Introduction
Osteosarcoma (OS) is the most commonly observed
primary bone tumor in children (1).
It is an aggressive malignant tumor, and >80% of patients
treated with surgery alone exhibit distant metastases (2). Standard therapy is typically multimodal,
consisting of neoadjuvant chemotherapy and subsequent amputation or
limb-sparing reconstructive surgeries, in combination with adjuvant
chemotherapy (3). CDDP, a common
antitumor drug, has been found to be effective against cancer cells
derived from solid tumors, such as osteosarcoma, hepatoma and
thymoma. In clinical treatment, CDDP is a common choice for
osteosarcoma chemotherapy (4).
Advances in molecular technologies have facilitated
an increased understanding of the mechanisms underlying
carcinogenesis, primarily focusing on the recognized model of
multistage carcinogenesis with underlying progressive genetic
changes, which induce malignant transformation (5). Recently, the mechanisms underlying
autophagy have gained increasing attention.
Autophagy describes the bulk degradation of proteins
and organelles, and is an essential process for effective cellular
maintenance, viability, differentiation and mammalian development
(6). In mammals, autophagy has been
observed in numerous tissues, and has been demonstrated to possess
significant associations with neurodegenerative diseases,
cardiomyopathies, tumors and apoptosis, as well as bacterial and
viral infections (7). It has been
proposed that autophagy may be capable of protecting certain cancer
cells from anticancer therapies by blocking apoptotic pathways,
whereas other cancer cells are observed to undergo autophagic cell
death following anticancer therapy (8). The opening of the mitochondrial
permeability transition pore induces autophagy and apoptosis, and a
small quantity of mitochondria spontaneously depolarize and induce
autophagy (9). In this way, autophagy
eliminates dysfunctional mitochondria, so that cells are protected
from apoptosis.
Whilst several signaling pathways are implicated in
autophagic cell death, the PI3K-AKT-mTOR signaling pathway appears
to play a pivotal role. 3-MA, an inhibitor of class III PI3K, has
been used to suppress autophagy (10). Microtubule-associated protein light
chain 3II (LC3II) is increasingly being used to monitor levels of
autophagy; the expression levels of LC3II have been observed to
correlate with the number of autophagosomes, and therefore this is
potentially a useful method (11).
In the present study, MG63 OS cell proliferation
inhibition ratios were evaluated following the application of
various drugs. In particular, the present study focused on
apoptosis, the formation of autophagosomes under various conditions
and the expression of characteristic autophagy genes, including
LC3II and the apoptosis-activated protein caspase-3.
Materials and methods
Cell culture and experimental
design
The MG63 human OS cell line was cultured in
RPMI-1640 supplemented with 10% heat-inactivated fetal bovine serum
(FBS; both from HyClone, Logan, UT, USA), 1% penicillin (50
U/ml)/streptomycin (50 µg/ml) (Gibco Life Technologies, Carlsbad,
CA, USA) and 5 µg/ml Plasmocin™ prophylactic (InvivoGen, San Diego,
CA, USA) in a humidified atmosphere with 5% CO2, in a
water-jacketed incubator at 37°C. The MG63 cell line was obtained
from the China Center for Typical Culture Collection (Wuhan
University, Wuhan, Hubei, China).
CDDP and 3-MA were purchased from Sigma-Aldrich (St.
Louis, MO, USA). MG63 cells were divided into four groups: Control
group, cisplatin (CDDP) group, 3-methyladenine (3-MA) group and
CDDP + 3-MA group. Control group cells were treated with
phosphate-buffered saline (PBS; Sigma-Aldrich). The CDDP group was
exposed to CDDP (10 µg/ml), the 3-MA group was treated with 3-MA (6
mM) and the CDDP + 3-MA group was treated with CDDP (10 µg/ml) and
3-MA (6 mM). Cells were exposed to the drugs for 24 h.
