Introduction
Bortezomib (also identified as PS-341 or Velcade®)
is the first proteasomal inhibitor (PI) to be utilized for cancer
therapy; it is used to treat multiple myeloma and mantle cell
lymphoma (1). Bortezomib has been
demonstrated to inhibit tumor neoangiogenesis, a requirement for
cancer progression and metastasis (2). This inhibition is accomplished through
the upregulation of proapoptotic proteins and the suppression of
pathways responsible for antiapoptotic gene expression (3). Bortezomib has also been demonstrated to
obstruct hypoxia adaptation in tumors by repressing the activity of
hypoxia-inducible factor (HIF)-1, a transcription factor (4,5). HIF-1 is
a heterodimer, composed of an oxygen-regulated α and a
constitutively expressed β subunit.
HIF-1 is one of three related heterodimeric
hypoxia-inducible factors (HIF-1, −2 and −3), which possess a
common β subunit and differing α subunits. The α subunit protein in
each case is rapidly degraded under normoxic conditions, but is
stable under hypoxic conditions. HIF-1 and HIF-2 have been revealed
to play important roles in the survival of hypoxic cells in solid
tumors (6). HIF-1α is structurally
similar to HIF-2α; these two subunits share 48% amino acid sequence
identity and are regulated in a similar manner (7). However, despite these similarities, the
heterodimeric HIF-1 and HIF-2 proteins exhibit distinct functional
roles in cancer. They also transactivate a number of common as well
as distinct downstream target genes (8). For example, HIF-1, but not HIF-2,
specifically regulates the transcription of carbonic anhydrase 9
(CA9) (9) and phosphoglycerate
kinase (PGK) (8,10). By contrast, other hypoxia-inducible
genes, including glucose transporter-1 (GLUT-1) and
erythropoietin (EPO), are targets of HIF-1α and HIF-2α
(8,10–12). In
addition to differing in terms of target genes, the expression
levels of the α subunits of HIF-1 and HIF-2 also vary in cells and
tissues. In neuroblastoma cells expressing HIF-1α and HIF-2α in
normoxic levels of oxygen, the HIF-1α protein has been demonstrated
to be expressed at a much lower level compared with HIF-2α
(13).
To date, mechanistic studies of the effects of
bortezomib on HIF have predominantly focused only on the inhibition
of proteasomal degradation of HIF-1α (5,14).
Previously, our group and others demonstrated that bortezomib
treatment led to an accumulation of HIF-1α; however, the
corresponding increased level of heterodimeric HIF-1 was inactive
(4,15). To the best of our knowledge, no
studies have investigated the effects of bortezomib on HIF-2
transcriptional activity. Therefore, in the present study, the
effects of bortezomib treatment on the stabilization of HIF-2α and
corresponding HIF-2 activity were examined using cancer cell lines
known to express both HIF-1α and HIF-2α, and a cancer cell line
expressing only HIF-2α.
Materials and methods
Human cell lines and culture
Osteosarcoma (Saos-2), breast carcinoma (MCF-7) and
renal clear cell carcinoma (786-O) cell lines were obtained from
the American Type Culture Collection (Manassas, VA, USA) and
maintained in Dulbecco's modified Eagle's medium supplemented with
10% fetal bovine serum (GE Healthcare, Pasching, Austria). Cells
were grown in normoxic conditions (21% O2) in a
humidified Forma 311 CO2 incubator (Thermo Forma,
Marietta, OH, USA), or hypoxic conditions (0.5% O2) in a
Galaxy 48R incubator (New Brunswick™, Eppendorf, Hamburg, Germany).
The PI bortezomib (Millennium Pharmaceuticals, Inc., Cambridge, MA,
USA) was dissolved in dimethyl sulfoxide. For bortezomib treatment,
cells were initially seeded at 6.6×104
cells/cm2 for 24 h. Cells were pre-treated with
bortezomib for 30 min, and then exposed to normoxia or hypoxia for
24 h in the presence of the drug (4).
IC20 concentrations of bortezomib (0.5, 0.2 and 0.17 µM
for Saos-2, MCF-7 and 786-O, respectively) were used. Samples were
harvested on ice using radioimmunoprecipitation assay buffer
(Thermo Fisher Scientific, Inc., Rockford, IL, USA) containing
EDTA-free protease inhibitor cocktail (Roche, Mannheim, Germany).
