Introduction
MicroRNAs (miRNAs) are a class of single-stranded,
non-coding RNA molecules that are 18–22 nucleotides in length, are
encoded by endogenous genes and are transcribed from genomic DNA
(1). However, miRNAs are not directly
translated into proteins; instead, they control protein synthesis
by binding their complementary sequence to the 3′ untranslated
region (UTR) of the target gene messenger RNA (mRNA) molecule and
inducing mRNA degradation or inhibition of translation, which
subsequently inhibits protein synthesis (2). Therefore, miRNAs are capable of
regulating protein-coding genes (1).
Previous research has demonstrated that miRNAs participate in a
series of critical physiological processes, including development,
hematopoiesis, organogenesis, apoptosis and cell proliferation, and
miRNAs have also been associated with the incidence of multiple
diseases, including the development of tumors (3). Endometrial cancer is a common malignant
tumor of the female reproductive system, which has a significant
impact on the quality of life of patients and may result in
mortality. Endometrial cancer may be classified as type I or type
II, according to certain clinical and pathological characteristics.
The initiation of type I endometrial cancer is associated with
long-term exposure to increased levels of estrogen stimulation;
therefore type I endometrial cancer is known as estrogen-dependent
endometrial cancer (4). The
initiation of type II endometrial cancer is not associated with
estrogen stimulation and it is therefore referred to as
estrogen-independent endometrial cancer (4). Ishikawa (ISK) cells are a highly
differentiated endometrial adenocarcinoma cell line that express
the estrogen receptor (ER), and are considered to be a classic
ER-positive cell line, exhibiting typical type I characteristics of
endometrial cancer (5). Therefore,
ISK cells are frequently used for in vitro experiments
examining the role of estrogen and the ER in endometrial cancer.
Conversely, the HEC-1B cell line, which consists of
intermediately-differentiated endometrial adenocarcinoma cells, is
ER-negative or expresses the ER at reduced levels, exhibiting
typical type II endometrial cancer characteristics (6). These two cell types are widely used for
basic endometrial cancer research (5). Type II endometrial cancer is less
frequently observed in patients compared with type I, and accounts
for 20–30% of total cases (7).
However, type II endometrial cancer has been revealed to be
associated with a poorer prognosis, and to differ significantly
from type I endometrial cancer, in terms of histological
differentiation, myometrial invasion, lymph node metastasis and
recurrence rate (8). In addition, ER
and progesterone receptor expression has been observed to differ
significantly between type I and II endometrial cancer, and is
considered to be a critical marker for the assessment of prognosis
(9).
Previous studies on ISK and HEC-1B endometrial
cancer cell lines have mainly focused on hormone and mRNA
expression levels. Previous research has revealed that miRNAs are
critical regulators of the progression of cancer; therefore the
abnormal miRNA expression observed in endometrial cancer has gained
attention (10–12). Myatt et al (10) identified that expression levels of the
tumor suppressor Forkhead box protein O1 (FOXO1) were significantly
downregulated in endometrial cancer cell tumor tissues compared
with normal endometrial tissues. miRNA (miR)-9, miR-27, miR-96,
miR-153, miR-182 and miR-183 were predicted, using bioinformatic
tools (miRNA_Targets; http://mamsap.it.deakin.edu.au/~amitkuma/mirna_targetsnew/find_gene.html),
to bind to the FOXO1 3′UTR and this hypothesis was subsequently
verified using reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) and northern blotting. These miRNAs are
associated with the downregulation of FOXO1 and therefore may be
involved in the development of endometrial cancer. Chung et
al (11) utilized RT-qPCR to
selectively screen for miR-205, which was observed to be expressed
at increased levels in endometrial adenocarcinoma-like cells.
Following endometrial cancer cell transfection with an miR-205
inhibitor, miR-205 expression was observed to be significantly
reduced and the protein expression of its predicted target gene
JPH4 was observed to be increased. This result indicated that
miR-205 may be involved in the initiation and development of
endometrial cancer. Snowdon et al (12) demonstrated that the expression of the
miRNA-200 family was significantly increased in endometrial cancer,
compared with the miRNA-200 family expression observed in atypical
endometrial hyperplasia. Furthermore, compared with studies
focusing on the expression of individual miRNAs, investigations
into the variation in the expression of miRNA clusters and families
have provided novel hypotheses for the study of miRNAs in
endometrial cancer (13).
Until recently, studies of miRNA expression in
endometrial cancer utilized conventional microarrays, which mainly
rely on sequence hybridization and qPCR. Next-generation sequencing
technology is capable of detecting novel miRNAs, and additionally
demonstrates increased sensitivity and a broad dynamic range
(14). A significant advantage of
deep sequencing is that the final sequencing results do not contain
saturation effects, which frequently occur during the analysis of
microarray results (14).
Although fluorescent qPCR possesses the advantages
of being sensitive, reproducible, quantitatively accurate and
rapid, it has additional disadvantages. Similarly to microarray
technology, fluorescent qPCR is only capable of detecting known
miRNAs. A significant disadvantage of fluorescent qPCR is that it
does not support high throughput detection of miRNAs. By contrast,
next-generation sequencing has successfully overcome these
disadvantages and is able to simultaneously achieve high throughput
analysis with increased sensitivity (15).
Materials and methods
Sample preparation and RNA
extraction
ISK and HEC-1B endometrial cancer cell lines were
obtained from the Obstetrics and Gynecology Research Institute of
Fudan University Women's Hospital (Shanghai, P.R. China). Cells
were maintained in Dulbecco's modified Eagle's medium supplemented
with 5% fetal bovine serum, 5 µg/ml bovine insulin, 100 U/ml
penicillin and 100 µg/ml streptomycin (all Solarbio Science &
Technology Co.,Ltd, Beijing, China) in a 5% CO2
atmosphere at 37°C. Total RNA was extracted from cells using the
mirVana™ miRNA isolation kit (Ambion Life Technologies, Carlsbad,
CA, USA). RNA concentrations were measured using a NanoDrop®
ND-1000 spectrophotometer (Thermo Fisher Scientific, Wilmington,
DE, USA).