MTT assay
MG63 cells were cultured until mid-log phase in
order to obtain a stock cell suspension that contained
1×108 cells/l. The stock cell suspension (100 µl) was
subsequently added to a 96-well plate. Following 24 h of culture,
cells were treated with the drugs, and subsequently incubated at
37°C in 5% CO2 for 24 h. Following incubation, 20 µl MTT
stock solution (5 mg/ml in PBS; Amresco LLC, Solon, OH, USA) was
added to each well. Cells were subsequently incubated at 37°C for 4
h, then centrifuged at 1,000 × g for 10 min at 37°C. Supernatant
was discarded, 150 µl dimethyl sulfoxide (Sigma-Aldrich) was added
and the plate was agitated for 10 min in the dark. Finally, the
optical density (OD) was detected using a microplate reader
(µQuant™; Bio-Tek Instruments, Inc., Winooski, VT, USA) at a
wavelength of 490 nm. The following formula was utilized in order
to calculate cell proliferation inhibition ratios: Inhibition ratio
= [1 - (ODexperimental samples/ODcontrol)] ×
100%.
Laser scanning confocal
microscopy
Cells in the four groups were incubated in culture
media with various supplements, as indicated. Following 24 h of
incubation, cells were plated on coverslips and fixed in 4%
formaldehyde (Sigma-Aldrich) in PBS for 20 min at 4°C, rinsed in
PBS and exposed to an incubation buffer (5% FBS in PBS) for an
additional 20 min at room temperature. Cells were subsequently
treated with 1:200 diluted polyclonal rabbit anti-human IgG
antibody against LC3II (#sc-28266; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) for 12 h in a humidifier at 4°C. Following rinsing
in the incubation buffer (twice for 5 min), the specimens were
incubated with 1:100-diluted goat anti-rabbit IgG-fluorescein
isothiocyanate (FITC) conjugate (#F-2765; Invitrogen Life
Technologies, Carlsbad, CA, USA) and propidium iodide (PI;
Sigma-Aldrich) for 1 h at room temperature, and subsequently rinsed
with PBS. Fluorescence was detected using a laser confocal
microscope (TCS SP2; Leica Microsystems GmbH, Wetzlar,
Germany).
Flow cytometry
Following treatment for 24 h with various drugs,
cells were collected by centrifugation at 1500 × g for 10 min at
4°C, fixed in 4% formaldehyde, rinsed with PBS and exposed to an
incubation buffer at 4°C. Cells were treated with 1:200 diluted
polyclonal rabbit anti-human antibody to LC3II for 12 h at 4°C.
Following rinsing with the incubation buffer, specimens were
exposed to 1:100 diluted goat anti-rabbit IgG-FITC conjugate for 1
h at room temperature and subsequently rinsed with PBS.
Apoptosis was quantified by combined staining with
Annexin V and PI using an Annexin V-FITC Apoptosis Detection kit
(MBL International Co., Woburn, MA, USA). Briefly, 24 h subsequent
to treatment with various drugs, cells were collected by
centrifugation at 1500 × g for 10 min at 4°C, and dissolved in 500
µl 1X binding buffer. Following the addition of 10 µl Annexin
V-FITC solution and 5 µl PI solution, cells were incubated for 15
min at room temperature in the dark.
Following incubation, cells were analyzed using a
flow cytometer (FACSort; BD Biosciences, Franklin Lakes, NJ, USA)
equipped with an argon laser and filter configuration for FITC/PI
dye combination. Light scattering and fluorescence signals were
subjected to linear and logarithmic amplifications, respectively.
At least 10,000 events were acquired and analyzed using Cell Quest
software version 5.1 (BD Biosciences). All the experiments were
performed at least three times for each condition. The data were
plotted on a logarithmic scale.
Western blot analysis
Cytosolic fraction proteins were separately
collected using an Apo Alert Cell Fractionation kit (Clontech
Laboratories, Inc., Mountainview, CA, USA). MG63 cells were
trypsinized (Sigma-Aldrich), collected, resuspended in ice-cold
wash buffer and then centrifuged at 700 × g for 5 min at 4°C.
Following removal of the supernatant, cells were resuspended in 200
µl ice-cold fractionation buffer mix and placed on ice for 10 min.
The homogenates were centrifuged at 10,000 × g for 10 min at 4°C,
and supernatants were transferred to a fresh tube. Supernatants
were collected as a cytosolic fraction. Protein samples (30 mg)
were electrophoresed on a 12.5% SDS gel (Clontech Laboratories,
Inc.) and transferred onto a Hybond enhanced chemiluminescence
(ECL) nitrocellulose membrane (Amresco, LLC, Solon, OH, USA).