Samples were probed using antibodies against HIF-1α (monoclonal
rabbit anti-human; cat. no. GTX61608; 1,1,000), HIF-2α (monoclonal
rabbit anti-human; cat. no. GTX103707: 1:1,000), GLUT-1 (polyclonal
rabbit anti-human; cat. no. GTX100684; 1,1,000), carbonic anhydrase
IX (CAIX; monoclonal mouse anti-human; cat. no. GTX70020; 1,1,000)
(all from Genetex, Inc., Irvine, CA, USA), EPO (polyclonal rabbit
anti-human; cat. no. sc-7956; 1,1,000; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) and β-actin (monoclonal mouse anti-human;
cat. no. A5316; 1:5,000; Sigma-Aldrich, St. Louis, MO, USA) for 1 h
at room temperature. The samples were washed three times with
Tris-buffered saline containing 0.1% Tween 20 (Amresco LLC, Solon,
OH, USA), then probed with horseradish peroxidase-conjugated
monoclonal horse anti-mouse (cat. no. 7076S; 1:5,000) or polyclonal
goat anti-rabbit (cat. no. 7074S; 1:5,000) IgG secondary antibodies
(Cell Signaling Technology, Inc., Danvers, MA, USA) for 1 h at room
temperature. Protein bands were detected using the SuperSignal West
Dura Extended Duration Substrate kit (Pierce Biotechnology, Inc.,
Rockford, IL, USA)and quantitated using ImageJ software (version
1.48; National Institutes of Health, Bethesda, MD, USA) as
previously described (16).
Reverse transcription-polymerase chain
reaction (RT-PCR)
RT-PCR was performed on 100 ng of RNA using the
Access RT-PCR system (Promega Corporation, Madison, WI, USA).
Specific primers for HIF-1α (4,17),
HIF-2α (18), CA9
(4), GLUT-1, EPO
(19) and β-actin (4) were used. The reaction system (Access
RT-PCR system; Promega Corporation) contained 1X AMV/Tfl Reaction
buffer, 10 mM dNTP mix, Tfl DNA polymerase (0.1 U), AMV RT (0.1 U),
25 mM MgSO4, 10 mM forward and reverse primers. PCR was
performed under the following conditions: 1 cycle of reverse
transcription at 45°C for 45 min, 1 cycle of predenaturation at
94°C for 2 min, followed by 30 cycles (with the exception of
β-actin, 25 cycles) at 95°C for 40 sec, 56°C for 40 sec followed by
72°C for 1 min with a final extension step at 72°C for 4 min.
RT-PCR products were then analyzed on 1.5% agarose gel and
quantitated using ImageJ 1.48 software.
Luciferase reporter assay
Transfection with a firefly luciferase reporter
construct driven by the hypoxia response elements (HREs) of
CA9, PGK and EPO was performed using the
pLuc-MCS vector (Agilent Technologies, Inc., Santa Clara, CA, USA)
and Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA,
USA) as previously described (20).
The HRE sequences are 5′-GGCTGTACGTGCATTGGAAACGAGAGCTG for
CA9, 5′-TTTGTCACGTCCTGCACGACGCG for PGK and
5′-GGCCCTACGTGCTGTCTCACACAGCCTGT for EPO. A
non-hypoxia-responsive plasmid, pRL-CMV (Promega Corporation),
expressing Renilla luciferase was used as the internal
control as described previously (20). Luciferase activities were determined
using a Dual-Luciferase® Reporter Assay System (Promega
Corporation) in a Sirius luminometer (Titertek-Berthold, Pforzheim,
Germany), according to the manufacturer's instructions. Data are
presented as the average ratio of firefly to Renilla
luciferase activities [± standard deviations [SD)] from at least
three independent experiments.
Statistical analysis
Experimental data were analyzed using the Student's
t-test (GraphPad Prism 5; GraphPad Software, Inc., La Jolla,
CA, USA) and expressed as the mean ± standard error of the mean
(SEM). P<0.05 was considered to indicate a statistically
significant difference.
Results and Discussion
Bortezomib attenuates HIF-1 but not
HIF-2 transcriptional activity
HIF-1α and HIF-2α subunits are closely related
(21), however their hypoxic
regulation, pattern of expression and specific target genes vary to
a certain degree (8). Previously, our
group demonstrated that bortezomib attenuated the transcriptional
activity of HIF-1 in a number of cancer cell lines (4). Since the accumulated inactive HIF-1
still formed a complex with the coactivator p300, the involvement
of a corepressor in its attenuated activity was proposed. p300 is
an important component of the transcriptional machinery which is
involved in the regulation of chromatin organization and
transcription initiation (4).
