Introduction
Cancer-initiating stem cells (CISC) represent a
minor cell subpopulation of heterogeneous breast cancer. CISC are
characterized by a unique self-renewal program, enhanced
tumorigenic potential and resistance to conventional therapeutic
interventions (1–4). Thus, reliable cell culture models for
clinical CISC may constitute useful experimental approaches for the
development of novel cancer stem cell-targeted therapies for the
treatment of chemoendocrine therapy-resistant clinical breast
cancer.
Gene expression profiling has facilitated the use of
targeted therapy for the treatment of certain molecular subtypes of
breast cancer that differ in the expression of hormone and/or
growth factor receptors, including luminal A and B, human epidermal
growth factor receptor (HER)-2-enriched, triple negative
(basal-like) and normal-like breast cancer (5). However, the long-term use of small
molecule inhibitor-based targeted chemoendocrine therapy is
frequently associated with de novo or acquired tumor
resistance, which compromises the therapeutic efficacy of the
treatment and promotes the progression of therapy-resistant disease
in patients affected by these molecular subtypes of breast cancer
(6–9).
Therapy-resistant CISC are considered to be responsible for the
progression of metastatic disease (2,4).
Therefore, experimental approaches that enable the isolation and
characterization of putative CISC are essential for the
identification of promising stem cell-targeted therapeutic lead
compounds. Several in vitro and in vivo assays have
been previously optimized for the isolation and characterization of
putative CISC, which are specific for the following cell
populations: i) Drug efflux positive side populations; ii)
drug-resistant phenotypes; iii) phenotypes expressing signature
cellular markers, including cluster of differentiation (CD)44, CD24
and aldehyde dehydrogenase (ALDH); iv) phenotypes expressing
signature nuclear transcription factors such as octamer-binding
transcription factor 4 (Oct-4), homeobox transcription factor NANOG
and c-Myc; v) phenotypes able to form non-adherent tumor spheroids
in vitro; and vi) phenotypes with enhanced in vivo
tumorigenic potential (10–12).
The aim of the present study was to establish
reliable cell culture models for several molecular subtypes of
breast cancer, in addition to demonstrating the efficacy of
prototypic chemoendocrine therapeutic agents on the established
cell models, and isolating putative CISC from these models.
The results demonstrated that the cell models
employed in the present study, which represent different molecular
subtypes of clinical breast cancer (namely luminal A,
HER-2-enriched and triple negative breast cancer), exhibited loss
of control of normal homeostatic growth, retention of cancer risk
and sensitivity to growth inhibition by prototypic chemoendocrine
therapeutic agents. Furthermore, the drug-resistant phenotypes
isolated from the aforementioned breast cancer models displayed
persistent cell proliferation in the presence of high doses of
chemotherapeutic agents, suggesting the presence of putative CISC
in these models.
Materials and methods
Experimental models
The human breast carcinoma-derived cell line
Michigan Cancer Foundation (MCF)-7 (estrogen receptor (ER)+,
progesterone receptor (PR)+ and HER-2−) (Michigan Cancer
Foundation, Detroit, MI, USA), the HER-2-transfectant tumorigenic
mammary epithelial cell line 184-B5/HER (ER−,
PR− and HER-2+) (Professor CW Welsch; Michigan State
University, East Lansing, MI, USA) and the breast adenocarcinoma
cell line MDA-MB-231 (ER−, PR− and
HER-2−) (American Type Culture Collection, Manassas, VA,
USA) were selected as representative models for three molecular
subtypes of clinical breast cancer, namely luminal A,
HER-2-enriched and triple negative breast cancer, respectively. The
184-B5 cell line (Professor CW Welsch; Michigan State University)
was used as the control. The cell lines were cultured in Dulbecco's
modified Eagle medium (DMEM)/F-12 (Sigma-Aldrich, St. Louis, MO,
USA) supplemented with 10% serum (Gibco; Thermo Fisher Scientific,
Waltham, MA, USA), 240 IU/ml insulin (Eli Lilly & Co.,
Indianapolis, IA, USA), 1 µM dexamethasone (Sigma-Aldrich), 10
ng/ml epidermal growth factor, 0.5 µg/ml hydrocortisone and 10
µg/ml transferrin (all obtained from Sigma-Aldrich). A total of
1.0×105 cells were seeded, incubated at 37°C in a humidified
atmosphere with 5% CO2, and routinely subcultured at a
1:4 ratio when the culture reached ~70–80% confluency (13–15).