Membranes were blocked by incubation with 4% skim milk in PBS with
Tween (PBST; Clontech Laboratories, Inc.) at 4°C overnight.
Following rinsing, membranes were incubated with rabbit anti-human
polyclonal anti-LC3II and rabbit anti-human monoclonal
anti-caspase-3 (#N791; Amresco) antibodies (1:1,000 dilution),
respectively, overnight at 4°C. Following five washes with PBST,
membranes were incubated with the secondary antibody (horseradish
peroxidase-conjugated anti-rabbit IgG antibody; Amresco) for 1 h at
room temperature. Following rewashing with PBST (5 times), the
membranes were analyzed using an ECL Western Blotting Substrate Kit
(Amresco). X-ray films were scanned on a flat-bed scanner, and
western blotting results were quantified using Photoshop Image
Analysis software CS3 (Adobe Systems, Inc., San Jose, CA, USA).
Statistical analysis
SPSS 10.0 package (SPSS, Inc., Chicago, IL, USA) was
utilized for statistical analysis. Data are expressed as the mean ±
standard error. Single-factor analysis of variance was performed
for each treatment group. P<0.05 was considered to indicate a
statistically significant difference. Experiments were repeated at
least three times for each condition.
Results
Inhibition of autophagy enhances
CDDP-induced cell death
A previous study revealed that CDDP promotes
apoptosis in OC cells (12). To
determine the cytotoxicity of CDDP and 3-MA on MG63 cells, the
present study investigated the effects of these drugs on cell
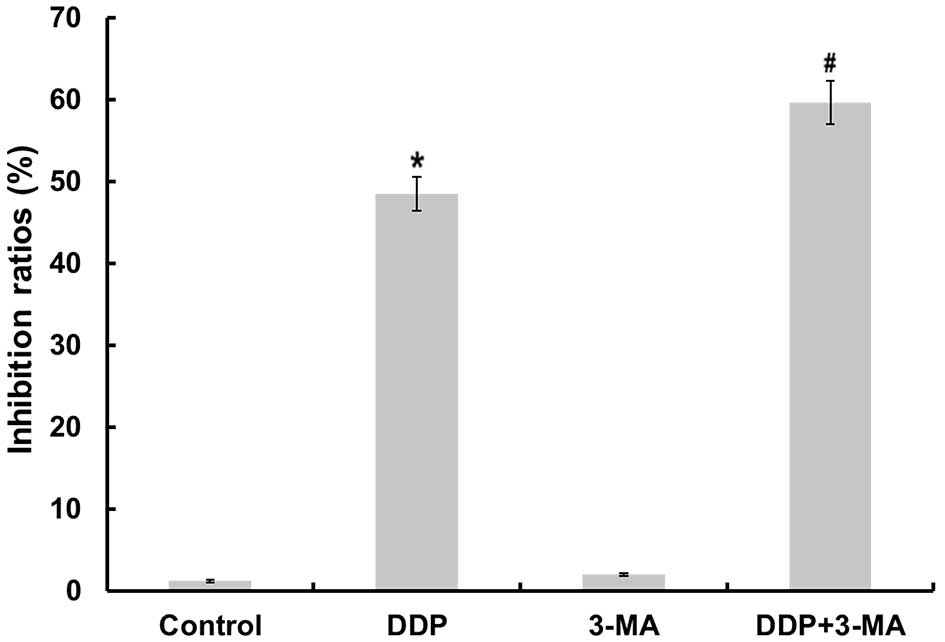
proliferation using an MTT assay. As illustrated in Fig. 1, CDDP significantly inhibited the
growth of MG63 cells (P<0.05). The 3-MA-treated cells also
demonstrated a slight change in the inhibition ratio of cell
proliferation. To determine if autophagy was involved in
CDDP-induced cell death, the present study examined whether 3-MA
was able to inhibit CDDP-induced cell death. The cell proliferation
inhibition ratio in the CDDP + 3-MA group was significantly
increased compared with that of the CDDP group (P<0.05). These
results suggest that 3-MA treatment increases the sensitivity of
MG63 cells to CDDP-induced cell death.