However, the exact mechanism involving the potential corepressor(s)
remains a topic of investigation. In the present study, the
attenuated effect of bortezomib on HIF-1 activity was reproduced in
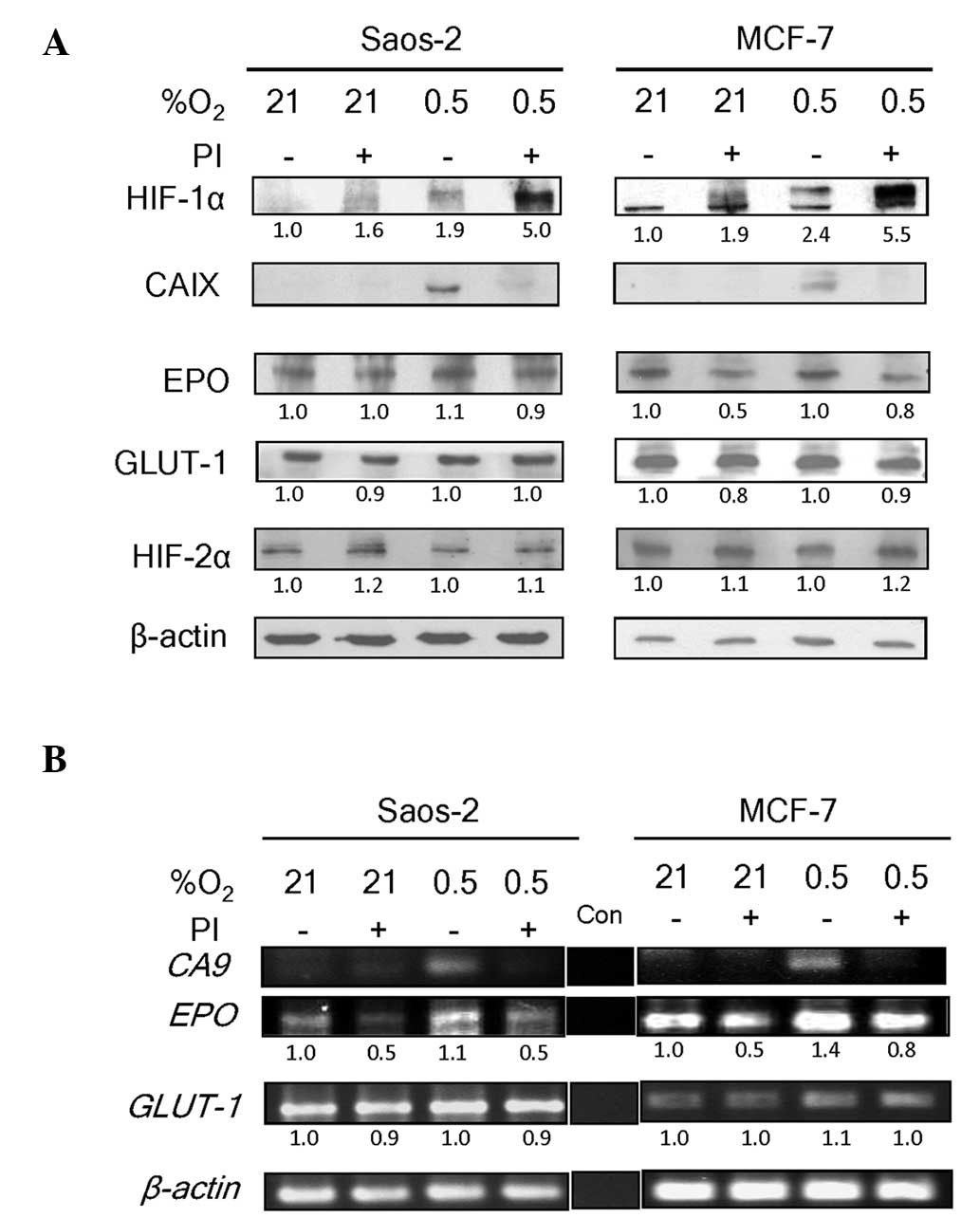
Saos-2 and MCF-7 cell lines. These cell lines express both HIF-1α
and HIF-2α proteins (4,22). Bortezomib treatment caused an
accumulation of HIF-1α protein under normoxia (21% O2),
as well as further accumulation under hypoxia (0.5% O2)
in the two cell lines (Fig. 1A). In
normoxic conditions, bortezomib-mediated HIF-1α stabilization
failed to cause upregulation of the CAIX protein, of which the
encoding gene, CA9, is a HIF-1 specific target (9). As expected, the hypoxia-induced
accumulation of HIF-1α in the absence of bortezomib was associated
with an increase in CAIX expression. This expression was absent in
the presence of bortezomib. These observations concur with those of
our previous study, which showed stabilization of inactive HIF-1
with bortezomib treatment in Saos-2 and MCF-7 cell lines (4).
