Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of malignant primary liver cancer and is the third
leading cause of cancer-related mortality worldwide (1). Although surgery remains the preferred
therapeutic strategy for HCC, tumor size, hepatic functional
reserve and/or portal hypertension may all limit the extent of
surgical resection (2). Chemotherapy
and internal radiation therapy are also used to treat liver cancer
(3), however, both can lead to the
damage of tissues and organs unaffected by cancer. The prognosis of
HCC is poor due to the development of resistance to current
chemotherapy regimens through the downregulation of various
signaling pathways; in particular, those that control cell
proliferation and survival, such as nuclear factor κB (NF-κB)
(4). Thus, the development of novel,
effective therapeutic strategies for HCC are required to improve
the prognosis of this disease.
Tumor necrosis factor (TNF)-related
apoptosis-inducing ligand (TRAIL) belongs to the TNF superfamily
(5) and possesses a number of
anti-cancer properties (6,7). For example, TRAIL can induce apoptosis
in tumor cells by binding to the plasma membrane death receptors
(DRs) TRAIL-R1 (DR4) and TRAIL-R2 (DR5) (8). Therefore, TRAIL is a potential candidate
for cancer treatment (9). However,
HCC cells are intrinsically resistant to TRAIL-induced cell death
(10). This resistance to TRAIL is a
major clinical challenge that leads to failure of treatment, poor
prognosis and reduced survival of patients with HCC (10). Previous studies have demonstrated that
TRAIL can activate the NF-κB signaling pathway, which activates
genes that encode various key anti-apoptotic proteins, such as
B-cell lymphoma-extra large (Bcl-xL) and inhibitor of apoptosis
proteins (IAPs). These proteins contribute to TRAIL resistance
(10,11). Thus, overcoming NF-κB-associated
survival signals may enhance the antitumor effect of TRAIL in HCC
cells.
Glycogen synthase kinase-3 (GSK-3) is a
multifunctional serine/threonine protein kinase that participates
in numerous cellular processes, including protein synthesis,
glycogen metabolism, mitosis and apoptosis. Additionally, GSK-3 is
involved in various signaling pathways, such as the Wnt/β-catenin
signaling pathway (12). Two major
GSK-3 isoforms (GSK-α and GSK-3β) have been identified in mammals;
they are encoded by distinct genes and perform different functions
(13,14). Mice with a homozygous deletion of the
GSK-3β gene experience massive hepatocyte apoptosis during
embryogenesis, leading to premature death (15). Furthermore, previous studies have
demonstrated that GSK-3β is important in cell survival through its
ability to regulate the NF-κB signaling pathway in hepatocytes
(16).
In consideration of the role of NF-κB target genes
on TRAIL-induced apoptosis, the present study aims to evaluate the
effect of GSK-3β on TRAIL-induced cell death and examine the
mechanism by which GSK-3β inhibition sensitizes HCC cells to
TRAIL-induced apoptosis.
Materials and methods
Cell culture
HL7702, SMMC7721, HuH-7, HuH-6 and HepG2 Human HCC
cell lines were purchased from the American Type Culture Collection
(Manassas, VA, USA). Cells were grown at 37°C in a 5%
CO2 humidified atmosphere, and cultured as a monolayer
in RPMI-1640 medium (HyClone, Logan, UT, USA) supplemented with 100
U/ml penicillin, 100 µg/ml streptomycin, 2 mmol/l glutamine and 10%
fetal bovine serum. HCC cell growth was observed and recorded
regularly.
Reagents and antibodies
Annexin V-R-phycoerythrin (PE) and propidium iodide
(PI) were purchased from Invitrogen Life Technologies (Carlsbad,
CA, USA), and GSK-3β inhibitor (SB216763) was purchased from
Sigma-Aldrich (St. Louis, MO, USA). Rabbit anti-human monoclonal
antibodies raised against GSK-3β (cat no. 12456; 1:1,000), TRAIL
(cat. no. 3219; 1:500), GAPDH (cat. no. 2118; 1:2,000) and
β-catenin (cat no. 8480; 1:1,000) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Rabbit anti-human
monoclonal antibodies raised against Bcl-xL (cat no. ab2568;
1:1,000) and cellular IAP2 (cIAP2; cat no. ab32059; 1:1,000) were
purchased from Abcam (Cambridge, UK). Recombinant Ad5.TRAIL, short
hairpin (sh) GSK-3β, shBcl-xL and short shcIAP2 constructs were
obtained from the Central Laboratory, The Affiliated Hospital,
Qingdao University (Qingdao, China).