Small RNA library generation and
sequencing
Small RNA libraries for deep sequencing analysis
were prepared from total RNA using the Applied Biosystems Small RNA
Expression kit (Thermo Fisher Scientific, Waltham, MA, USA).
Briefly, ~500 ng total RNA was hybridized and ligated with the 5′
and 3′ adapter A and adapter B overnight. Subsequently, template
bead preparation and emulsion PCR were performed using the Applied
Biosystems SOLiD emulsion PCR and SOLiD buffer kits (Thermo Fisher
Scientific), according to the manufacturer's instructions. Briefly,
the oil phase was prepared: A total of 400 µl Tween 80
(Sigma-Aldrich, St. Louis, MO, USA) and 1.8 ml SP80 (Sigma-Aldrich)
were added to 37.8 ml mineral oil (Sigma-Aldrich) and mixed
thoroughly. Next, 80 µl P1 primer-labeled beads (DNA sequence,
5′-TTTTTTCCACTACGCCTCCGCTTTCCTCTCTATGGGCAGTCGGTGAT-3′; Applied
Biosystems; Thermo Fisher Scientific) were added to the aqueous
phase [consisting of 10X PCR Buffer (280 µl), 100 mM dNTP Mix (392
µl), 1M MgCl2 (70 µl), 10 µM ePCR Primer 1 (11.2 µl),
500 µM ePCR Primer 2 (16.8 µl), 500 pM prepared library (2.8 µl),
nuclease-free water (1647.2 µl) and 5 U/µl AmpliTaq Gold® DNA
Polymerase, UP (300 µl)] to a final volume of 2,800 µl. The DNA
primer sequences of the forward ePCR Primer 1 and reverse ePCR
Primer 2 were 5′-CCACTACGCCTCCGCTTTCCTCTCTATG-3′ and
5′-CTGCCCCGGGTTCCTCATTCT-3′, respectively. The emulsion was
produced using the ULTRA-TURRAX®Tube Drive (IKA®-Werke GmbH &
Co. KG, Staufen, Germany). The aqueous phase (2,800 µl) was added
to 9 ml oil phase gradually, and 100 µl emulsion was gently
dispensed into each well of a 96-well PCR plate. ePCR was performed
using the Applied Biosystems GeneAmp® PCR System 9700 (Thermo
Fisher Scientific) under the following conditions: Initial
denaturation step at 95°C for 5 min, followed by 60 cycles of 93°C
for 15 sec, 62°C for 30 sec and 72°C for 75 sec, with a final
extension step at 72°C for 7 min. Following PCR, the emulsion was
broken by the addition of two volumes of 2-butanol (294810-1L;
Sigma-Aldrich) and the template beads were washed three times with
wash solution (10 mM Tris-HCl, 50 mM KCl, 2 mM EDTA and 0.01% (v/v)
Triton X-100) and suspended in Applied Biosystems TE buffer (10 mM
Tris-HCl and 1 mM EDTA; Thermo Fisher Scientific) for further
sequencing. Finally, deposition was performed according to the
standard protocol.
Sequence processing and
annotation
The SOLiD System Small RNA Analysis Pipeline tool
(RNA2MAP, version 0.5.0; Life Technologies, Carlsbad, CA, USA) was
used to analyze the miRNA sequencing data, according to the
following protocol. Initially, two mismatches for the sequencing
data were defined, the data from the present study was compared
with the available known RNA library (www.mirbase.org) and subsequently, known RNA sequences
were eliminated to prevent interference with the comparison with
the miRNA library. Processed reads resulting from the above method
were compared with the known miRNA precursor library (miRBase 20.0;
www.mirbase.org) (16). Comparison data for each miRNA was
collected and a statistical analysis of the length of the reads
that matched the precursors was undertaken. Finally, the remaining
reads were matched to the genome to identify potential novel
miRNAs.
Normalization of read counts
It was assumed that unique sequence reads of <10
copies may represent sequencing errors; therefore, these reads were
eliminated from the total sequencing data. To simplify the
comparison of miRNA expression variation in various samples, the
total copy number in each sample was standardized and uniformly set
to 1,000,000 for each sample.
Statistical analysis
The characteristics of study subjects were compared
using Student's t-test. To show the differential expression level
of specific miRNA between the ISK and HEC-1B. The data were
normalized and log2-transformed. GraphPad Prism 5.0 software
(GraphPad Software Inc., La Jolla, CA, USA) was used for all
statistical analyses. P<0.05 was considered to indicate a
statistically significant difference.