Notably, the levels of two other HIF-regulated
proteins, EPO (11,12) and GLUT-1 (8,10) were
only minimally reduced by bortezomib treatment (Fig. 1A). EPO and GLUT-1 are
regulated by HIF-1 as well as HIF-2 (8,10–12). As their expression patterns in
response to bortezomib treatment differed from that of CA9,
a HIF-1-specific target gene, we hypothesized that their continued
expression in the presence of bortezomib was due to the lack of an
inhibitory effect of the drug on HIF-2 activity. To test this, the
level of HIF-2α in the samples was examined. HIF-2α was found to be
expressed constitutively at high levels in Saos-2 and MCF-7 cells
under the normoxic and hypoxic conditions used in the present study
(Fig. 1A). This was consistent with
the patterns of HIF-2α protein levels under normoxic conditions
reported in another study (13).
HIF-2α is less efficiently degraded via the
prolyl-4-hydroxylase-mediated proteasomal degradation pathway
compared with HIF-1α under physiological oxygen conditions
(13). In the present study, the
basal levels of HIF-2α in Saos-2 and MCF-7 cells were not increased
by hypoxia, indicating that the constitutive levels approached
saturation. In accordance with this, bortezomib treatment only
marginally increased the level of HIF-2α protein under normoxic or
hypoxic conditions.
Bortezomib does not inhibit HIF-2
transcriptional activity in 786-O cells
The effect of bortezomib on the transcriptional
activities of HIF-1 was observed (4).
To confirm that bortezomib interfered only with transcriptional
activities of HIF-1 and not HIF-2, RT-PCR was performed using
CA9-, EPO- and GLUT-1-specific primers. A band
representing the CA9 transcript, which is exclusively under
HIF-1 regulation (9), was visible
only in hypoxic conditions in the absence of bortezomib (Fig. 1B). No CA9 band was observed in
the normoxic conditions (without bortezomib), which was in
accordance with the absence of HIF-1α (Fig. 1A). In all bortezomib-treated samples,
despite the accumulation of HIF-1α (Fig.
1A), the CA9 band was almost absent. The levels of
EPO and GLUT-1 transcripts, however, were clearly
visible even under normoxia (Fig.
1B). As these genes are under the regulation of HIF-1 and HIF-2
(8,10–12), the
result is consistent with a lack of effect of bortezomib on the
functional status of constitutively expressed HIF-2α. The modest
decrease in EPO band intensity is likely to reflect the inhibition
