notch pathway and its abundant associated genes
comprise a complicated network, which plays a significant role in
the progressive growth of tumor cells in multiple cancer types
(5). The Notch pathway alterations
are prevalent and significantly associated with poor outcomes,
including early recurrence in ovarian carcinoma (6,7). We
speculate that Notch pathway associated molecular signatures may be
useful for characterizing ovarian carcinomas at high risk of
recurrence. In the present study, a 10-gene Notch pathway signature
is defined that may assist in improved predictions of recurrence in
ovarian carcinoma patients.
Materials and methods
Datasets
Three ovarian carcinoma gene expression datasets
[The Cancer Genome Atlas (TCGA), GSE9891 and GSE30161] with
documented recurrence information were selected for analysis in the
present study (8–10). The expression data together with the
curated and documented clinical metadata were extracted by the R
curated Ovarian Data Bioconductor package, as previously described
(11). Microarray platforms used in
these datasets were Affy HT U133a (TCGA) and Affy U133 Plus 2.0
(GSE9891 and GSE30161). TCGA dataset comprises the whole-genome
mRNA expression data of 522 ovarian carcinoma samples. The GSE9891
dataset comprises the gene expression microarray data of 275
ovarian carcinoma samples, including 40 early-stage and 257
late-stage tumors. The GSE30161 dataset was generated from 58
late-stage ovarian cancer samples, and 5 arrays were excluded due
to the lack of complete recurrence information.
Finding of genes correlated with
recurrence in TCGA ovarian carcinoma dataset
The expression data of a subset of 81 Notch
pathway-associated genes (including core Notch pathway members,
Notch pathway target genes, genes that crosstalk with Notch pathway
and other genes involved in Notch pathway) were selected from TCGA
ovarian carcinoma dataset. Cox proportional hazards model was used
to test whether the gene expression of a particular gene
significantly influenced recurrence using the BRB-Arraytools
software (12). The tests were
performed at a significance threshold of univariate tests of 0.05
using permutation tests. The number of permutation tests was set as
10,000.
Recurrence-free survival (RFS) and
overall survival (OS) time prediction based on the supervised
principal components method using the 10-Notch pathway gene
signature
After subseting the gene expression data using the
10-Notch pathway gene signature, recurrence and survival risk
prediction in TCGA, GSE9891 and GSE30161 datasets were performed
based on principal components using the BRB-ArrayTools software.
10-fold cross validation was selected, and the number of principal
components was set as 2. The prognostic index percentile was used
to separate arrays into high- and low-risk groups.
Statistical analysis
Distributions of RFS and OS were assessed using the
Kaplan-Meier curve method and evaluated by the log-rank test.
Multivariate analyses of prognostic factors were based on the Cox
proportional hazards model. The receiver operating characteristic
(ROC) curve was constructed using R package survival ROC and
determined by permutation testing. The difference in
clinicopathological characteristics between the high- and low-risk
subgroups was determined by χ2 test. All the statistical
analyses were performed with Medcalc 11.4 Software unless otherwise
specified. P<0.05 was considered to indicate a statistically
significant difference.
Results
We hypothesized that Notch pathway genes correlated
with recurrence may be clinically useful to differentiate between
high- and low-risk ovarian carcinoma tumors. To investigate this,
the BRB-Arraytool package was used to find Notch pathway genes with
expression that was correlated with RFS time by univariate tests.
As shown in Table I, using a
significance threshold at 0.05, a list of 10 Notch pathway genes
was determined to be associated with RFS time in TCGA ovarian
carcinoma dataset.
 | Table I.Notch pathway genes correlated with
recurrence in The Cancer Genome Atlas ovarian carcinoma
dataset. |
Table I.