CDDP treatment induces autophagy in
MG63 cells
Autophagy is an alternative form of cell death to
apoptosis, characterized by the generation of autophagic vesicles,
and the degradation of cytoplasmic components and organelles
(13). LC3II, a novel autophagosome
marker, was utilized in the present study (14). In order to confirm the role of
CDDP-induced autophagy, the present study investigated the
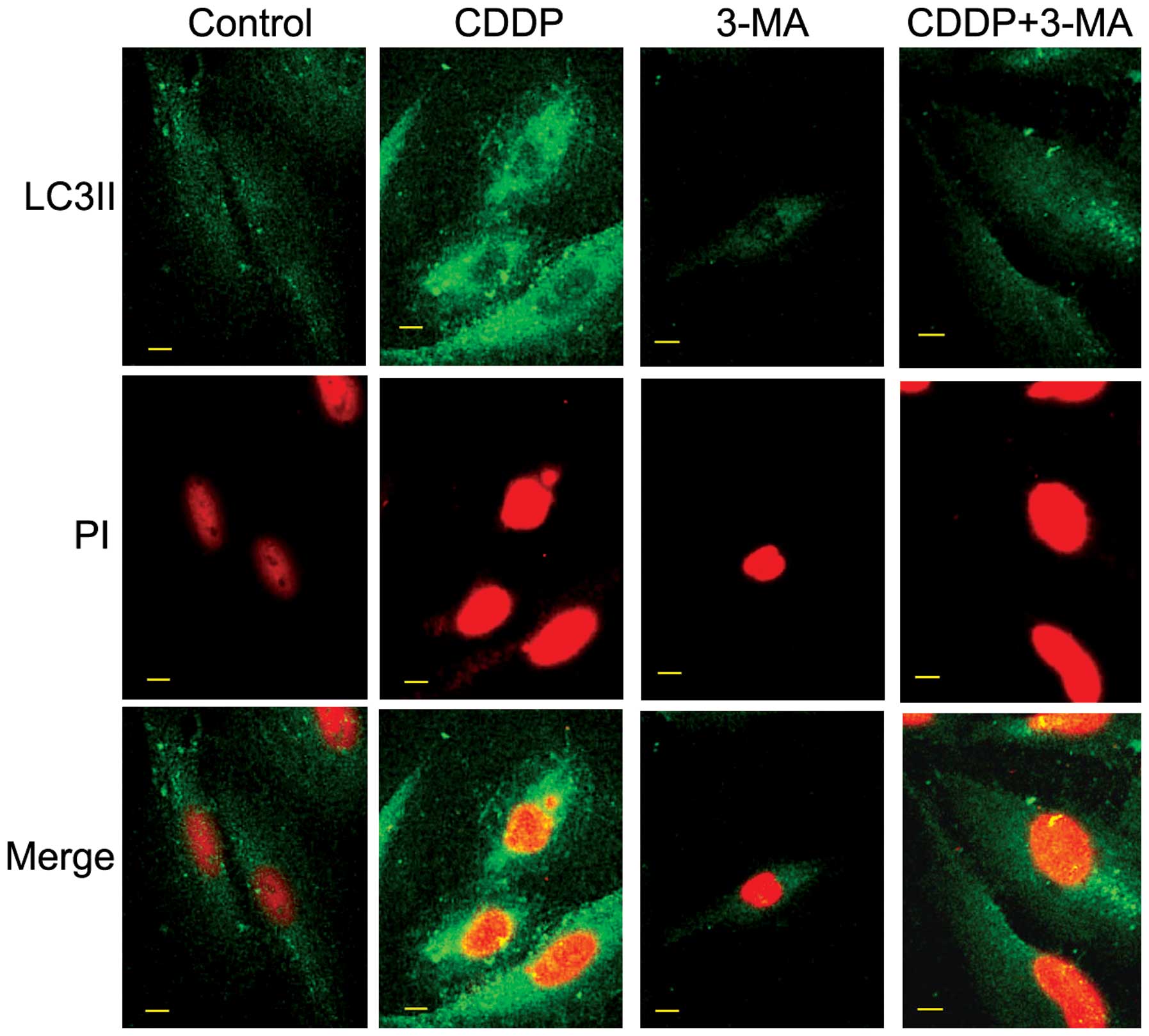
expression of LC3II in MG63 cells using confocal microscopy. As
illustrated in Fig. 2, the
fluorescence intensity of LC3II was upregulated following CDDP
treatment, indicating that CDDP may induce autophagy. Inhibitor of
autophagy 3-MA partially obstructed the expression of LC3II in MG63
cells.