Results
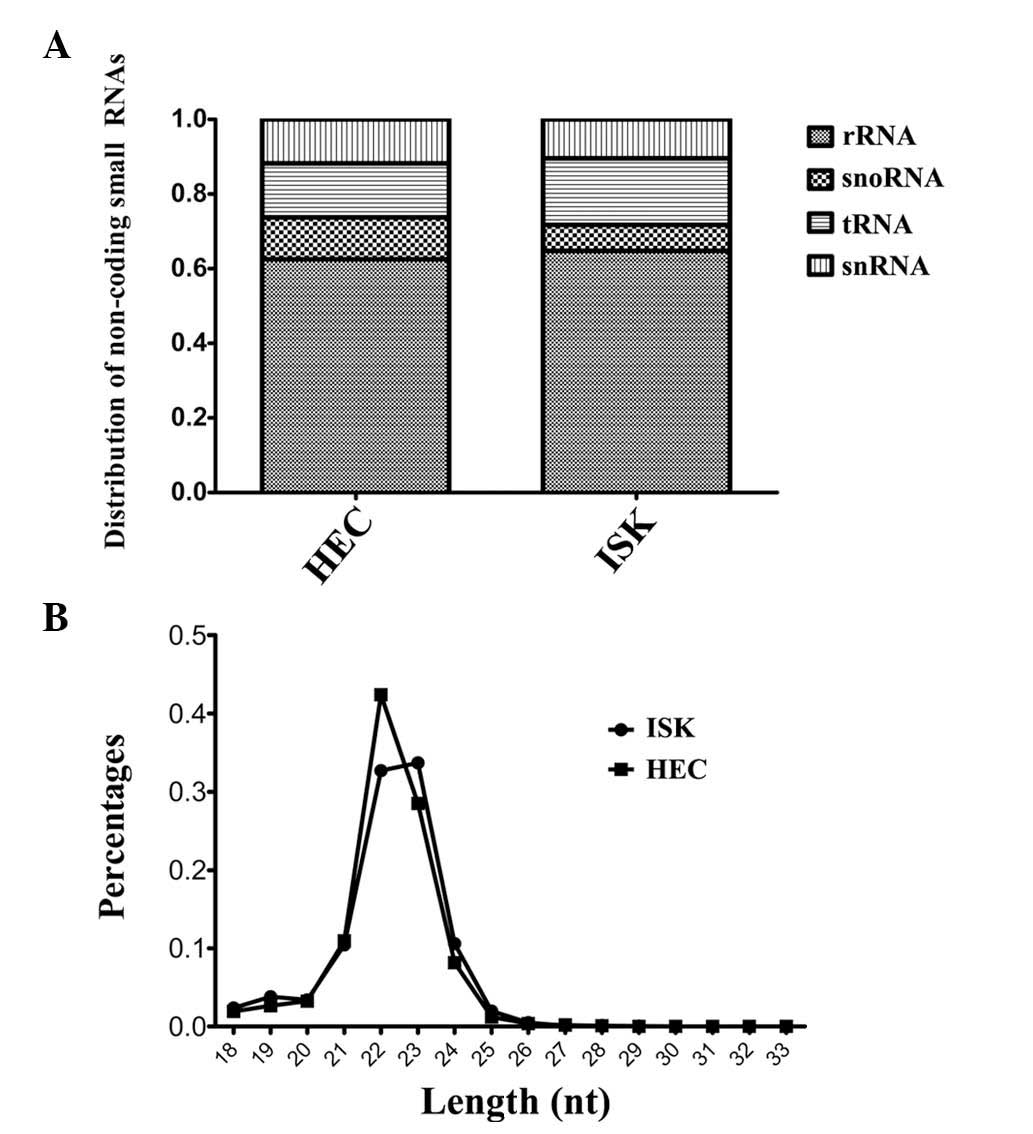
Overview of sequencing data
The raw sequencing data was classified into the
following six databases: Ribosomal RNA (rRNA), small nucleolar RNA,
transfer RNA (tRNA), small nuclear RNA, miRNA and genome. The
results indicated that the proportion of miRNAs among the total
non-coding small RNAs was similar for HEC-1B and ISK cells (77 vs.
78%, respectively). Excluding miRNA, rRNA was the most abundant RNA
observed among the non-coding small RNAs, and was revealed to
represent 62 and 64% of non-coding small RNAs in HEC-1B and ISK
cells, respectively. The distribution of small RNAs in each sample
is illustrated in Fig. 1A. In
conclusion, small RNA sequencing results demonstrated that miRNAs
and rRNAs were the most predominant non-coding small RNAs detected
in the HEC-1B and ISK cell lines and the proportions of these RNAs,
compared with the total observed non-coding small RNA population,
were comparable between the two cell lines.
The length distribution of the miRNAs in the
sequencing data was analyzed. It was revealed that in the HEC-1B
and ISK cell lines, miRNAs with lengths of 22 and 23 nucleotides
(nt) appeared as the two highest peaks. The overall length
distribution indicated that the authenticity of the miRNA
expression profile was maximally preserved during the process of
sequencing library preparation (Fig.
1B). However, in the ISK cell line, the proportion of miRNAs
with lengths of 22 and 23 nt was similar, whereas the proportion of
22 nt miRNAs was greater than that of 23 nt miRNAs in the HEC-1B
cell line.
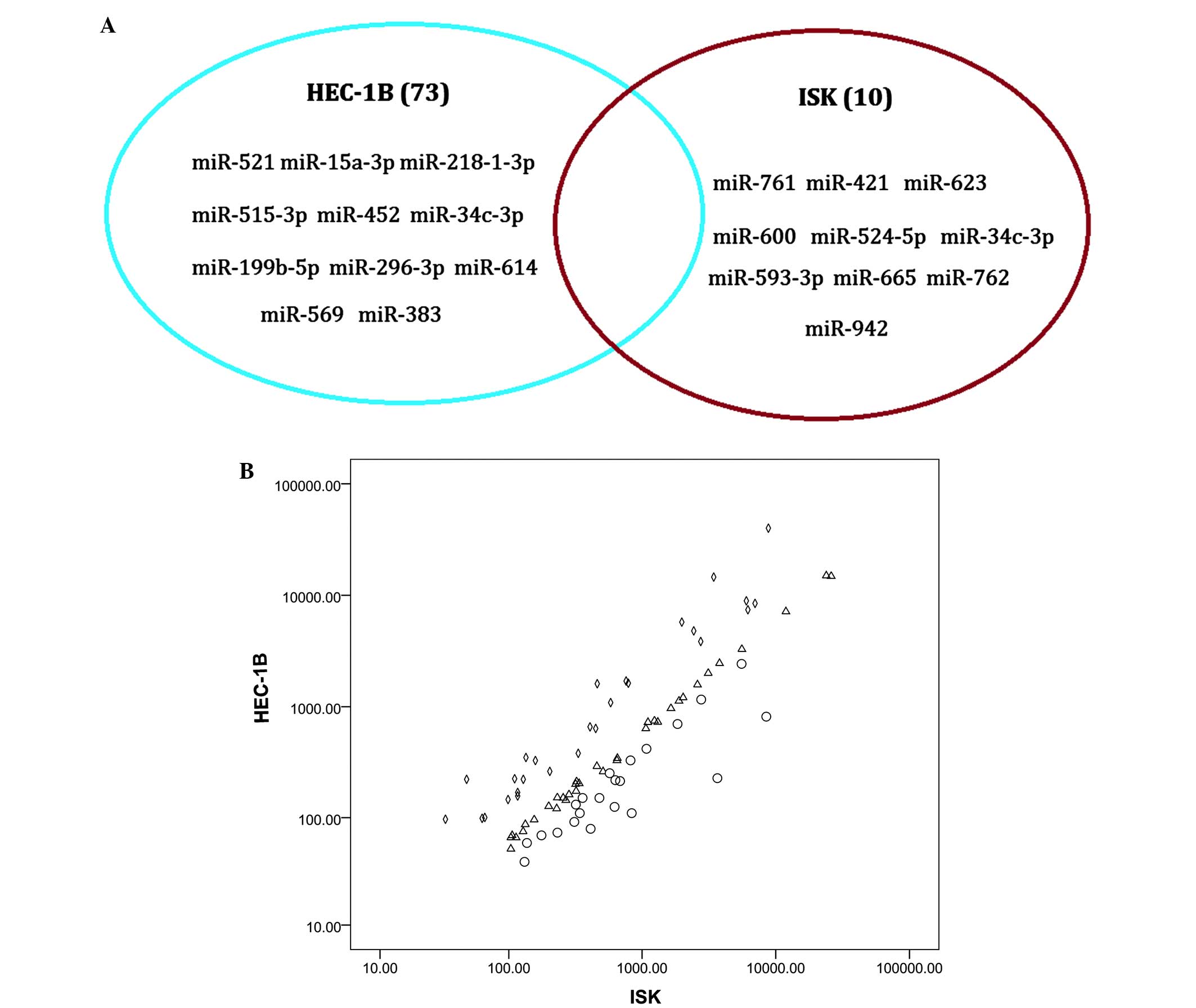
Differential expression of miRNAs
between the HEC-1B and ISK cell lines
The primary focus of the present study was to
examine the differential expression of miRNAs between an
estrogen-dependent (ISK) and an estrogen-independent endometrial
cancer cell line (HEC-1B). Mature miRNAs with copy numbers >10
were considered to be representative of a valid read. According to
this definition, 381 miRNAs in the HEC-1B cell line and 318 miRNAs
in the ISK cell line were detected. Furthermore, 73 unique miRNAs
were identified to be present in the HEC-1B cell line and absent in
ISK cell line, whereas 10 miRNAs were expressed in the ISK cell
line and absent in HEC-1B cells (Fig.