Notch pathway genes correlated with
recurrence in The Cancer Genome Atlas ovarian carcinoma
dataset.
| Notch pathway
genes | Gene names | Accession no. | Hazard ratio | FDR | Permutation
P-value |
|---|
| FZD4 | Frizzled family
receptor 4 | NM_012193 | 0.786 | 0.0466 | 0.0010 |
| HES1 | Hairy and enhancer of
split 1, (Drosophila) | NM_005524 | 0.874 | 0.259 | 0.0091 |
| PSEN2 | Presenilin 2
(Alzheimer disease 4) | NM_000447 | 0.471 | 0.259 | 0.0094 |
| JAG2 | Jagged 2 | NM_002226 | 0.819 | 0.286 | 0.0241 |
| PPARG | Peroxisome
proliferator-activated receptor gamma | NM_005037 | 0.741 | 0.286 | 0.0284 |
| FOS | FBJ murine
osteosarcoma viral oncogene homolog | NM_005252 | 1.094 | 0.286 | 0.0266 |
| HEY1 |
Hairy/enhancer-of-split related with YRPW
motif 1 | NM_001040708 | 0.879 | 0.286 | 0.0298 |
| CDC16 | Cell division cycle
16 | NM_001078645 | 1.202 | 0.286 | 0.0296 |
| MFNG | MFNG O-fucosylpeptide
3-β-N-acetylglucosaminyltransferase | NM_001166343 | 1.323 | 0.286 | 0.0357 |
| EP300 | E1A-binding protein
p300 | NM_001429 | 0.818 | 0.321 | 0.0429 |
Using a principal components model based on these 10
Notch pathway genes as a signature, a predictor was generated and
TCGA ovarian cancer samples were classified into high-risk (n=263)
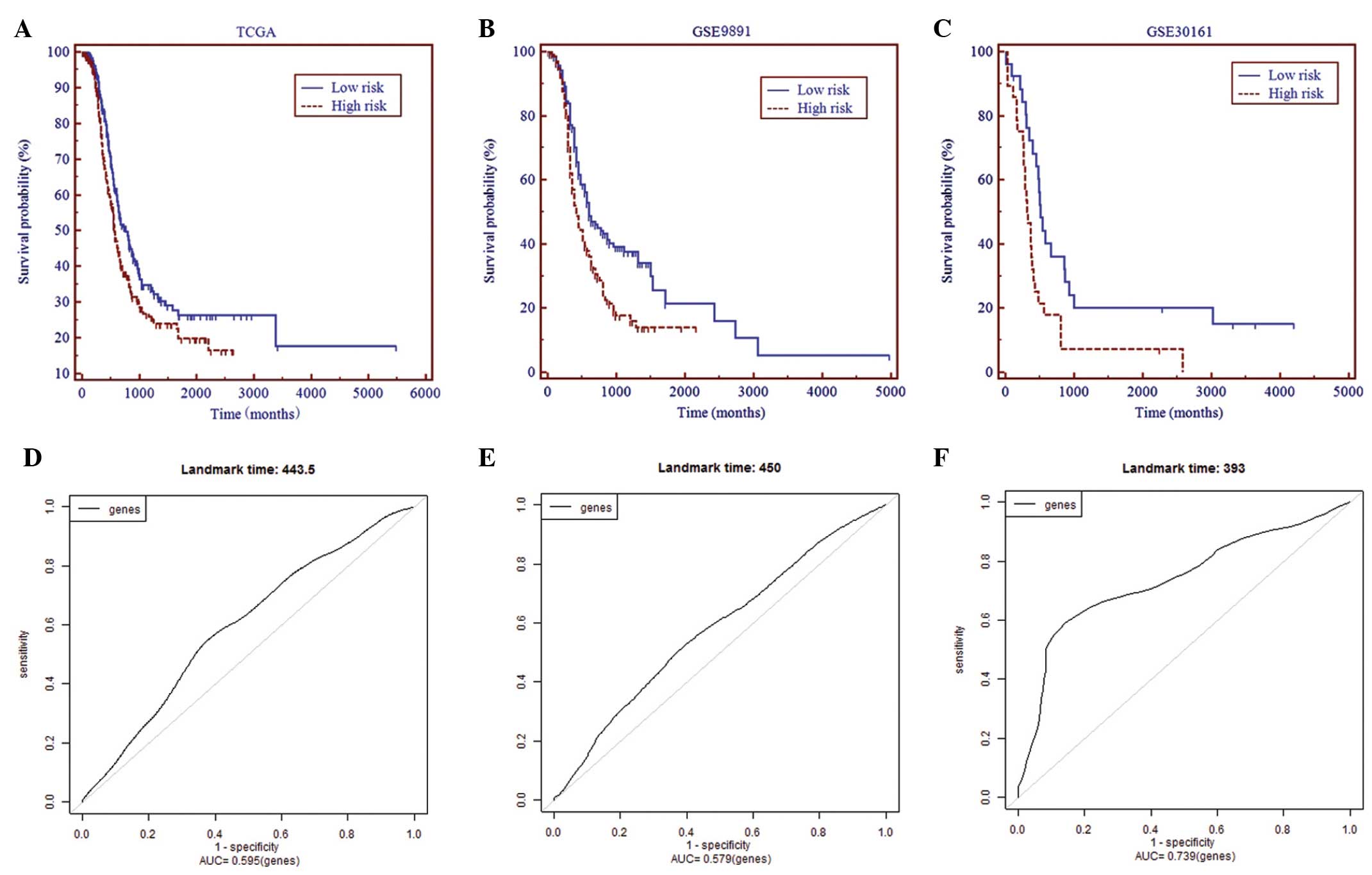
and low-risk (n=259) subgroups. As shown in Fig. 1A, in TCGA dataset, the high-risk
ovarian carcinomas demonstrated a significantly shorter RFS time
than the low-risk cases (hazard ratio, 1.3656; 95% confidence
interval, 1.0739–1.7364; P=0.0104). In order to evaluate the
performance of this novel gene signature, its performance in
predicting recurrence in two other independent ovarian carcinoma
datasets, GSE9891 and GSE30161, was further validated. As shown in
Fig. 1B and C, the signature was
independently predictive of recurrence in the two validation
datasets. In addition, this gene signature gave a significant value
for the area under the curve when discriminating between high- and