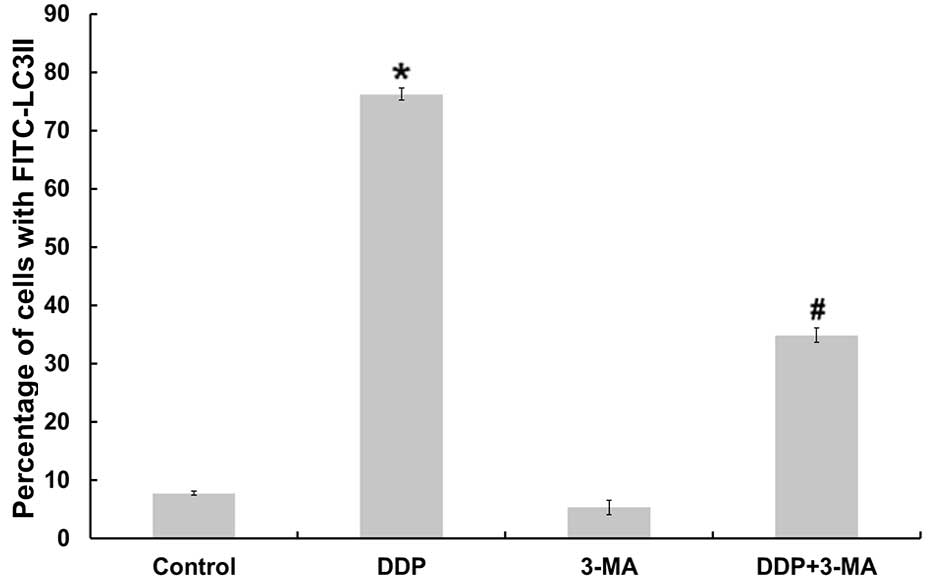
In order to further determine whether CDDP was
capable of inducing autophagy, the present study monitored the
expression profile of LC3II in cultured MG63 cells using flow
cytometry. As illustrated in Fig. 3,
MG63 cells produced significantly more LC3II following CDDP
treatment compared with the control group cells (P<0.05). By
contrast, following co-treatment with CDDP and 3-MA, the
fluorescence intensity of LC3II was downregulated compared with
that of cells treated with CDDP alone (P<0.05).
Inhibition of autophagy increases
CDDP-induced apoptosis of MG63 cells
To investigate the association between autophagy and
apoptosis in MG63 cells treated with CDDP, MG63 cells were
co-treated with CDDP and 3-MA and the degree of apoptosis was
assessed. The percentage of cells undergoing apoptosis was
quantified using flow cytometry. Apoptotic (Annexin
V+/PI−) and necrotic (Annexin
V+/PI+) cells were distinguished on the basis
of double-labeling for Annexin V-FITC and PI (Fig. 4A). As indicated in Fig. 4B, untreated control MG63 cells
demonstrated slight fluorescent staining. By contrast, treatment
with CDDP alone for 24 h induced a significant increase in the
levels of apoptosis in MG63 cells compared with control cells
(P<0.05), and CDDP and 3-MA co-treatment significantly increased
the percentage of apoptotic MG63 cells compared with that of cells
treated with CDDP alone (P<0.05). Following 24 h of incubation,
there was no significant increase in the number of necrotic
(Annexin V+/PI+) and damaged (Annexin
V−/PI+) cells between the untreated control
group and the 3-MA-treated group. Thus, the results of the present
study demonstrated that inhibition of autophagy by 3-MA
significantly increased the apoptotic effects of CDDP in MG63
cells.