2A).
The ten most abundant miRNAs in the HEC-1B and ISK
cell lines were identified (Table I)
and those that were included in the lists from the two cell types
included Homo sapiens (hsa)-miR-19b, hsa-miR-23b,
hsa-miR-17, hsa-let-7a, hsa-miR-18a and hsa-miR-30d. However,
expression of these miRNAs varied significantly between the two
samples; miR-141 was observed to be highly expressed in the HEC-1B
cell line but was not abundantly expressed in the ISK cell
line.
 | Table I.Ten most abundantly expressed miRNAs
in HEC and ISK cell lines. |
Table I.
Ten most abundantly expressed miRNAs
in HEC and ISK cell lines.
| A, HEC |
|
|---|
|
|---|
| Most abundantly
expressed | All isomiRs |
|---|
| hsa-miR-141 | hsa-miR-141 |
| hsa-miR-17 | hsa-miR-19b |
| hsa-miR-130a | hsa-miR-23b |
| hsa-miR-18a | hsa-miR-17 |
| hsa-miR-200a | hsa-let-7a |
| hsa-miR-93 | hsa-miR-200a |
| hsa-miR-19b | hsa-miR-191 |
| hsa-miR-30d | hsa-miR-18a |
| hsa-miR-210 | hsa-miR-130a |
| hsa-miR-23b | hsa-miR-30d |
|
| B, ISK |
|
|
| Most abundantly
expressed | All isomiRs |
|
| hsa-miR-18a | hsa-miR-19b |
| hsa-miR-17 | hsa-miR-18a |
| hsa-miR-30d | hsa-miR-191 |
| hsa-miR-19b | hsa-miR-17 |
| hsa-miR-93 | hsa-miR-23b |
| hsa-miR-130a | hsa-miR-93 |
| hsa-miR-191 | hsa-miR-103 |
| hsa-let-7g | hsa-let-7a |
| hsa-miR-23b | hsa-miR-20a |
In addition, to understand the differential
expression of miRNAs between the HEC-1B and ISK cell lines, the
miRNA reads were normalized and the log2 values of each
miRNA in the ISK and HEC-1B cells were calculated and compared
(Fig. 2B).
As miRNAs with copy numbers <10 reads may
represent sequencing errors, the differential expression of miRNAs
with copy numbers >10 reads was compared. It was observed that
following normalization, there were 34 miRNAs in the HEC-1B cell
line which differed >1.5-fold compared with those in the ISK
cell line, and there were 105 miRNAs in the ISK cell line which
differed >1.5-fold compared with those in the HEC-1B cell
line.
Notably, when TAM online software (17) (http://cmbi.bjmu.edu.cn/tam) was used to analyze
miRNAs with expression levels that were significantly different
between the ISK and HEC-1B cell lines, it was revealed that there
were 18 upregulated miRNAs in the ISK cell line that were
correlated with hormone regulation (P=1.25×10−3, where
P<0.01 was considered to represent a significant correlation).
However, in the HEC-1B cell line, there were only 4 upregulated
miRNAs that were correlated with hormone regulation (P=0.36). These
results may indicate that there are a number of miRNAs associated
with hormone regulation in the ISK cell line. These results support
the findings of previous studies, as the ISK cell line is
associated with ER expression, which is known to possess a critical
role in hormonal regulation (18).