GSK-3β inhibitor (SB216763)
SB216763 is potent and selective ATP-competitive
GSK-3 inhibitor. It is equally effective at inhibiting human GSK-3α
and GSK-3β (17). HepG2 and HuH-7
cells (1×105 cells/well) were seeded in 96-well plates
and incubated with Dulbecco's modified Eagle's medium (DMEM;
HyClone) for 24 h. The cells were then pretreated with 10 µM
SB216763 in 10% fetal bovine serum and 1 µmol/l DMEM for 24 h at
37°C in an atmosphere of 5% CO2.
Construction of the pGenesil-GSK-3β
short interfering (si)RNA
Three pairs of shRNAs were used for screening to
obtain the most effective downregulation of the gene fragment.
Non-targeting siRNA (Wuhan Genesil Biotechnology Co., Ltd., Wuhan,
China) was used as the negative control. Using siRNA design
software (http://sirna.wi.mit.edu/), the
following three coding regions corresponding to the target GSK-3β
were selected as siRNA target sequences in the enzyme site of the
GFP-tagged pGenesil-1 vector (pGenesil-1-GFP; Wuhan Genesil
Biotechnology Co., Ltd.): 5′-ACTGGTCGCCAT-CAAGAAA-3′ (471–489 bp);
5′-GAAAGCTA-GATCACTGTAA-3′ (536–554 bp); and
5′-GCCACT-GATTATACCTCTA-3′ (922–940 bp). In addition, an unrelated
sequence was designed for the negative control:
5′-TTCTCCGAACGTCTCACGT-3′. Two oligonucleotides encoding the target
shRNA and its complementary sequence were annealed and ligated, and
the pGenesil-1-GFP vector was cleaved by BamHI and
HindIII (Wuhan Genesil Biotechnology Co., Ltd.). Then the
products of both reactions were recovered and purified. The shRNA
oligonucleotide fragment and the pGenesil-1-GFP vector were ligated
using T4 ligase (Takara Bio, Inc., Shiga, Japan), and the
recombinant plasmids were termed pGenesil-GFP GSK-3β shRNA 1–3.
Next, pGenesil-GFP-GSK-3β shRNAs 1–3 (8 µg) were transfected into
HepG2 cells, respectively, using 20 µl Lipofectamine 2000 reagent
(Invitrogen Life Technologies), according to the manufacturer's
instructions. The most effective shRNA expression cassette,
p-Genesil-GSK-3β shRNA 1, was selected as described previously
(18), and excised from the
pGenesil-1-GFP vector by BamHI and MluI, and ligated
into a pLV-mCMV-ZsGreen-PGK-puro shuttle vector (Wuhan Genesil
Biotechnology Co., Ltd.), termed pLV-GSK-3β shRNA. Plasmids were
purified with a MaxPrep kit (Wuhan Genesil Biotechnology Co., Ltd.)
and successful ligations were verified by sequencing (Sangon
Biotech Co., Ltd., Shanghai, China). Recombinant lentiviral vectors
were produced by co-transfecting HepG2 cells with the lentiviral
expression plasmid and packaging plasmids using the calcium
phosphate method. Briefly, 8 µg shRNA plasmid DNA, 5 µg lentiviral
helper-1, 6 µg lentiviral helper-2 plasmids were mixed with sterile
ddH2O to a final volume of 450 µl and mixed with 50 µl
of 2.5 M CaCl2. Following transfection, infectious media
containing shRNA lentiviral vectors was harvested at 48 and 72
h.
Cell viability assay
The effect of shGSK-3β and shRNA on HCC cell
viability was measured using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. The HepG2 cell line, which is derived from
well-differentiated hepatocellular carcinoma, can be grown
successfully on a large scale, secretes numerous plasma proteins,
and is composed of adherent epithelial-like cells that grow as
monolayers and in small aggregates; therefore, these cells were
used to investigate cell viability. HepG2 cells (5×103
cells/well) were seeded in 96-well plates in triplicate, and
infected with shGSK-3β and negative control shRNA at a multiplicity
of infection (MOI) of 10. Control cells were treated with DMEM.