Growth assays
Population doubling time, saturation density, cell
cycle progression, cellular apoptosis, anchorage-independent (AI)
colony formation and tumorigenic potential were evaluated on the
aforementioned cell lines, following previously published protocols
(14–18). Population doubling times were
determined from the number of viable cells counted every 24 h
during the exponential growth phase of the cell culture, which
lasted 7 days. For the saturation density assay, 1.0×105 cells were
seeded, and the number of viable cells counted 7 days later was
considered to represent the saturation density. The viable cell
number for population doubling and saturation density was
determined via trypan blue exclusion test (Sigma-Aldrich) using a
hemocytometer (Sigma-Aldrich) (14–18). Cell
cycle progression and cellular apoptosis were determined by flow
cytometry analysis of cells at G1, S, G2/M
and sub G0 phases of the cell cycle. Briefly, cells were
stained with 50 µg the propidium iodide (Calbiochem, La Jolla, CA,
USA) and sorted using the EPICS 752 flow cytometer (Beckman
Coulter, Miami, FL, USA). The data from the cell cycle phase
distribution was analyzed using the multi-cycle MPLUS software
(Phoenix Flow Systems, San Diego, CA, USA) following published
protocols (14,15). The data were represented as
G1:S+G2/M and S+G2/M:sub
G0 ratios. These ratios provided information about the
relative proportion of quiescent vs. proliferative cells, and
proliferative vs. apoptotic cells, respectively. To evaluate the
capacity of the different cell lines to form AI colonies, 1,000
cells/well were seeded, and subsequently the cells were suspended
in 0.33% soft agar, overlaid on a 0.6%-agar basement matrix, and
maintained in culture for 21 days. AI colony formation was then
determined by the number of AI colonies derived from these cells.
The in vivo tumorigenic potential of the different cell
lines was determined following subcutaneous transplantation of
1.0×106 cells, suspended in 0.2 ml phosphate-buffered saline, in
6–8 week old female BALB/c athymic ‘nude’ mice (Charles River
Laboratories, Newark, DE, USA). The mice were sacrificed by
CO2 asphyxiation, 3–5 weeks following transplantation
when the tumors had reached a diameter of 1 cm. The data were
presented as tumor incidence and latency rates.
Chemotherapeutic agents
The selective ER modulator tamoxifen (TAM), the
synthetic retinoid 4-(hydroxyphenyl) retinamide (4-HPR) and the
anthracycline doxorubicin (DOX; all obtained from Sigma-Aldrich)
were selected as test compounds, based on their efficacy as
clinical chemoendocrine therapeutic agents (6–8). Stock
solutions of these agents at 100 mM concentration were prepared by
dissolving the drugs in 100% ethanol (Thermo Fisher Scientific). In
order to obtain pharmacologically relevant final concentrations of
these compounds, the stock solutions were serially diluted in the
culture medium. Dose response experiments, performed within the
pharmacologically achievable ranges, identified the values
corresponding to the inhibitory concentration (IC)50 and
IC90 for these drugs. The IC90 represented
the maximum cytostatic dose, which was defined as the highest
concentration of the test compound that resulted in a number of
viable cells equal to or greater than the initial seeding
density.
Drug-resistant phenotype
The drug-resistant phenotypes were isolated from the
cell subpopulation that survived and exhibited progressive growth
in the presence of a concentration of the corresponding
chemoendocrine therapeutic agent equivalent to its IC90.
This surviving population was expanded in the presence of high
doses of the corresponding chemoendocrine therapeutic drug for a
minimum of five passages prior to the experiments.
Statistical analysis
Cell culture experiments were performed in duplicate
(N=6/treatment group), while AI colony formation experiments were
performed in triplicate (N=18/treatment group). Cell culture data
are presented as the mean ± SD. In vivo transplantation
experiments were performed using 10 mice/cell line. Statistically
significant differences between the control and the experimental
data points were assessed by two-sample t-test, using GraphPad
Prism software, version 5.0 (GraphPad Software Inc., La Jolla, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Status of homeostatic growth control
and cancer risk
The data summarized in Table I (17,19)
compare the values obtained for a series of quantitative biomarker
end points measured in non-tumorigenic 184-B5 cells vs. breast
cancer-derived MCF-7, 184-B5/HER and MDA-MB-231 cells. The
tumorigenic cells exhibited a ≥55% reduction in population doubling
time, 19–47% increase in saturation density, 39–74% reduction in
the G1:S+G2/M ratio and 7.0–9.5 fold increase
in the S+G2/M:sub G0 ratio. Additionally,
unlike the non-tumorigenic 184-B5 cells, the tumorigenic cells
exhibited AI growth in vitro and retained their tumor
development ability in vivo (Table II) (17,19).
 | Table I.Loss of control of homeostatic growth
in cell culture models representing different molecular subtypes of
clinical breast cancer. |
Table I.