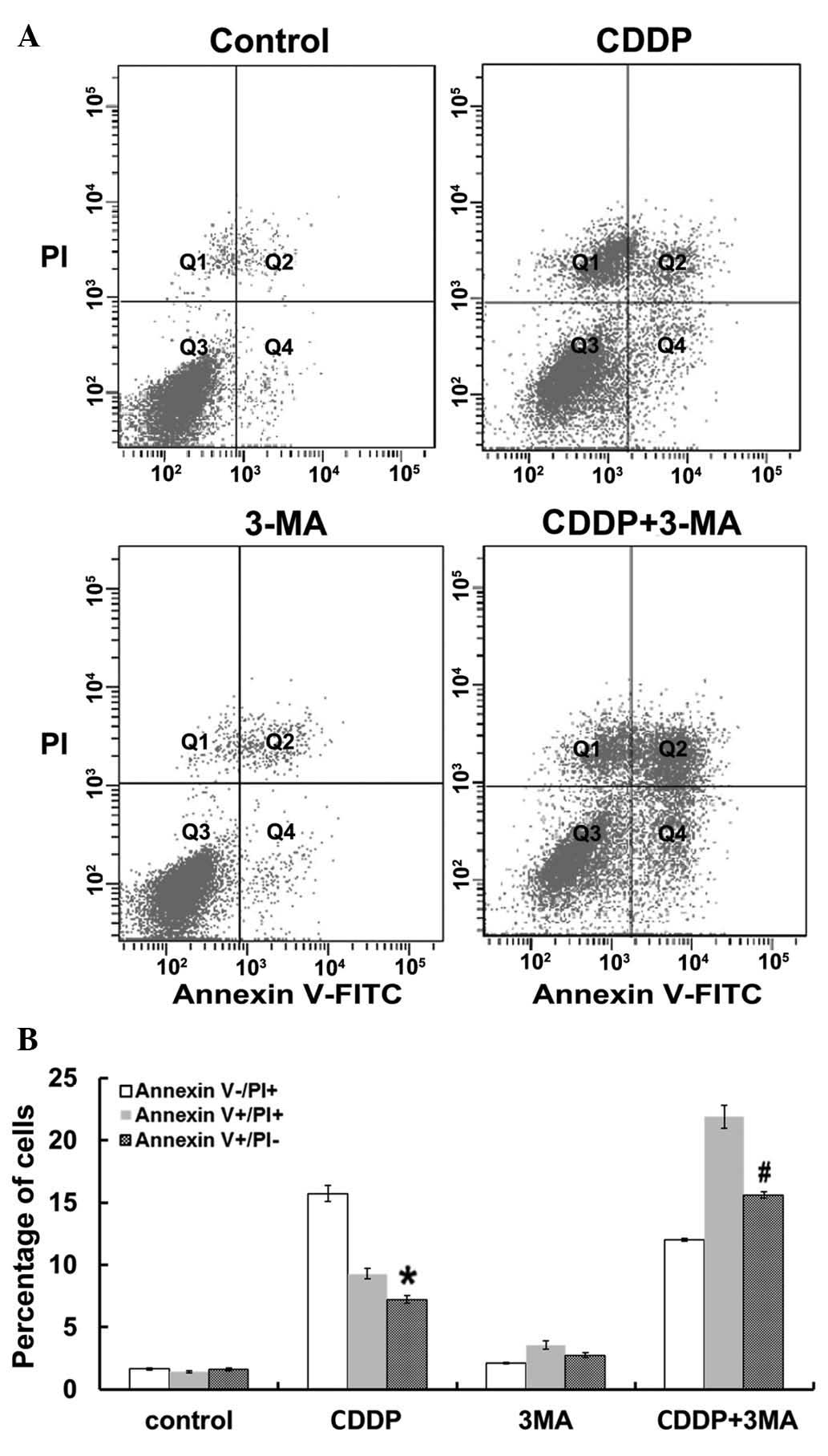 | Figure 4.Flow cytometric analysis of MG63 cell
apoptosis. (A) Representative plots of Annexin V vs. PI
fluorescence for control cells and cells treated with CDDP, 3-MA
and CDDP + 3-MA for 24 h. Q1, Annexin V−/PI+,
damaged cells; Q2, Annexin V+/PI+, necrotic
cells; Q3, Annexin V−/PI−, viable cells; and
Q4, Annexin V+/PI−, apoptotic cells. (B)
Percentage of cells labeled with Annexin
V+/PI− (apoptotic cells), Annexin
V−/PI+ (damaged cells) and Annexin
V+/PI+ (necrotic cells) following 24 h of
treatment. Data are presented as the mean ± standard error. CDDP
significantly increased the percentage of Annexin
V+/PI− (apoptotic cells) compared with that
of control cells (*P<0.05). 3-MA + CDDP co-treatment
significantly increased the percentage of Annexin
V+/PI− (apoptotic cells) compared with that
of cells treated with CDDP alone (#P<0.05). There was
no significant difference between the untreated control and 3-MA
groups (P>0.05). PI, propidium iodide; CDDP, cisplatin; 3-MA,
3-methyladenine; FITC, fluorescein isothiocyanate. |
Differential effects of CDDP and 3-MA
may be due to the expression of LC3II and caspase-3 in MG63
cells
The present study investigated the protein
expression of LC3II and caspase-3 in MG63 cells. As illustrated in
Fig. 5, western blot analyses
revealed that the expression levels of LC3II and caspase-3 proteins
were increased in MG63 cells following CDDP treatment. Furthermore,
reduced levels of LC3II protein and increased levels of caspase-3
protein were detected in the CDDP + 3-MA co-treatment group,
compared with those of the CDDP-treated group (P<0.05). The
results of the present study suggest that the differential effects
of CDDP and 3-MA on MG63 cells may be due to the expression of
proapoptotic and autophagic proteins.
Discussion
The results of the present study revealed that
autophagy protected MG63 human OC cells from CDDP-induced
apoptosis, and inhibition of autophagy mediated by 3-MA enhanced
the sensitivity of MG63 cells to the apoptosis inducer CDDP. CDDP
is an alkylating agent that reacts with DNA and cellular proteins,
and the magnitude of cell death is well correlated with tumor
response to CDDP (15). The molecular
mechanisms through which CDDP induces apoptosis are DNA