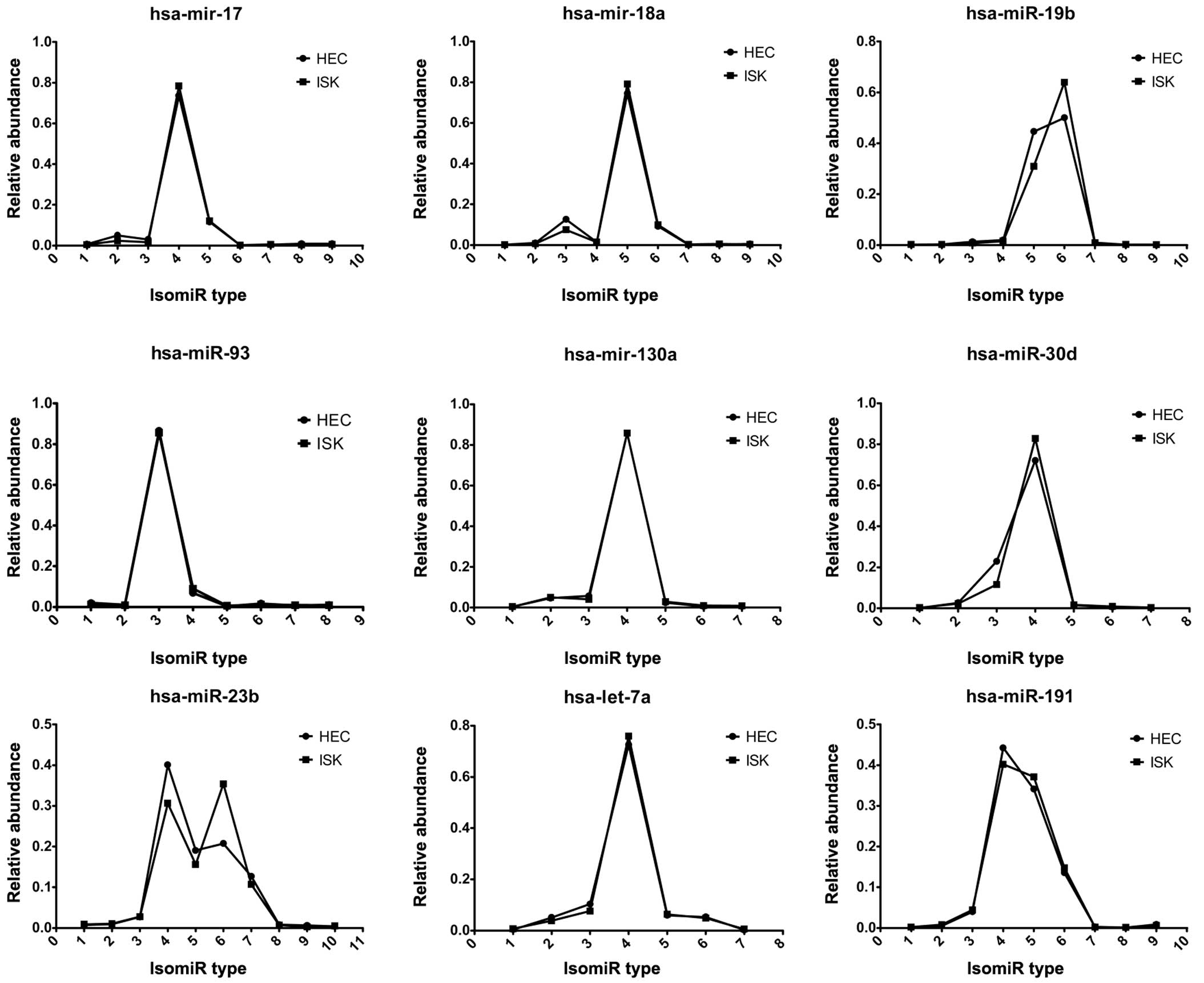
Expression patterns of miRNA variants
in HEC-1B and ISK cells
Multiple isomiRs, including isomiRs with 3′
additions, were detected from a given miRNA locus (Fig. 3). These nine abundant miRNAs
demonstrated similar numbers of miRNA variants with comparable
expression levels. Furthermore, the isomiR expression patterns of
hsa-miR-17, hsa-mir-18a and hsa-miR-19b, which are encoded by the
miR-17 cluster in HEC-1B and ISK cells, were highly consistent
(Fig. 3).
Differential expression of miRNA
clusters and families
Previous studies have demonstrated that miRNA genes
are arranged as clusters on the chromosome (19,20). The
members of an miRNA cluster typically share sequence similarities
(21) and coordinate the regulation
of similar biological processes (22). When members of a gene cluster exhibit
sequence similarity and homologous regulation, they form miRNA gene
families, which further increase the diversity of miRNA gene
cluster distribution (23). Previous
studies have generally focused on the expression and regulation of
single miRNAs, while studies investigating the expression and
function of clustered miRNAs are infrequent and studies examining
the expression profile of miRNA clusters at the genome-wide level
are particularly limited. However, the expression patterns of
homologous miRNAs arranged as clusters are typically coordinated
(22).
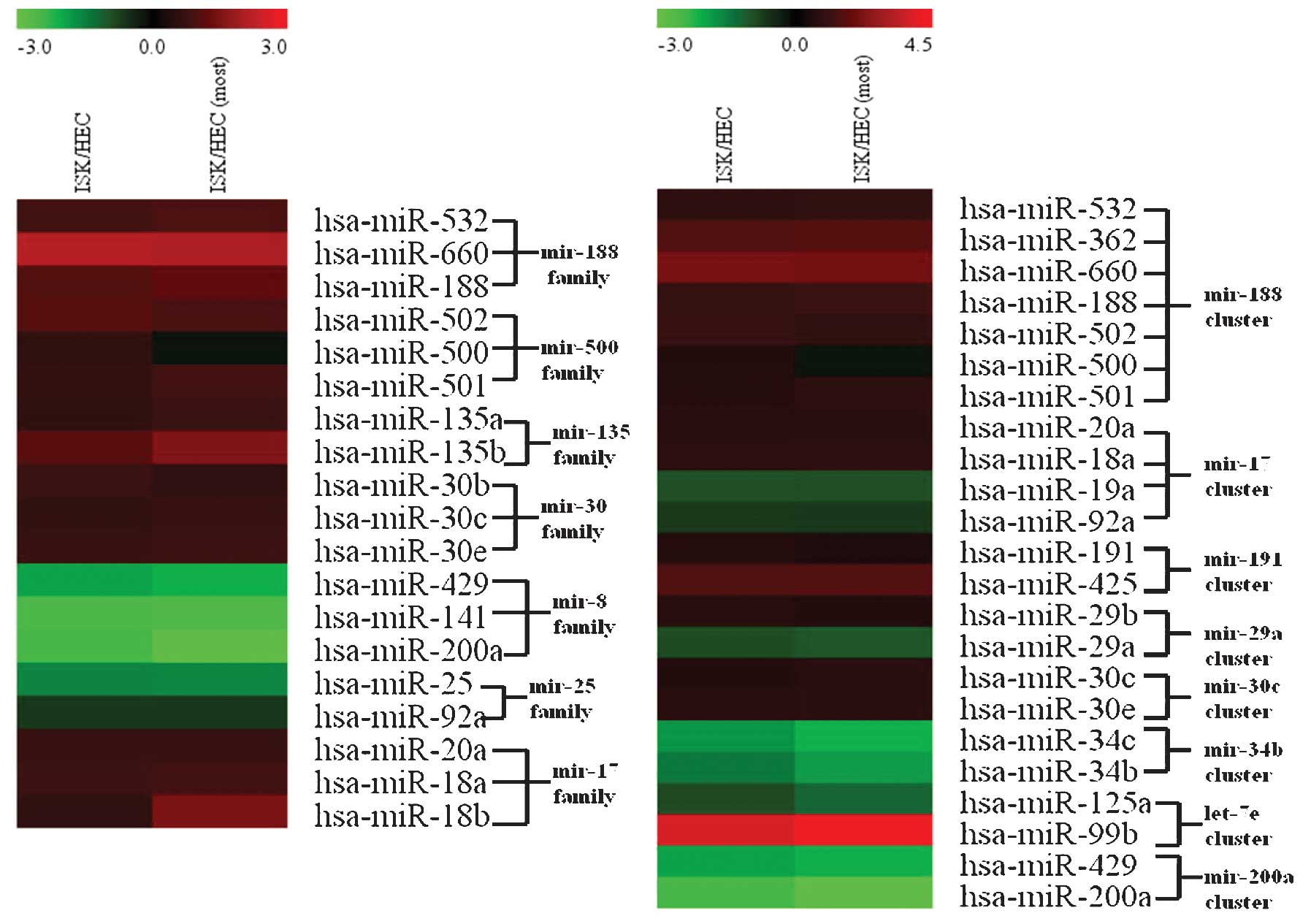
miRNAs exhibiting significantly different expression
patterns between the ISK and HEC-1B cell lines were classified into
clusters and families. miRNA clusters and families were considered
distinct if there were ≥2 miRNA members in the cluster or family
that exhibited significantly different expression levels between
ISK and HEC-1B cells.
Based on the cluster comparison definition used in
the present study, it was observed that there were 5 miRNA clusters
in the ISK cell line in which expression levels were significantly
upregulated compared with those of the HEC-1B cell line, and 3
miRNA clusters in which expression levels were significantly
downregulated in ISK cells as compared with HEC-1B cells. In
addition, there were also 5 miRNA families in the ISK cell line for
which expression levels were significantly upregulated compared
with the HEC-1B cell line and 2 miRNA families for which expression
levels were significantly downregulated. Notably, expression levels
of all 7 miRNA members in the hsa-mir-188 cluster were
significantly upregulated (P=1.12×10−6), which
demonstrated that these 7 miRNAs exhibited expression patterns
consistent with each other. The presence of similar expression
levels of miRNA members within an miRNA cluster indicated a
co-transcription process and suggested that clustered miRNAs
possess identical expression levels. Notably, the hsa-mir-17
cluster exhibited an inconsistent expression pattern; in the ISK
cell line, hsa-mir-20a and hsa-mir-18a were upregulated, while
hsa-mir-19a and hsa-mir-92a were downregulated. This suggested that
clustered miRNAs may additionally possess an independent
transcription process and subsequent post-transcriptional
regulation may induce miRNA species within the same miRNA cluster
to exhibit differential expression patterns. It was also
hypothesized that the observed inconsistent expression patterns
among members of miRNA clusters may partially result from the