Loss of control of homeostatic growth
in cell culture models representing different molecular subtypes of
clinical breast cancer.
|
| Cell culture
model |
|---|
|
|
|
|---|
| Biomarker end
point | 184-B5 | MCF-7 | 184-B5/HER | MDA-MB-231 |
|---|
| Population doubling
time (h)a | 34.0 | 15.2 | 15.0 | 15.0 |
| Saturation density
(×105 cells)b | 22.3±1.2 | 26.6±1.7 | 32.8±1.5 | 32.9±2.3 |
|
G1:S+G2/M
ratioc |
2.3±0.4 |
1.4±0.2 |
0.8±0.1 |
0.6±0.3 |
|
S+G2/M:sub G0
ratioc |
1.6±0.3 | 12.8±2.4 | 10.8±1.6 | 16.8±3.2 |
| Table II.Gain of cancer risk in cell culture
models representing different molecular subtypes of clinical breast
cancer. |
Table II.
Gain of cancer risk in cell culture
models representing different molecular subtypes of clinical breast
cancer.
|
| Cell culture
model |
|---|
|
|
|
|---|
| Biomarker end
point | 184-B5 | MCF-7 | 184-B5/HER | MDA-MB-231 |
|---|
| AI colony
formationa |
|
|
|
|
|
Incidence | 0/18 | 18/18 | 18/18 | 18/18 |
| Mean
colony number | – | 30.9±2.4 | 23.0±2.6 | 38.9±1.6 |
| Tumor
formationb |
|
|
|
|
|
Incidence | 0/10 | 10/10 | 10/10 | 10/10 |
| Latency
(weeks) | 24 | 3–5 | 3–5 | 3–5 |
Drug-resistant CISC
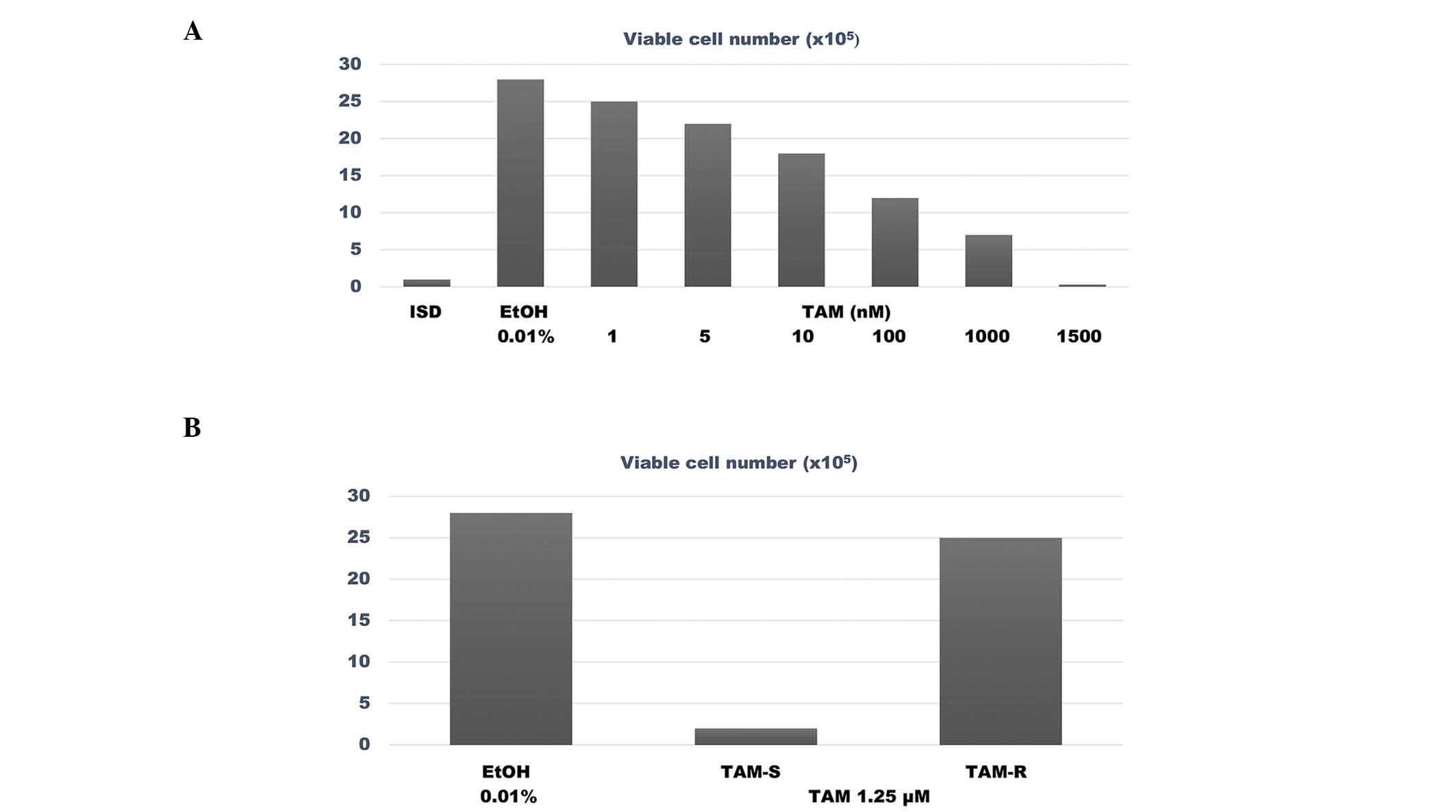
The data presented in Fig.