crosslinking, inhibition of DNA replication and RNA transcription
(16). In the present study, it was
demonstrated that CDDP specifically induced apoptosis via caspase-3
activation in MG63 cells.
Although a number of researchers have hypothesized
potential mechanisms, the signaling pathways of the target
molecules of CDDP that lead to cell death have not yet been
elucidated (17). Previous studies
have revealed that numerous chemotherapeutic agents are capable of
inducing autophagic cell death. The alkylating agent temozolomide
is capable of killing malignant glioma cells via the process of
autophagic cell death (18).
Comparably, resveratrol is capable of inducing autophagic cell
death in ovarian cancer cells (19).
Autophagy is characterized by the appearance of
abundant cytoplasmic autophagic vacuoles, and an increase in the
size of the endoplasmic reticulum and Golgi apparatus (20). The LC3II protein, located in the
autophagosomal membrane, may be used as a general marker for the
autophagic membrane (21). 3-MA is an
inhibitor of the class III phosphatidylinositol 3-kinases and is
known to be involved in the initial phase of autophagy (22). A previous study investigating 3-MA
demonstrated that it was capable of increasing the sensitivity of
HT-29 colon cancer cells to apoptosis induced by a cyclooxygenase
inhibitor, sulindac sulfide (23). In
the present study, 3-MA was used in combination with CDDP, and the
results revealed that the sensitivity to chemotherapy may be
increased by the downregulation of autophagy. The findings of the
aforementioned studies suggest that autophagy may inhibit apoptosis
by sequestrating mitochondrial death-promoting factors, for example
cytochrome c (24). However,
the mechanism of autophagic activity, which promotes the protection
of cells from apoptosis remains to be elucidated.
Recent advances in our understanding of the
molecular mechanisms underlying anticancer therapy-induced
autophagy and apoptosis, have provided significant information for
studying tumor responses, in terms of the signal transduction
pathways of cell death (25). This
understanding potentially facilitates the design of targeted
therapies for the promotion of cancer cell sensitivity to
anticancer treatments.