low-risk ovarian carcinomas in all three datasets (TCGA, P=0.0167;
GSE9891, P=0.04; GSE30161, P=0.01) (Fig.
1D–F).
Next, the association between the predicted
recurrence risk subgroups and the known prognostic factors was
analyzed. In GSE9891, but not the other two datasets, the high
recurrence risk subgroup exhibited a significant association with
stage and debulking level (Table
II). The multivariate Cox proportional hazards regression
analyses found that the prognostic value on recurrence of the
10-Notch gene signature was independent of other known predictors
in all three datasets (Table
III).
| Table II.Difference in clinicopathological
characteristics between high- and low-risk recurrence subgroups
defined by a 10-Notch pathway gene signature in 3 ovarian carcinoma
gene expression datasets.a |
Table II.
Difference in clinicopathological
characteristics between high- and low-risk recurrence subgroups
defined by a 10-Notch pathway gene signature in 3 ovarian carcinoma
gene expression datasets.a
| Characteristics | Low risk | High risk | P-value |
|---|
| TCGA dataset |
|
| 0.9589 |
| Number | 259 | 263 |
|
| Grade |
|
|
|
| 1–2 | 33 | 34 |
|
| 3 | 224 | 220 |
|
| Stage |
|
| 0.6486 |
|
Early | 18 | 22 |
|
| Late | 240 | 239 |
|
| Debulking |
|
| 0.1067 |
|
Optimal | 184 | 167 |
|
|
Suboptimal | 52 | 68 |
|
| GSE9891 dataset |
|
|
|
| Number | 142 | 133 | 0.0114 |
| Grade |
|
|
|
| 1–2 | 68 | 44 |
|
| 3 | 71 | 89 |
|
| Stage |
|
| 0.0008 |
|
Early | 31 |
9 |
|
| Late | 111 | 123 |
|
| Debulking |
|
| 0.8884 |
|
Optimal | 41 | 41 |
|
|
Suboptimal | 83 | 77 |
|
| GSE30161 dataset |
|
|
|
| Number | 26 | 28 | 1.0000 |
| Grade |
|
|
|
| 1–2 | 11 | 10 |
|
| 3 | 14 | 15 |
|
| Debulking |
|
| 0.9485 |
|
Optimal | 14 | 15 |
|
|
Suboptimal | 10 | 13 |
|
| Table III.Multivariate Cox proportional hazards
regression analyses on recurrence-free survival time in 3 ovarian
carcinoma gene expression datasets. |
Table III.