1A summarize the results of the dose response experiment
performed with TAM in MCF-7 cells, which identified the
IC50 and IC90 for TAM as 94.30 nM and 1.25
µM, respectively. The data presented in Fig. 1B compare the survival rate of
TAM-sensitive (TAM-S) vs. TAM-resistant (TAM-R) MCF-7 cells in the
presence of a concentration of TAM equivalent to its
IC90. The number of viable cells treated with TAM was
2.0±1.0×105 cells in the TAM-S group, vs. 25.0±1.5×105 cells in the
TAM-R group. Thus, compared with the TAM-S phenotype, the TAM-R
phenotype exhibited an 11.5 fold higher number of viable cells
(P=0.001).
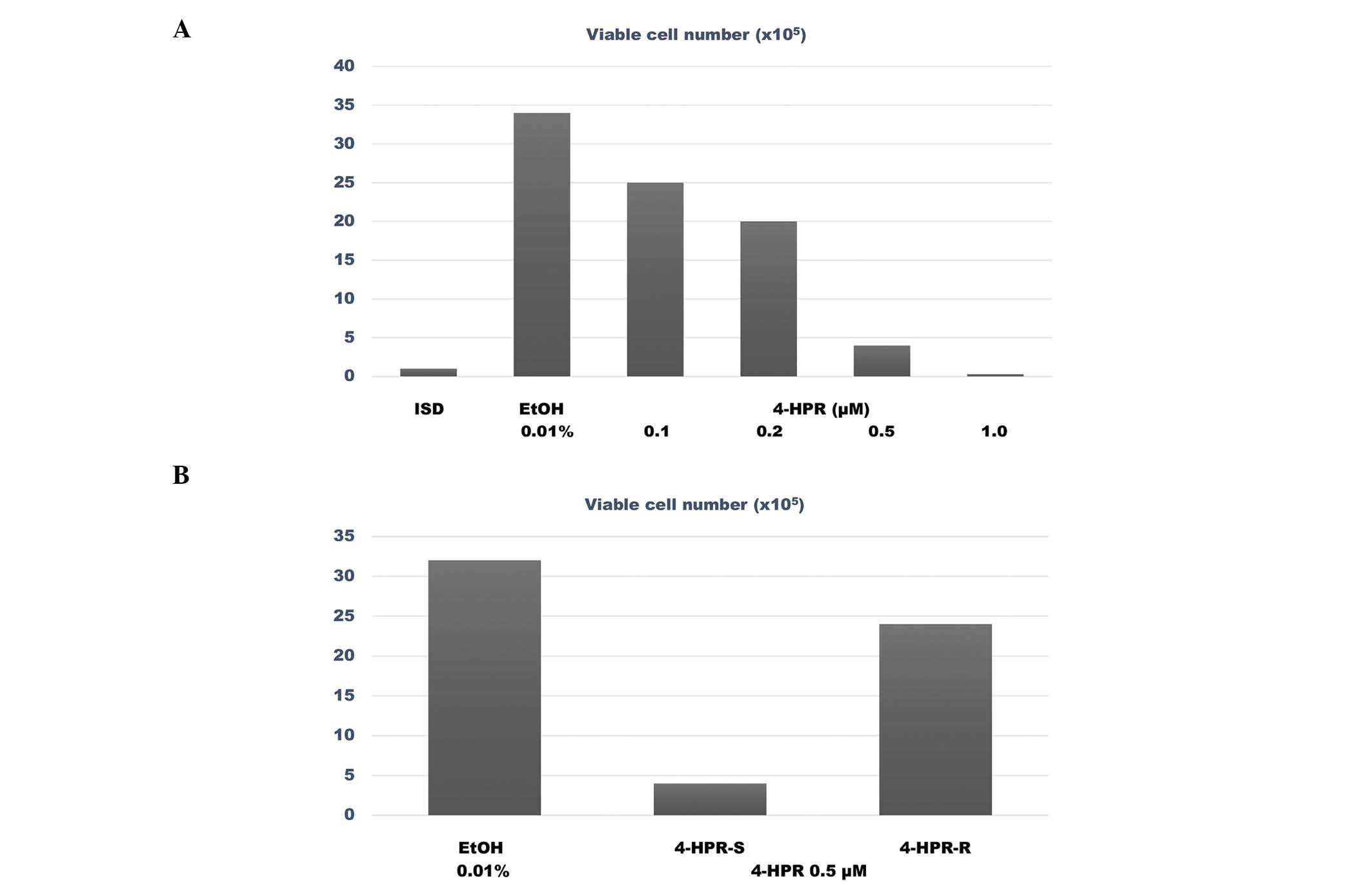
The data presented in Fig.