of HIF-1 by bortezomib, as EPO is regulated by both HIF-1 and
HIF-2. These varying effects of bortezomib concur with the concept
that HIF-1 and HIF-2 have non-redundant roles in the regulation of
their target genes (23). Therefore,
the suppression of HIF-1 activity may not directly affect HIF-2
activity. Other cell lines are currently being investigated by our
group to address the possibility of cell-type specific aspects of
the findings.
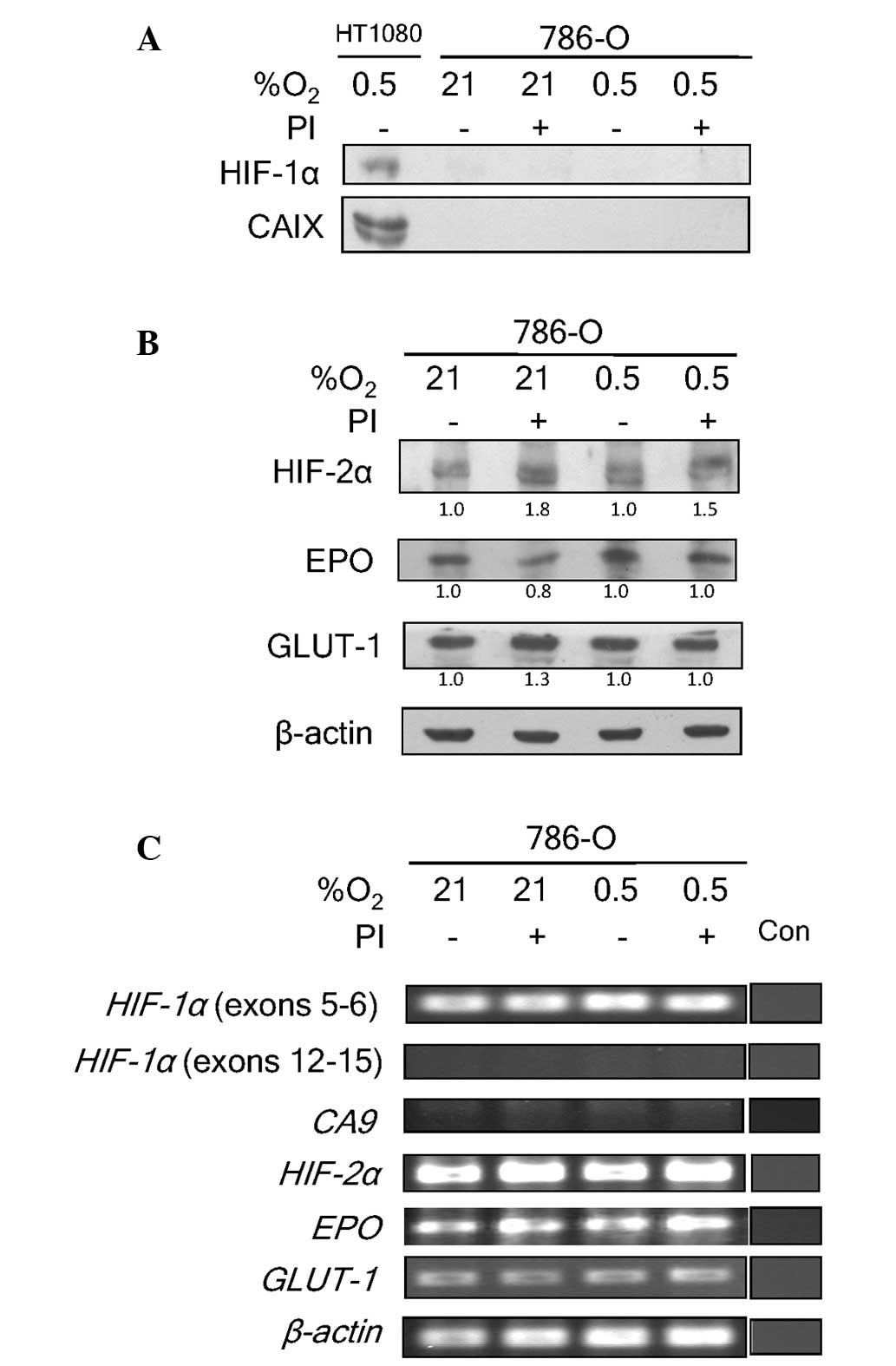
To confirm that bortezomib did not attenuate the
activity of HIF-2, the HIF-1α-deficient 786-O cell line was also
examined (24). This cell line is
devoid of the Von Hippel-Lindau (VHL) tumor suppressor (8), and therefore has a constitutive
stabilization of HIF-2α. VHL forms a complex with elongin-B,
elongin-C and cullin-2 to function as an E3 ubiquitin ligase for
ubiquitination and degradation of hydroxylated HIF-α proteins
(4). Since these cells express HIF-2α
and not HIF-1α, they allow the investigation of the effects of
bortezomib on HIF-2 exclusively. Predictably, no HIF-1α protein
expression was detected in 786-O cells (Fig. 2A). The absence of HIF-1α in 786-O was
associated with a lack of CAIX expression. The addition of
bortezomib caused a marginal increase in HIF-2α expression under
normoxic and hypoxic conditions (Fig.
2B). This increase, however, did not significantly influence
the expression level of EPO or GLUT-1 proteins, which are also
HIF-2 target genes. These data further strengthen the hypothesis
that bortezomib does not interfere with HIF-2 transcriptional
activity, as it does with HIF-1.
At the genetic level, the absence of exons 12–15 for
HIF-1a was confirmed in 786-O cells (Fig. 2C). The absence of these exons has been
previously reported (17). Exons 5–6,
however, were still present. The lack of functioning HIF-1α
resulted in the absence of transcriptional activation of CA9
by HIF-1 in the cells (Fig. 2C).
HIF-2α transcript levels, by contrast, were not
significantly affected by hypoxia or bortezomib treatment. The
transcript levels of EPO and GLUT-1 also remained
unaltered. The lack of functional HIF-1α in 786-O implied that
EPO and GLUT-1 expression was being regulated solely
by HIF-2 in this cell line. These data clearly demonstrate a lack
of influence of bortezomib on HIF-2 transcriptional activity. They
further confirm the assertion of the differential effects of
bortezomib on HIF-1 and HIF-2 transcriptional activities.