increased sensitivity of high-throughput technology.
Furthermore, members within miRNA families typically
demonstrated similar expression levels (Fig. 4) compared with members of miRNA
clusters. Inconsistent expression patterns were absent from miRNA
members within each evaluated miRNA family, for example the mir-17
family. Due to the fact that the miRNA species within an miRNA gene
family were homologous, their similar expression levels may be
indicative of their closely associated functions.
The miR-17-92 gene cluster is widely expressed in
humans and mice, and represents a potentially oncogenic miRNA
cluster. This gene cluster may induce tumors by inhibiting the
expression of genes that regulate oncogenes and the cell cycle
(24). It is of note that certain
miRNA sequences in the miR-17-92 gene cluster (hsa-mir-20a and
hsa-mir-18a) were observed to be highly similar to the miRNA
sequences in the miR-106a gene cluster, which may be grouped into
the hsa-mir-17 family. These miRNAs may therefore possess similar
biological functions and may regulate identical target genes. The
sequences of the additional members of the miR-17-92 gene cluster
(hsa-mir-19a and hsa-mir-92a) exhibited variation compared with
miRNA sequences within the hsa-mir-17 family.
Castellano et al (25) reported that the expression levels of
pri-mir-17-92 in ERα-positive breast cancer cells were
significantly upregulated compared with those of ERα-negative
breast cancer cells. The miRNA that is transcribed from this miRNA
gene cluster downregulates the expression of the target gene ERα
and is also able to inhibit the protein expression of AIBI, a p160
family member and co-activator that is required for transcription.
The results of the present study exhibited a similar phenomenon; in
the ERα-positive ISK cell line, the expression of certain mature
miRNA species within the miR-17-92 cluster was upregulated, however
the expression of a small number of miRNA species in the miR-17-92
cluster was downregulated. Therefore, the results of the present
study appeared to indicate that the non-homologous miRNA species in
the miR-17-92 cluster not only had differential expression levels,
induced by additional post-transcriptional regulation, but
additionally may perform distinct biological functions.
It was also revealed that for the miR-200 family in
particular, the expression levels of miR-141 in the HEC-1B cell
line were significantly increased (Fig.
4). Notably, miR-141 is highly expressed in a number of common
epithelial cancers (26). This
suggests that the miR-200 family, which was highly expressed in the
HEC-1B cell line, may promote cancer cell invasion and metastasis
and may have the potential to be used as a biomarker for the
differentiation of endometrial cancer subtypes.
Discussion
In the present study, high-throughput sequencing
technology was used to screen for the differential expression of
miRNAs in ISK and HEC-1B cells. The results of the present study
suggested that although the two cell types investigated were
endometrial cancer cell lines, their miRNA expression profiles
varied significantly. Of the known human miRNAs, there were 139
miRNAs that were differentially expressed between these two types
of cell, and a number of these miRNAs have previously been
demonstrated to be associated with tumors (27).