2A summarize the results of the dose response experiment
conducted with 4-HPR in 184-B5/HER cells. This experiment
identified the IC50 and IC90 for 4-HPR as
0.31 and 0.49 µM, respectively. The data presented in Fig. 2B compare the survival rate of
4-HPR-sensitive (4-HPR-S) vs. 4-HPR-resistant (4-HPR-R) 184-B5/HER
cells in the presence of a concentration of 4-HPR equivalent to its
IC90. The number of viable cells in the 4-HPR-treated
4-HPR-S group was 4.0±2.0×105 cells, while in the
4-HPR-treated 4-HPR-R group the number of viable cells was
24.0±1.1×105 cells. Therefore, the 4-HPR-R cells
exhibited a 5 fold higher number of viable cells, compared with the
4-HPR-S cells (P=0.02).
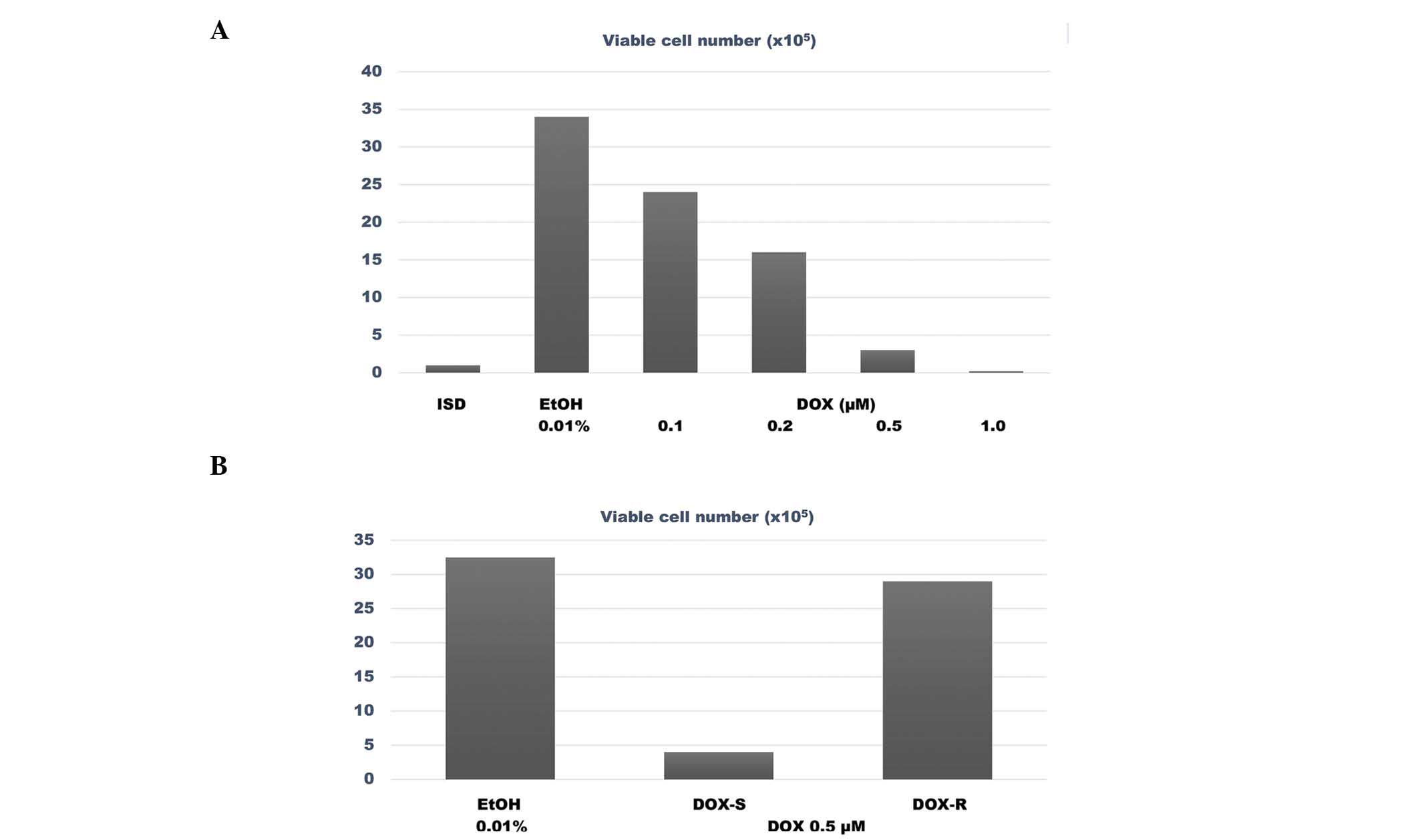
The data presented in Fig.