Multivariate Cox proportional hazards
regression analyses on recurrence-free survival time in 3 ovarian
carcinoma gene expression datasets.
| Covariate | P-value | Exp(b) | 95% CI of Exp(b) |
|---|
| TCGA dataset |
|
|
|
|
Grade | 0.5624 | 1.1112 | 0.7792–1.5848 |
|
Stage | 0.0155 | 2.0291 | 1.1473–3.5887 |
|
Debulking | 0.6801 | 0.9404 | 0.7032–1.2575 |
| Predicted
risk | 0.0083 | 1.4119 | 1.0944–1.8215 |
| GSE9891 dataset |
|
|
|
|
Grade | 0.6509 | 0.9377 | 0.7106–1.2373 |
|
Stage | 0.0001 | 5.0027 | 2.3040–10.8624 |
|
Debulking | 0.0010 | 0.5829 | 0.4228–0.8035 |
|
Predicted risk | 0.0382 | 1.3984 | 1.0200–1.9171 |
| GSE30161
dataset |
|
|
|
|
Grade | 0.7870 | 1.0997 | 0.5537–2.1841 |
|
Debulking | 0.0160 | 0.4235 | 0.2113–0.8488 |
|
Predicted risk | 0.0088 | 2.3312 | 1.2413–4.3781 |
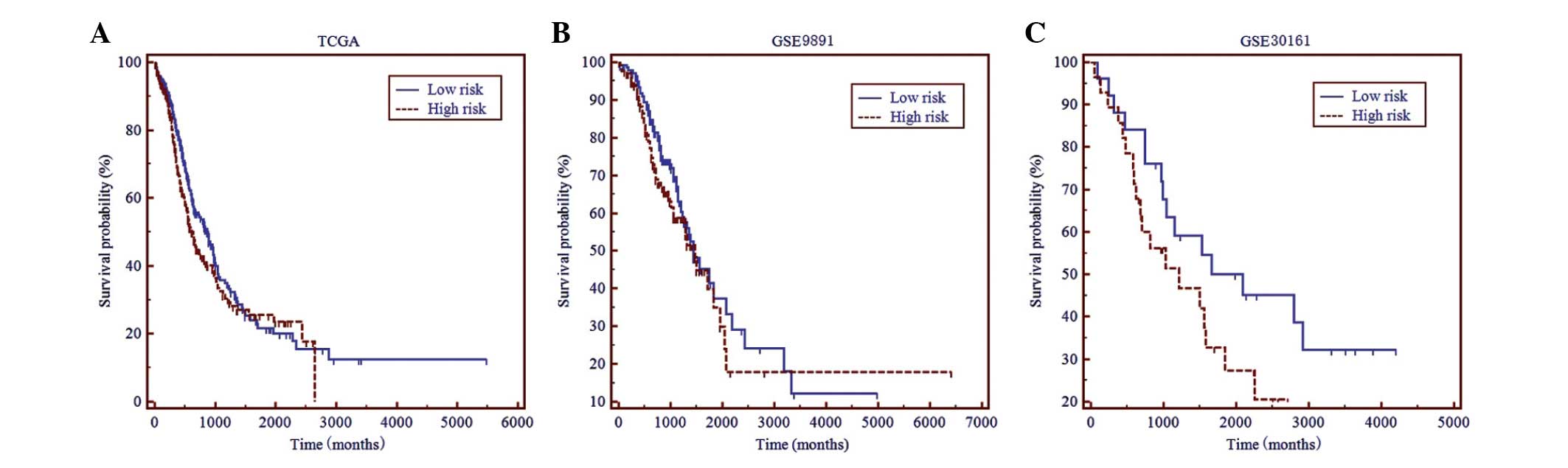
Finally, it was evaluated whether this 10-gene
signature could predict OS time for ovarian carcinomas. In contrast
to RFS, no significant difference was found in OS time between the
high- and low- risk subgroups in all three datasets (Fig. 2).