Bortezomib inhibits the activation of
exogenously-introduced promoters of HIF-1, but not HIF-2 target
genes
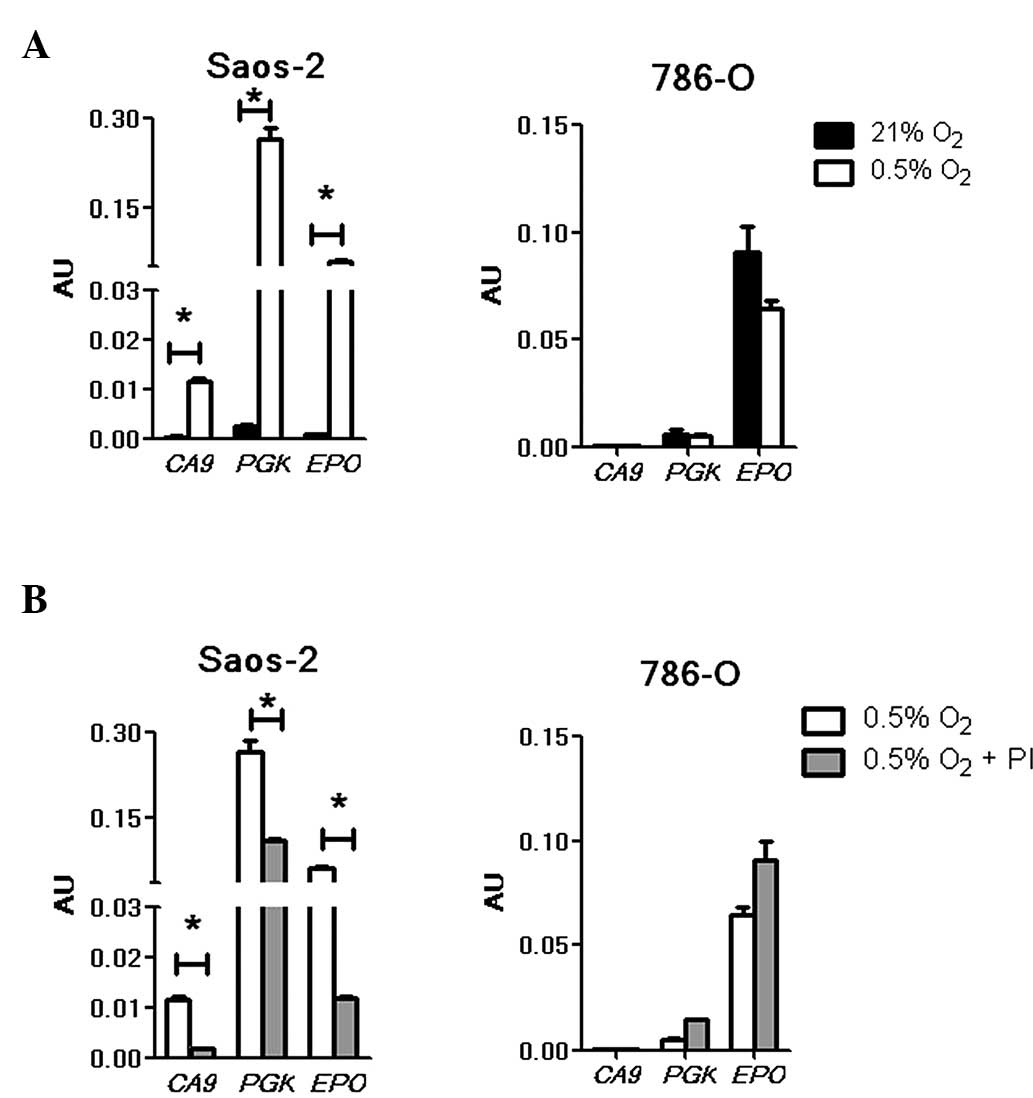
To investigate the effects of bortezomib treatment
on exogenously introduced HREs of HIF-1 and HIF-2 target genes, a
Dual-Luciferase® Reporter Assay was performed using selected
plasmid constructs (4,20) carrying a CA9 (regulated by
HIF-1) or an EPO (regulated by HIF-1 and HIF-2) HRE. As the
available CA9 reporter construct produced low luciferase
signals (20), a HRE construct of
another HIF-1-specific target gene, PGK, was also included
(8,10). All constructs were responsive to
hypoxic stimuli in Saos-2 cells (Fig.
3A, left panel). The hypoxia-induced luciferase signal driven
by the CA9 HRE was low compared with that of PGK, as
previously documented (20). In
contrast to Saos-2, no activation of the CA9 and PGK
HRE constructs was observed in the hypoxic 786-O cells (Fig. 3A, right panel), indicating the absence
of functional HIF-1 in the cells. Under normoxia and hypoxia, no
signal was detected for CA9; however, a low level PGK
signal was observed. The presence of PGK expression in 786-O cells
has been previously reported (8,10) and, in
accordance with the current findings, a basal level of expression
that was not enhanced by hypoxia was documented. Unlike the
CA9 and PGK reporter constructs, EPO produced
high luciferase signals under both normoxic and hypoxic conditions.