3A summarize the results from the dose response experiment
performed with DOX in MDA-MB-231 cells, wherein 0.2 and 0.5 µM were
identified as the IC50 and IC90 of DOX,
respectively. The data presented in Fig.
3B compare the survival ability of DOX-sensitive (DOX-S) vs.
DOX-resistant (DOX-R) MDA-MB-231 cells in the presence of a
concentration of DOX equivalent to its IC90. The number
of viable cells among the DOX-treated DOX-S cells was
4.0±2.2×105 cells, vs. 29.0±2.0×105 cells
among the DOX-treated DOX-R cells. Thus, the DOX-R cells exhibited
a 6.2 fold higher number of viable cells, compared with the DOX-S
cells (P=0.02).
The data presented in Table III summarize the results of the AI
growth assay conducted to compare the AI growth of drug-sensitive
vs. drug-resistant phenotypes in the presence of maximum cytostatic
concentrations of the corresponding chemotherapeutic agent. The
results demonstrated that the TAM-R, 4-HPR-R and DOX-R phenotypes,
which were seeded at an initial cell density ≤10 fold lower than
that of their corresponding parental cell lines, exhibited a 5.6
(P=0.02), 5.4 (P=0.02) and 4.4 (P=0.03) fold increase in the number
of AI colonies, respectively, compared with the drug-sensitive
controls.
| Table III.Anchorage-independent colony
formation in drug-resistant phenotypes. |
Table III.
Anchorage-independent colony
formation in drug-resistant phenotypes.
|
|
Anchorage-independent colony
numbera |
|---|
|
|
|
|---|
| Chemotherapy
status | MCF-7 | 184-B5/HER | MDA-MB-231 |
|---|
|
Drug-sensitiveb | 3.8±1.4 |
3.5±0.9 |
4.1±1.1 |
|
Drug-resistantb | 25.3±2.5 | 22.3±2.5 | 22.1±1.8 |
| Increase
(fold) |
5.6 |
5.4 |
4.4 |
Discussion
The aim of the present study was to develop reliable
cell culture models for putative CISC in three molecular subtypes
of clinical breast cancer, namely luminal A, HER-2-enriched and
triple negative breast cancer. The results of the experiments
conducted to determine the status of homeostatic growth control and
cancer risk in the cell culture models for the aforementioned
molecular subtypes of clinical breast cancer indicated that,
compared with the non-tumorigenic triple negative 184-B5 cells, the
tumorigenic ER+/PR+/HER2− luminal
A, ER−/PR−/HER-2+ HER-2-enriched
and ER−/PR−/HER-2− triple negative
cell culture models exhibited hyperproliferation, aberrant cell
cycle progression and downregulated cellular apoptosis. Taken
together, these data suggest that the breast carcinoma-derived cell
culture models used in the present study had lost control of their
normal homeostatic growth, as evidenced by hyperproliferation,
aberrant cell cycle progression and downregulated cellular
apoptosis. These findings are consistent with those previously
reported in the literature (13–18,20). In
addition, contrary to the non-tumorigenic control cells, the breast
carcinoma-derived cells appeared to have retained an enhanced risk
for tumor development, as indicated by their ability to exhibit AI
growth in vitro and tumor development in vivo
following subcutaneous transplantation. Thus, AI growth represents
a specific and sensitive in vitro surrogate biomarker for
cancer risk (13,17–20).
Previous studies on luminal A, HER-2-enriched and
triple negative models of breast cancer have demonstrated that
hyperproliferation and cancer risk are reduced in these cell models
by several mechanistically distinct pharmacological agents and
naturally occurring dietary components, demonstrating the
susceptibility of these models to effective chemopreventive or
therapeutic treatment options (13–18). These
aspects prompted the application of cell culture models for the
characterization of CISC in the different molecular subtypes of
breast cancer analyzed in the present study.
Phenotypic resistance to chemoendocrine therapy has
been effectively used for the isolation of putative drug-resistant
CISC (2,4,10–12). In the present study, the experiments
designed to isolate drug-resistant phenotypes from the models for
luminal A, HER-2-enriched and triple negative breast cancer
utilized TAM, 4-HPR and DOX, respectively, for the selective
expansion of the resistant phenotypes, since these agents have been
used for the clinical management of the aforementioned molecular
subtypes of breast cancer (6–8). The data derived from these experiments
clearly demonstrated persistent growth of drug-resistant cells in
the presence of maximum cytostatic concentrations of the
corresponding chemotherapeutic agents, thereby providing
experimental evidence for the potential clinical translatability of
the in vitro data obtained in the present study.
Furthermore, the status of cellular markers specific for stem
cells, including CD44, CD24 and ALDH, and nuclear transcription
factors such as Oct-4, NANOG and c-Myc (10–12), may
provide a robust characterization of the putative drug-resistant
phenotypes isolated from the breast cancer models used in the
present study.