Discussion
In the current study, 10 Notch pathway genes were
identified to significantly correlate with recurrence in ovarian
carcinoma. This 10-gene signature could classify ovarian carcinoma
into high- and low-risk recurrence subgroups, and showed a
significant performance to predict recurrence in independent
cohorts. Moreover, the prognostic value of this gene signature is
independent of the common clinicopathological predictors of ovarian
cancers.
Prognostic gene signatures based on microarray data
have been recently developed to identify subgroups with a more
aggressive phenotype or poor outcomes in ovarian carcinoma. For
example, Gillet et al (13)
found 3 multidrug resistance gene signatures with a statistically
significant correlation with OS and progression-free survival time.
Cheon et al (14) identified
and validated a 10-gene signature regulated by transforming growth
factor-β signaling that is associated with poor OS time in patients
with high-grade serous ovarian cancer. Compared with the previous
gene signatures, the present 10-gene classifier exhibited a better
performance on predicting recurrence, but not OS time for ovarian
carcinoma. Moreover, for the first time, a Notch pathway gene
signature is reported to have clinical significance in the
prognosis prediction for patients with ovarian carcinoma. The use
of this 10-Notch pathway gene signature should be investigated in
prospective patient cohorts in the future.
Of the 10 Notch pathway genes identified in the
present study, the functional and clinical significance has only
been investigated in FZD4, HES1, PPARG and FOS. Only FZD4 has been
found to be associated with recurrence in a previous study. Dai
et al (15) found that DNA
methylation and reduced expression of FZD4 are indicators of early
disease relapse in ovarian tumors, which is consistent with the
present finding that FZD4 gene expression is negatively associated
with recurrence in ovarian carcinoma. Increased expression of HES1
and PPARG were validated to be predictors for poor OS in ovarian
cancers (16,17). High expression of FOS was
significantly associated with advanced clinical stage and
chemoresistance (18). Considering
that this 10-gene signature has clinical significance in
classifying different subgroups with high and low recurrence risk,
it is also possible that these Notch pathway genes may act as
important mediators during ovarian carcinogenesis, and may
represent novel therapeutic targets. Therefore, the biological
significance of these 10 Notch signaling genes also deserves
further investigation.
In conclusion, in the present study, a novel Notch
pathway gene signature that is useful for predicting recurrence in
ovarian cancers was developed. If prospectively validated, it would
provide a reference for informing treatment decisions for patients
with ovarian carcinoma. The biological significance of this
signature and its potential as a biomarker also deserve further
investigation in future studies.
References
|
1
|
Lowe KA, Chia VM, Taylor A, O'Malley C,
Kelsh M, Mohamed M, Mowat FS and Goff B: An international
assessment of ovarian cancer incidence and mortality. Gynecol
Oncol. 130:107–114. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Foley OW, Rauh-Hain JA and del Carmen MG:
Recurrent epithelial ovarian cancer: an update on treatment.