Clustered miRNA genes may coordinate the regulation
of numerous critical biological processes, including embryonic
development, the cell cycle and cell proliferation (28). As these miRNAs may inhibit the
expression of numerous proteins involved in tumor development, they
possess crucial roles during organogenesis, as well as in the
initiation and development of human diseases, including cancer
(29). miRNA clusters possess a more
complex regulatory network, associated with expression, compared
with the regulation of individual miRNAs. The miRNA members of
certain gene clusters are functionally associated and may target or
regulate identical genes or the same gene family, or target various
protein components of the same signaling pathway (30). The sequences of members in the
miR-17-92 gene cluster are highly conserved across vertebrates, and
the miR-17-92 gene cluster has received extensive attention as has
been suggested to be involved in mammalian organ development, which
is a function thought to be closely associated with the initiation
of solid tumor development (24).
Members of the miR-17-92 gene cluster are expressed at high levels
in numerous tumor cells, including those of B-cell lymphoma, as
well as lung, liver, bladder, colon, stomach and pancreatic cancer.
Furthermore, members of the miR-17-92 gene cluster are thought to
induce tumorigenesis mainly by inhibiting the expression of genes
that regulate oncogenes and the cell cycle (31).
The unique distribution and expression pattern of
miRNA gene clusters has suggested that they may possess important
regulatory functions. As numerous miRNAs are distributed in
clusters, the majority of these gene clusters are transcribed in a
single transcript and co-regulate a number of biological processes.
Thus, the investigation of miRNA gene clusters, rather than the
study of the expression of single miRNAs, may present a novel
research strategy. As high-throughput sequencing technology has
been widely used in miRNA research, it is possible that studies
regarding the expression and function of gene clusters will further
improve miRNA research.
In the present study, it was revealed that the
hsa-miR-17 cluster exhibited an inconsistent pattern of expression,
and it was also identified that the expression of the miR-200
family was significantly upregulated, particularly the expression
of miR-141, in the HEC-1B cell line. These findings not only
provide novel potential biomarkers for differentiating between type
I and type II endometrial cancer, but also reveal the miRNA
alterations associated with the underlying mechanisms of
endometrial cancer.
Following a review of previous literature regarding
endometrial cancer and miRNA expression (Table II), it was also identified that
expression of the miR-200 family was significantly increased in
endometrial cancer. It was observed that the expression levels of
hsa-miR-200a, hsa-miR-429 and hsa-miR-141 in the miR-200 family
were significantly increased in the HEC-1B cell line compared with
the ISK cell line. This finding was also verified by sequencing
(Table II).
| Table II.Comparison of upregulated miRNAs
identified in previous studies with the results of the present
study. |
Table II.
Comparison of upregulated miRNAs
identified in previous studies with the results of the present
study.
| Study | Increased miRNA
expression | Tissue | Technique | Ref |
|---|
| Chung et
al | miR-103, miR-106a,
miR-107, miR-10a, miR-130b, miR-141, miR-151, miR-155, miR-17-5p,
miR-182, miR-183, miR-184, miR-191, miR-194, miR-200a,
miR-200c | EEC compared with
normal endometrium | RT-PCR | (11) |
| Snowdon et
al | miR-141, miR-146a,
miR-18a, miR-200a, miR-200b, miR-200c, miR-203, miR-205, miR-210,
miR-421, miR-429 | EEC compared with
normal endometrium | Microarray &
RT-PCR | (12) |
| Present study | miR-141, miR-200a,
miR-92a, let-7b, miR-429, miR-125a miR-25, miR-124, miR-185
miR-29a, miR-34c, miR-519a | HEC cell line
compared with ISK cell line | Deep
sequencing |
|
Notably, the miR-200 family possesses the potential
to be used as a biomarker for differentiating between the HEC-1B
and ISK cell lines, and may also be closely associated with the
distinct pathogenic mechanisms of these two cell types. A previous
study demonstrated that the expression of the miR-200 family may be
highly correlated with the epithelial phenotype of cancer cells
(32). The study used two markers to
classify 60 types of cell line into two groups: One group consisted
of cells with an epithelial phenotype (marked by E-cadherin) and
the other consisted of cells with a mesenchymal phenotype
(indicated by vimentin). This previous study revealed that the
expression of miR-200 family members was positively correlated with
an epithelial phenotype and that these miRNAs demonstrated high
expression levels in epithelial-type tumor cells. Recent studies
also demonstrated that the overexpression of miR-200 family members
in mouse breast cancer cell lines stimulated macroscopic metastases
(33,34). Based on the characteristics of the
HEC-1B and ISK cell lines, the miR-200 family may be used as a
biomarker for differentiating between endometrial cancer subtypes.
In addition, miR-200 family members that were expressed at
increased levels in HEC-1B cells, particularly miR-141, may also be
involved in myometrial invasion and lymph node metastasis (33).
As the function and targets of numerous miRNAs
remain unclear, the alteration of cell phenotypes due to the
differential expression of miRNAs remains to be fully elucidated.
Thus, further research may be required in this area. The roles of
the miR-17-92 cluster and the miR-200 family in the regulation of
signaling pathways in the HEC-1B and ISK cell lines remain to be
elucidated by further experiments.
Furthermore, based on high-throughput sequencing
data, the protocol for how to combine the characteristics of miRNAs
that are distributed throughout the genome in clusters remains to
be elucidated. However, the analysis of the expression profiles of
specific types of miRNAs, including sense/antisense miRNAs located
in specific positions in the genome or miRNAs possessing homologous
sequences remains a notable direction for future research.
The present study identified potential novel
biomarkers as certain representative members of hsa-mir-17 cluster
and the miR-200 family: has-miR-20a, has-miR-18a, hsa-miR-19a,
has-miR-20a in the hsa-mir-17 cluster and hsa-miR-429, hsa-miR-141,
hsa-miR-200a in the miR-200 family.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 61271055).
References
|
1
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2006. View Article : Google Scholar
|
|
3
|
Bushati N and Cohen SM: microRNA
functions. Annu Rev Cell Dev Biol. 23:175–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bokhman JV: Two pathogenetic types of
endometrial carcinoma. Gynecol Oncol. 15:10–17. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Treeck O, Diedrich K and Ortmann O: The
activation of an extracellular signal-regulated kinase by
oestradiol interferes with the effects of trastuzumab on HER2
signalling in endometrial adenocarcinoma cell lines. Eur J Cancer.