Targeted therapy for luminal A, HER-2-enriched and
triple negative breast cancer, includes selective ER modulators and
aromatase inhibitors, HER-2-targeted small molecule inhibitors and
poly (ADP-ribose) polymerase inhibitors, respectively (21–24).
However, these treatment options are frequently associated with
de novo or acquired tumor drug resistance, which has been
attributed to the upregulation of survival pathways associated with
the overexpression of phosphoinositide 3-kinase/Akt/mammalian
target of rapamycin; the upregulation of proliferative pathways
associated with the overexpression of rat sarcoma/rapidly
accelerated fibrosarcoma/mitogen-activated protein
kinases-extracellular signal-regulated kinase; and the
downregulation of apoptotic pathways associated with the modulation
of the expression of anti- or pro-apoptotic proteins in the
aforementioned molecular subtypes of breast cancer (2,9,22,23,25,26).
Thus, the analysis of the expression levels of molecules that have
been previously documented to be associated with survival,
proliferative and apoptotic pathways in putative CISC is likely to
further aid in the characterization of CISC phenotypes.
Long-term chemoendocrine therapy is frequently
associated with de novo or acquired tumor resistance in
breast cancer, which compromises the therapeutic efficacy of the
treatment, and CISC are widely considered to be the cell type
responsible for recurrence of the metastatic cancer phenotype
(1–4,9). Previous
studies have demonstrated that small molecule inhibitors selective
for the Wnt/β-catenin signaling pathway preferentially inhibit the
growth of breast CISC by inhibiting the binding of β-catenin to T
cell factor and downregulating the insulin-like growth factor-1
signaling pathway (27). Therefore,
further studies on stem cell-specific mechanistic pathways,
including the Wnt/β-catenin, notch and hedgehog signaling pathways,
may aid in the identification of novel therapeutic targets for the
treatment of breast cancer (2,3,27–29).
In conclusion, future studies that employ reliable
assays for the isolation and characterization of CISC and focus on
the biology of cancer stem cells (particularly the molecular,
genetic and endocrine mechanisms responsible for the survival of
CISC), should collectively represent valuable approaches for
identifying novel stem cell-targeted therapeutic options for the
treatment of breast cancer.
Acknowledgements
The present study was funded by the National Cancer
Institute (Bethesda, MD, USA; grants/contracts nos. CA-44741,
CA-29502 and CN-75029-63) and the Department of Defense (Fort
Detrick, MD, USA; grant no. DAMD17–94-J-4208). The author would
like to acknowledge former colleagues Dr Meena Katdare, Dr Dan
Sepkovic, Dr H. Leon Bradlow, Dr George Y.C. Wong and Dr Michael P.
Osborne, for their active collaborations.
References
|
1
|
Stingl J and Caldas C: Molecular
heterogeneity of breast carcinomas and the cancer stem cell
hypothesis. Nat Rev Cancer. 7:791–799. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dean M, Fojo T and Bates S: Tumour stem
cells and drug resistance. Nat Rev Cancer. 5:275–284. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lobo NA, Shimono Y, Qian D and Clarke MF:
The biology of cancer stem cells. Annu Rev Cell Dev Biol.
23:675–699. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Patel SA, Ndabahaliye A, Lim PK, Milton R
and Rameshwar P: Challenges in the development of future treatments
for breast cancer stem cells. Breast Cancer (Dove Med Press).
2:1–11. 2010.PubMed/NCBI
|
|
5
|
Sørlie T, Perou CM, Tibshirani R, et al:
Gene expression patterns of breast carcinomas distinguish tumor
subclasses with clinical implications. Proc Natl Acad Sci USA.
98:10869–10874. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Veronesi U, De Palo G, Costa A, Formelli
F, Marubini E and Del Vecchio M: Chemoprevention of breast cancer
with retinoids. J Natl Cancer Inst Monogr. 12:93–97.
1992.PubMed/NCBI
|
|
7
|
Fisher B, Costantino JP, Wickerham DL,
Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N,
Atkins J, et al: Tamoxifen for prevention of breast cancer: Report
of the National Surgical Adjuvant Breast and Bowel Project P-1
Study. J Natl Cancer Inst. 90:1371–1388. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cleator S, Heller W and Coombes RC:
Triple-negative breast cancer: Therapeutic options. Lancet Oncol.
8:235–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Musgrove EA and Sutherland RL: Biological
determinants of endocrine resistance in breast cancer. Nat Rev
Cancer. 9:631–643. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tanei T, Morimoto K, Shimazu K, Kim SJ,
Tanji Y, Taguchi T, Tamaki Y and Noguchi S: Association of breast
cancer stem cells identified by aldehyde dehydrogenase 1 expression
with resistance to sequential Paclitaxel and epirubicin-based
chemotherapy for breast cancers. Clin Cancer Res. 15:4234–4241.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van Phuc P, Nhan PL, Nhung TH, Tam NT,
Hoang NM, Tue VG, Thuy DT and Ngoc PK: Downregulation of CD44
reduces doxorubicin resistance of CD44CD24 breast cancer cells.