Although a minimal reduction was noted under hypoxia, this was not
statistically different from the normoxic results (P>0.05).
These data were consistent with the previously reported
constitutive expression of HIF-2α, and thereby constitutively
active HIF-2, in this cell line (8).
To examine the effects of bortezomib on these
HRE-driven luciferase signals, Saos-2 and 785-O cells were treated
with the drug. In agreement with previous studies (4,20), the
hypoxia-induced signals for CA9, PGK and EPO
were markedly suppressed in the presence of bortezomib in hypoxic
Saos-2 cells (P<0.05; Fig. 3B,
left panel). These results indicate that HIF-1 transactivation of
the CA9 and PGK (HIF-1 target genes), as well as
EPO (a HIF-1 and HIF-2 target gene) promoters were likely
suppressed by the drug. In agreement with a previous report
(4), the repression of CA9 and
PGK was not absolute. In 786-O cells, there was no
detectable signal from the CA9 HRE construct under either
treatment condition (Fig. 3B, right
panel). Additionally, a non-hypoxia-inducible background reading
for PGK (10) was observed and
was not reduced by the drug treatment. EPO HRE-driven
luciferase expression, which was high in hypoxic conditions, was
also not significantly affected by the drug. This result is further
indication of the concept that bortezomib attenuates only HIF-1 and
not HIF-2 transcriptional activities. This distinction may have
profound clinical implications for certain cancer types. For
example, overexpression of HIF-2α has a protumorigenic effect when
HIF-1 activity is lacking (25).
Furthermore, recent evidence demonstrated that HIF-1 is involved in
the inhibition of cell proliferation via a non-transcriptional
mechanism, while HIF-2 enhances cell proliferation (26). In line with our findings, it is
tempting to speculate that bortezomib also inhibits this specific
HIF-1 action, which would account for the reduced efficacy of
bortezomib in certain cancers with differing levels of HIF-1/HIF-2.
Our group is currently investigating this possibility.
In conclusion, using the Saos-2, MCF-7 and 786-O
cell lines as models of cells with differing levels of HIF-1α and
HIF-2α, the present study demonstrated the inhibitory effect of
bortezomib on HIF-1 but not HIF-2 transcriptional activities. Even
though the molecular mechanisms that underlie such specificity are
yet to be elucidated, information obtained in the current study
will contribute towards a further understanding of the therapeutic
efficacy of bortezomib, and potentially of other PI drugs, for
cancer cells that express HIF-1α and/or HIF-2α; a case in point is
renal clear cell carcinoma (17).
Acknowledgements
This work was supported by the Malaysian government
grants 04-01-11-1159RU, 09-05-IFN-BPH-009, 02-01-04-SF1269 and
05-02-12-2010RU.
References
|
1
|
Richardson PG, Mitsiades C, Hideshima T
and Anderson KC: Bortezomib: Proteasome inhibition as an effective
anticancer therapy. Annu Rev Med. 57:33–47. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roccaro AM, Hideshima T, Raje N, Kumar S,
Ishitsuka K, Yasui H, Shiraishi N, Ribatti D, Nico B, Vacca A, et
al: Bortezomib mediates antiangiogenesis in multiple myeloma via
direct and indirect effects on endothelial cells. Cancer Res.
66:184–191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen D, Frezza M, Schmitt S, Kanwar J and
Dou QP: Bortezomib as the first proteasome inhibitor anticancer
drug: Current status and future perspectives. Curr Cancer Drug
Targets. 11:239–253. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kaluz S, Kaluzová M and Stanbridge EJ:
Proteasomal inhibition attenuates transcriptional activity of
hypoxia-inducible factor 1 (HIF-1) via specific effect on the
HIF-1alpha C-terminal activation domain. Mol Cell Biol.
26:5895–5907. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shin DH, Chun YS, Lee DS, Huang LE and
Park JW: Bortezomib inhibits tumor adaptation to hypoxia by
stimulating the FIH-mediated repression of hypoxia-inducible
factor-1. Blood. 111:3131–3136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Talks KL, Turley H, Gatter KC, Maxwell PH,
Pugh CW, Ratcliffe PJ and Harris AL: The expression and
distribution of the hypoxia-inducible factors HIF-1alpha and
HIF-2alpha in normal human tissues, cancers and tumor-associated
macrophages. Am J Pathol. 157:411–421. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tian H, McKnight SL and Russell DW:
Endothelial PAS domain protein 1 (EPAS1), a transcription factor
selectively expressed in endothelial cells. Genes Dev. 11:72–82.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu CJ, Wang LY, Chodosh LA, Keith B and
Simon MC: Differential roles of hypoxia-inducible factor 1alpha
(HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell
Biol. 23:9361–9374. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaluz S, Kaluzová M, Liao SY, Lerman M and
Stanbridge EJ: Transcriptional control of the tumor- and
hypoxia-marker carbonic anhydrase 9: A one transcription factor
(HIF-1) show? Biochim Biophys Acta. 1795:162–172. 2009.PubMed/NCBI
|
|
10
|
Hu CJ, Sataur A, Wang L, Chen H and Simon
MC: The N-terminal transactivation domain confers target gene
specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha.