References
|
1
|
Desandes E: Survival from adolescent
cancer. Cancer Treat Rev. 33:609–615. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Biermann JS, Adkins D, Benjamin R, Brigman
B, Chow W, Conrad EU III, Frassica D, Frassica FJ, George S, Healey
JH, et al: National Comprehensive Cancer Network: Bone cancer. J
Natl Compr Canc Netw. 5:420–437. 2007.PubMed/NCBI
|
|
3
|
Sakamoto A and Iwamoto Y: Current status
and perspectives regarding the treatment of osteo-sarcoma:
Chemotherapy. Rev Recent Clin Trials. 3:228–231. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yamamoto N and Tsuchiya H: Chemotherapy
for osteosarcoma - where does it come from? What is it? Where is it
going? Expert Opin Pharmacother. 14:2183–2193. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shen HM and Tergaonkar V: NFkappaB
signaling in carcinogenesis and as a potential molecular target for
cancer therapy. Apoptosis. 14:348–363. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Monastyrska I and Klionsky DJ: Autophagy
in organelle homeostasis: Peroxisome turnover. Mol Aspects Med.
27:483–494. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gewirtz DA: Autophagy as a mechanism of
radiation sensitization in breast tumor cells. Autophagy.
3:249–250. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lemasters JJ, Nieminen AL, Qian T, Trost
LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA,
Brenner DA, et al: The mitochondrial permeability transition in
cell death: A common mechanism in necrosis, apoptosis and
autophagy. Biochim Biophys Acta. 1366:177–196. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Z, Shao Z, Xiong L, Che B, Deng C
and Xu W: Expression of Beclin1 in osteosarcoma and the effects of
down-regulation of autophagy on the chemotherapeutic sensitivity. J
Huazhong Univ Sci Technolog Med Sci. 29:737–740. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu J, Dang Y, Su W, Liu C, Ma H, Shan Y,
Pei Y, Wan B, Guo J and Yu L: Molecular cloning and
characterization of rat LC3A and LC3B - two novel markers of
autophagosome. Biochem Biophys Res Commun. 339:437–442. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Seki K, Yoshikawa H, Shiiki K, Hamada Y,
Akamatsu N and Tasaka K: Cisplatin (CDDP) specifically induces
apoptosis via sequential activation of caspase-8, −3 and −6 in
osteosarcoma. Cancer Chemother Pharmacol. 45:199–206. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Suzuki K, Kubota Y, Sekito T and Ohsumi Y:
Hierarchy of Atg proteins in pre-autophagosomal structure
organization. Genes Cells. 12:209–218. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Halpain S and Dehmelt L: The MAP1 family
of microtubule-associated proteins. Genome Biol. 7:2242006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nooter K and Stoter G: Molecular
mechanisms of multidrug resistance in cancer chemotherapy. Pathol
Res Pract. 192:768–780. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ogawa M: Anticancer drugs and
pharmacologic actions. Nihon Rinsho. 55:1017–1023. 1997.(In
Japanese). PubMed/NCBI
|
|
17
|
Park YP, Kim KD, Kang SH, Yoon Y, Park JW,
Kim JW and Lee HG: Human telomerase reverse transcriptase (hTERT):
A target molecule for the treatment of cisplatin-resistant tumors.
Korean J Lab Med. 28:430–437. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kanzawa T, Germano IM, Komata T, Ito H,
Kondo Y and Kondo S: Role of autophagy in temozolomide-induced
cytotoxicity for malignant glioma cells. Cell Death Differ.
11:448–457. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Opipari AW Jr, Tan L, Boitano AE, Sorenson
DR, Aurora A and Liu JR: Resveratrol-induced autophagocytosis in
ovarian cancer cells. Cancer Res. 64:696–703. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xie Z and Klionsky DJ: Autophagosome
formation: Core machinery and adaptations. Nat Cell Biol.
9:1102–1109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tanida I, Ueno T and Kominami E: LC3
conjugation system in mammalian autophagy. Int J Biochem Cell Biol.
36:2503–2518. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang YP, Liang ZQ, Gu ZL and Qin ZH:
Molecular mechanism and regulation of autophagy. Acta Pharmacol
Sin. 26:1421–1434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bauvy C, Gane P, Arico S, Codogno P and
Ogier-Denis E: Autophagy delays sulindac sulfide-induced apoptosis
in the human intestinal colon cancer cell line HT-29. Exp Cell Res.
268:139–149. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Herman-Antosiewicz A, Johnson DE and Singh
SV: Sulforaphane causes autophagy to inhibit release of cytochrome
c and apoptosis in human prostate cancer cells. Cancer Res.
66:5828–5835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|