Onco Targets Ther. 4:71–78. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang F, Song C, Ma Y, Tang L, Xu Y and
Wang H: Effect of fibroblasts on breast cancer cell mammosphere
formation and regulation of stem cell-related gene expression. Int
J Mol Med. 28:365–371. 2011.PubMed/NCBI
|
|
13
|
Suto A, Bradlow HL, Kubota T, Kitajima M,
Wong GY, Osborne MP and Telang NT: Alteration in proliferative and
endocrine responsiveness of human mammary carcinoma cells by
prototypic tumor-suppressing agents. Steroids. 58:215–219. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Telang NT, Katdare M, Bradlow HL, Osborne
MP and Fishman J: Inhibition of proliferation and modulation of
estradiol metabolism: Novel mechanisms for breast cancer prevention
by the phytochemical indole-3-carbinol. Proc Soc Exp Biol Med.
216:246–252. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jinno H, Steiner MG, Nason-Burchenal K,
Osborne MP and Telang NT: Preventive efficacy of receptor class
selective retinoids on HER-2/neu oncogene expressing preneoplastic
human mammary epithelial cells. Int J Oncol. 21:127–134.
2002.PubMed/NCBI
|
|
16
|
Mukherjee B, Telang N and Wong GYC: Growth
inhibition of estrogen receptor positive human breast cancer cells
by Taheebo from the inner bark of Tabebuia avellandae tree. Int J
Mol Med. 24:253–260. 2009.PubMed/NCBI
|
|
17
|
Telang N and Katdare M: Epithelial cell
culture models for the prevention and therapy of clinical breast
cancer (Review). Oncol Lett. 3:744–750. 2012.PubMed/NCBI
|
|
18
|
Telang N, Li G, Sepkovic D, Bradlow HL and
Wong GY: Comparative efficacy of extracts from Lycium barbarum bark
and fruit on estrogen receptor positive human mammary carcinoma
MCF-7 cells. Nutr Cancer. 66:278–284. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Telang N: Cellular metabolism of estradiol
in models for select molecular subtypes of clinical breast cancer.
J Steroids Hormon Sci. 5:1–5. 2014. View Article : Google Scholar
|
|
20
|
Telang NT, Li G, Sepkovic DW, Bradlow HL
and Wong GY: Anti-proliferative effects of Chinese herb Cornus
officinalis in a cell culture model for estrogen receptor-positive
clinical breast cancer. Mol Med Rep. 5:22–28. 2012.PubMed/NCBI
|
|
21
|
National Comprehensive Cancer Network:
NCCN Clinical practice guidelines in oncology: Breast Cancer.
1(2010)http://www.nccn.org2010.Accessed.
July 05–2011
|
|
22
|
Johnston SRD and Dowsett M: Aromatase
inhibitors for breast cancer: Lessons from the laboratory. Nat Rev
Cancer. 3:821–831. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Baselga J and Swain SM: Novel anticancer
targets: Revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer.
9:463–475. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Romond EH, Perez EA, Bryant J, Suman VJ,
Geyer CE Jr, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman
PA, et al: Trastuzumab plus adjuvant chemotherapy for operable
HER-2-positive breast cancer. N Engl J Med. 353:1673–1684. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ibrahim YH, García-García C, Serra V, He
L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J,
et al: PI3K inhibition impairs BRCA1/2 expression and sensitizes
BRCA-proficient triple-negative breast cancer to PARP inhibition.
Cancer Discov. 2:1036–1047. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Martin HL, Smith L and Tomlinson DC:
Multidrug-resistant breast cancer: Current perspectives. Breast
Cancer (Dove Med Press). 6:1–13. 2014.PubMed/NCBI
|
|
27
|
Hoffmeyer K, Raggioli A, Rudloff S, Anton
R, Hierholzer A, Del Valle I, Hein K, Vogt R and Kemler R:
Wnt/β-catenin signaling regulates telomerase in stem cells and
cancer cells. Science. 336:1549–1554. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Anastas JN and Moon RT: WNT signalling
pathways as therapeutic targets in cancer. Nat Rev Cancer.
13:11–26. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jang GB, Hong IS, Kim RJ, Lee SY, Park SJ,
Lee ES, Park JH, Yun CH, Chung JU, Lee KJ, et al: Wnt/β-catenin
small molecule inhibitor CWP232228 preferentially inhibits the
growth of breast cancer stem-like cells. Cancer Res. 75:1961–1702.
2015. View Article : Google Scholar
|