Oncology (Williston Park). 27:288–294. 2982013.PubMed/NCBI
|
|
3
|
Geurts SM, de Vegt F, van Altena AM, van
Dijck JA, Tjan-Heijnen VC, Verbeek AL and Massuger LF: Considering
early detection of relapsed ovarian cancer: a review of the
literature. Int J Gynecol Cancer. 21:837–845. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Santillan A, Garg R, Zahurak ML, Gardner
GJ, Giuntoli RL II, Armstrong DK and Bristow RE: Risk of epithelial
ovarian cancer recurrence in patients with rising serum CA-125
levels within the normal range. J Clin Oncol. 23:9338–9343. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takebe N, Nguyen D and Yang SX: Targeting
notch signaling pathway in cancer: Clinical development advances
and challenges. Pharmacol Ther. 141:140–149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rose SL, Kunnimalaiyaan M, Drenzek J and
Seiler N: Notch 1 signaling is active in ovarian cancer. Gynecol
Oncol. 117:130–133. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jung SG, Kwon YD, Song JA, Back MJ, Lee
SY, Lee C, Hwang YY and An HJ: Prognostic significance of Notch 3
gene expression in ovarian serous carcinoma. Cancer Sci.
101:1977–1983. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cancer Genome Atlas Research Network, .
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tothill RW, Tinker AV, George J, Brown R,
Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro
B, et al: Novel molecular subtypes of serous and endometrioid
ovarian cancer linked to clinical outcome. Clin Cancer Res.
14:5198–5208. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ferriss JS, Kim Y, Duska L, Birrer M,
Levine DA, Moskaluk C, Theodorescu D and Lee JK: Multi-gene
expression predictors of single drug responses to adjuvant
chemotherapy in ovarian carcinoma: Predicting platinum resistance.
PLoS One. 7:e305502012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ganzfried BF, Riester M, Haibe-Kains B,
Risch T, Tyekucheva S, Jazic I, Wang XV, Ahmadifar M, Birrer MJ,
Parmigiani G, et al: CuratedOvarianData: Clinically annotated data
for the ovarian cancer transcriptome. Database (Oxford).
2013:bat0132013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Simon R, Lam A, Li MC, Ngan M, Menenzes S
and Zhao Y: Analysis of gene expression data using BRB-ArrayTools.
Cancer Inform. 3:11–17. 2007.PubMed/NCBI
|
|
13
|
Gillet JP, Wang J, Calcagno AM, Green LJ,
Varma S, Bunkholt Elstrand M, Trope CG, Ambudkar SV, Davidson B, et
al: Clinical relevance of multidrug resistance gene expression in
ovarian serous carcinoma effusions. Mol Pharm. 8:2080–2088. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cheon DJ, Tong Y, Sim MS, Dering J, Berel
D, Cui X, Lester J, Beach JA, Tighiouart M, Walts AE, et al: A
collagen-remodeling gene signature regulated by TGF-β signaling is
associated with metastasis and poor survival in serous ovarian
cancer. Clin Cancer Res. 20:711–723. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dai W, Teodoridis JM, Zeller C, Graham J,
Hersey J, Flanagan JM, Stronach E, Millan DW, Siddiqui N, Paul J,
et al: Systematic CpG islands methylation profiling of genes in the
wnt pathway in epithelial ovarian cancer identifies biomarkers of
progression-free survival. Clin Cancer Res. 17:4052–4062. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang X, Fu Y, Chen X, Ye J, Lü B, Ye F, Lü
W and Xie X: The expressions of bHLH gene HES1 and HES5 in advanced
ovarian serous adenocarcinomas and their prognostic significance: A
retrospective clinical study. J Cancer Res Clin Oncol. 136:989–996.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ivan C, Hu W, Bottsford-Miller J, Zand B,
Dalton HJ, Liu T, Huang J, Nick AM, Lopez-Berestein G, Coleman RL,
et al: Epigenetic analysis of the Notch superfamily in high-grade
serous ovarian cancer. Gynecol Oncol. 128:506–511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kang KW, Lee MJ, Song JA, Jeong JY, Kim
YK, Lee C, Kim TH, Kwak KB, Kim OJ and An HJ: Overexpression of
goosecoid homeobox is associated with chemoresistance and poor
prognosis in ovarian carcinoma. Oncol Rep. 32:189–198.
2014.PubMed/NCBI
|