39:1302–1309. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Boggess JF, Zhou C, Bae-Jump VL, Gehrig PA
and Whang YE: Estrogen-receptor-dependent regulation of telomerase
activity in human endometrial cancer cell lines. Gynecol Oncol.
103:417–424. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Doll A, Abal M, Rigau M, Monge M, Gonzalez
M, Demajo S and Reventos J: Novel molecular profiles of endometrial
cancer-new light through old windows. J Steroid Biochem Mol Biol.
108:221–229. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gründker C, Günthert AR and Emons G:
Hormonal heterogeneity of endometrial cancer. Adv Exp Med Biol.
630:166–188. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Uharcek P: Prognostic factors in
endometrial carcinoma. J Obstet Gynaecol Res. 34:776–783. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Myatt SS, Wang J, Monteiro LJ, Christian
M, Ho KK, Fusi L, Dina RE, Brosens JJ, Ghaem-Maghami S and Lam EW:
Definition of microRNAs that repress expression of the tumor
suppressor gene FOXO1 in endometrial cancer. Cancer Res.
70:367–377. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chung TK, Cheung TH, Huen NY, Wong KW, Lo
KW, Yim SF, Siu NS, Wong YM, Tsang PT, Pang MW, et al: Dysregulated
microRNAs and their predicted targets associated with endometrioid
endometrial adenocarcinoma in Hong Kong women. Int J Cancer.
124:1358–1365. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Snowdon J, Zhang X, Childs T, Tron VA and
Feilotter H: The microRNA-200 family is upregulated in endometrial
carcinoma. PLoS One. 6:e228282011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guo L, Yang Q, Lu J, Li H, Ge Q, Gu W, Bai
Y and Lu Z: A comprehensive survey of miRNA repertoire and 3′
addition events in the placentas of patients with pre-eclampsia
from high-throughput sequencing. PLoS One. 6:e210722011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Coppée JY: Do DNA microarrays have their
future behind them? Microbes Infect. 10:1067–1071. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Benes V and Castoldi M: Expression
profiling of microRNA using real-time quantitative PCR, how to use
it and what is available. Methods. 50:244–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Griffiths-Jones S, Saini HK, van Dongen S
and Enright AJ: miRBase: Tools for microRNA genomics. Nucleic Acids
Res. 36(Database): D154–D158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lu M, Shi B, Wang J, Cao Q and Cui Q: TAM:
A method for enrichment and depletion analysis of a microRNA
category in a list of microRNAs. BMC Bioinformatics. 11:4192010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bhat KP and Pezzuto JM: Resveratrol
exhibits cytostatic and antiestrogenic properties with human
endometrial adenocarcinoma (Ishikawa) cells. Cancer Research.
61:6137–6144. 2001.PubMed/NCBI
|
|
19
|
Guo L, Sun B, Sang F, Wang W and Lu Z:
Haplotype distribution and evolutionary pattern of miR-17 and
miR-124 families based on population analysis. PLoS One.
4:e79442009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo L, Yang S, Zhao Y, Zhang H, Wu Q and
Chen F: Global analysis of miRNA gene clusters and gene families
reveals dynamic and coordinated expression. Biomed Res Int.
2014:7824902014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aravin AA, Lagos-Quintana M, Yalcin A,
Zavolan M, Marks D, Snyder B, Gaasterland T, Meyer J and Tuschl T:
The small RNA profile during Drosophila melanogaster development.
Dev Cell. 5:337–350. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu J, Wang F, Yang GH, Wang FL, Ma YN, Du
ZW and Zhang JW: Human microRNA clusters: Genomic organization and
expression profile in leukemia cell lines. Biochem Biophys Res
Commun. 349:59–68. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ambros V, Bartel B, Bartel DP, Burge CB,
Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S,
Marshall M, et al: A uniform system for microRNA annotation. RNA.
9:277–279. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hayashita YI, Osada H, Tatematsu Y, Yamada
H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y and
Takahashi T: A polycistronic microRNA cluster, miR-17-92, is
overexpressed in human lung cancers and enhances cell
proliferation. Cancer Rese. 65:9628–9632. 2005. View Article : Google Scholar
|
|
25
|
Castellano L, Giamas G, Jacob J, Coombes
RC, Lucchesi W, Thiruchelvam P, Barton G, Jiao LR, Wait R, Waxman
J, et al: The estrogen receptor-alpha-induced microRNA signature
regulates itself and its transcriptional response. Proc Natl Acad
Sci USA. 106:15732–15737. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang D, Qiu C, Zhang H, Wang J, Cui Q and
Yin Y: Human microRNA oncogenes and tumor suppressors show
significantly different biological patterns: from functions to
targets. PLoS One. 5:e130672010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Altuvia YI, Landgraf P, Lithwick G,
Elefant N, Pfeffer S, Aravin A, Brownstein MJ, Tuschl T and
Margalit H: Clustering and conservation patterns of human
microRNAs. Nucleic Acids Res. 3:2697–2706. 2005. View Article : Google Scholar
|
|
29
|
Mendell JT: miRiad roles for the miR-17-92
cluster in development and disease. Cell. 133:217–222. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu J and Wong C: A computational screen
for mouse signaling pathways targeted by microRNA clusters. RNA.
14:1276–1283. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ventura AI, Young AG, Winslow MM, Lintault
L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D and
Stone JR: Targeted deletion reveals essential and overlapping
functions of the miR-17 through 92 family of miRNA clusters. Cell.
132:875–886. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gregory PAI, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A,
Petrocca F, Martinvalet D, Song E, Lim B and Lieberman J: miR-200
enhances mouse breast cancer cell colonization to form distant
metastases. PLoS One. 4:e71812009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pecot CVI, Rupaimoole R, Yang D, Akbani R,
Ivan C, Lu C, Wu S, Han HD, Shah MY and Rodriguez-Aguayo C: Tumour
angiogenesis regulation by the miR-200 family. Nat Commun.
4:24272013. View Article : Google Scholar : PubMed/NCBI
|