Mol Biol Cell. 18:4528–4542. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yoon D, Pastore YD, Divoky V, Liu E,
Mlodnicka AE, Rainey K, Ponka P, Semenza GL, Schumacher A and
Prchal JT: Hypoxia-inducible factor-1 deficiency results in
dysregulated erythropoiesis signaling and iron homeostasis in mouse
development. J Biol Chem. 281:25703–25711. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dioum EM, Chen R, Alexander MS, Zhang Q,
Hogg RT, Gerard RD and Garcia JA: Regulation of hypoxia-inducible
factor 2alpha signaling by the stress-responsive deacetylase
sirtuin 1. Science. 324:1289–1293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
HolmquistMengelbier L, Fredlund E,
Löfstedt T, Noguera R, Navarro S, Nilsson H, Pietras A,
VallonChristersson J, Borg A, Gradin K, et al: Recruitment of
HIF-1alpha and HIF-2alpha to common target genes is differentially
regulated in neuroblastoma: HIF-2alpha promotes an aggressive
phenotype. Cancer Cell. 10:413–423. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Birle DC and Hedley DW: Suppression of the
hypoxia-inducible factor-1 response in cervical carcinoma
xenografts by proteasome inhibitors. Cancer Res. 67:1735–1743.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kallio PJ, Wilson WJ, O'Brien S, Makino Y
and Poellinger L: Regulation of the hypoxia-inducible transcription
factor 1alpha by the ubiquitin-proteasome pathway. J Biol Chem.
274:6519–6525. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ch'ng WC, Stanbridge EJ, Yusoff K and
Shafee N: The oncolytic activity of Newcastle disease virus in
clear cell renal carcinoma cells in normoxic and hypoxic
conditions: The interplay between VHL and interferon-β signaling. J
Interferon Cytokine Res. 33:346–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shinojima T, Oya M, Takayanagi A, Mizuno
R, Shimizu N and Murai M: Renal cancer cells lacking
hypoxia-inducible factor (HIF)-1alpha expression maintain vascular
endothelial growth factor expression through HIF-2alpha.
Carcinogenesis. 28:529–536. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Freeburg PB, Robert B, St John PL and
Abrahamson DR: Podocyte expression of hypoxia-inducible factor
(HIF)-1 and HIF-2 during glomerular development. J Am Soc Nephrol.
14:927–938. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yeo EJ, Cho YS, Kim MS and Park JW:
Contribution of HIF-1alpha or HIF-2alpha to erythropoietin
expression: In vivo evidence based on chromatin
immunoprecipitation. Ann Hematol. 87:11–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kaluz S, Kaluzová M and Stanbridge EJ:
Rational design of minimal hypoxia-inducible enhancers. Biochem
Biophys Res Commun. 370:613–618. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ema M, Taya S, Yokotani N, Sogawa K,
Matsuda Y and Fujii-Kuriyama Y: A novel bHLH-PAS factor with close
sequence similarity to hypoxia-inducible factor 1alpha regulates
the VEGF expression and is potentially involved in lung and
vascular development. Proc Natl Acad Sci U S A. 94:4273–4278. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carroll VA and Ashcroft M: Role of
hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the
regulation of HIF target genes in response to hypoxia, insulin-like
growth factor-I, or loss of von Hippel-Lindau function:
Implications for targeting the HIF pathway. Cancer Res.
66:6264–6270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ratcliffe PJ: HIF-1 and HIF-2: Working
alone or together in hypoxia? J Clin Invest. 117:862–865. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Williams RD, Elliott AY, Stein N and
Fraley EE: In vitro cultivation of human renal cell cancer. I.
Establishment of cells in culture. Vitro. 12:623–627. 1976.
View Article : Google Scholar
|
|
25
|
Keith B, Johnson RS and Simon MC: HIF1α
and HIF2α: Sibling rivalry in hypoxic tumour growth and
progression. Nat Rev Cancer. 12:9–22. 2011.PubMed/NCBI
|
|
26
|
Huang LE: Biochemistry. How HIF-1α handles
stress. Science. 339:1285–1286. 2013. View Article : Google Scholar : PubMed/NCBI
|