After growing for 24, 48 and 72 h at 37°C in a 5% CO2
atmosphere, 20 µl of 5 mg/ml MTT [in phosphate-buffered saline
(PBS)] was added to each well and continually incubated for 4 h at
37°C in a CO2 incubator. The formazan granules obtained
from the cells were dissolved in 150 µl dimethylsulfoxide for 10
min. Cell viability was measured in terms of the optical density
using an enzyme-linked immune detector (Multiskan GO; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at a wavelength of 570 nm. Each
cell viability assay was performed in triplicate.
Flow cytometric analysis
HuH-7 cells were treated with 10 µM GSK-3β
inhibitor, Ad5.TRAIL, shBcl-xL and shcIAP2 alone or in combination
at an MOI of 10 for 48 h at 37°C in a 5% CO2 atmosphere.
Adherent and suspended cells were collected, washed in PBS and then
suspended in Annexin binding buffer (Immunotech, Marseille,
France). Subsequently, cells were stained with Annexin V-PE or PI
to distinguish between apoptotic and dead cells. All steps were
conducted in accordance with the Annexin binding buffer
manufacturer's instructions. Finally, the stained cells were
analyzed by flow cytometry using a FC 500 MPL Flow Cytometer
(Becton Dickinson, San Jose, CA, USA).
Western blot analysis
Cell samples were lysed in ice-cold lysis buffer
(Beyotime Institute of Biotechnology) with 1% phenylmethylsulfonyl
fluoride for 30 min and then centrifuged at 10,000 × g for 20 min
at 4°C. The protein concentration of the resulting supernatant was
determined using a bicinchoninic acid protein assay kit (Beyotime
Institute of Biotechnology). Proteins (50 µg) were separated by 12%
SDS-PAGE electrophoresis (Beyotime Institute of Biotechnology) and
subsequently transferred to polyvinylidene difluoride membranes.
Membranes were blocked with 5% non-fat dry milk in Tris-buffered
saline/Tween-20 (0.05%, v/v) for 2 h at room temperature and
incubated overnight at 4°C with the rabbit TRAIL, GSK-3β,
β-catenin, Bcl-xL, cIAP2 and GAPDH primary antibodies. The blots
were washed and incubated with a horseradish peroxidase-conjugated
secondary antibody (Agilent Technologies, Santa Clara, CA, USA) and
developed with the chemiluminescent substrate ECL Plus (Pierce
Biotechnology, Inc., Rockford, IL, USA). An autoradiograph was
obtained, and protein levels were measured using a Fluor-S scanner
for grayscale determination and Quantity One software for analysis
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Quantification by reverse
transcription quantitative-polymerase chain reaction (RT-qPCR)
Transfected and untreated cells were collected and
washed with PBS. Total RNA was extracted from the cells using
TRIzol® reagent (Invitrogen Life Technologies) and complementary
(c)DNA (1 µl) was generated using a PrimeScript® RT reagent kit
(Takara, Chiga, Japan) at a total volume of 20 µl, according to the
manufacturer's instructions. This cDNA was then used in each
amplification reaction. The primers used were as follows: Forward,
5′-CTGGGCTACACTGAGCACC-3′ and reverse, 5′-AAGTGGTCGTTGAGGGCAATG-3′
for GSK-3β; and forward, 5′-AGACGCTCCCTGTGATTTATGT-3′ and reverse,
5′-CCGATGGCAGATTCCAAAGG-3′ for GAPDH. Reactions were performed
using SYBR® Premix Ex Taq™ (Takara) under the following PCR
conditions: 95°C for 30 sec, followed by 40 cycles of 95°C for 5
sec and 60°C for 30 sec. Amplification specificity was also
confirmed by generating a melting curve for each sample. The
expression of proteins was assessed by normalization of the cycle
threshold (Ct) of these genes to that of the housekeeping gene
GAPDH. A Ct value was obtained from each amplification curve using
the software provided by the manufacturer (Roche Diagnostics GmbH,
Mannheim, Germany).
Statistical analysis
Student's t-test and one-way analysis of variance
were performed for continuous variables. The χ2 or
Fisher's exact test were used for categorical variables. Error bars
in all cases represent the standard error of the mean. All
statistical analyses were performed using SAS 9.0 software (SAS
Institute, Inc., Cary, NC, USA) and, using two-sided tests,
P<0.05 indicated a statistically significant difference.
Results
GSK-3β downregulation inhibits the
proliferation of HCC cells
Previous studies have reported that direct
downregulation of GSK-3 protein expression inhibits glioma cell
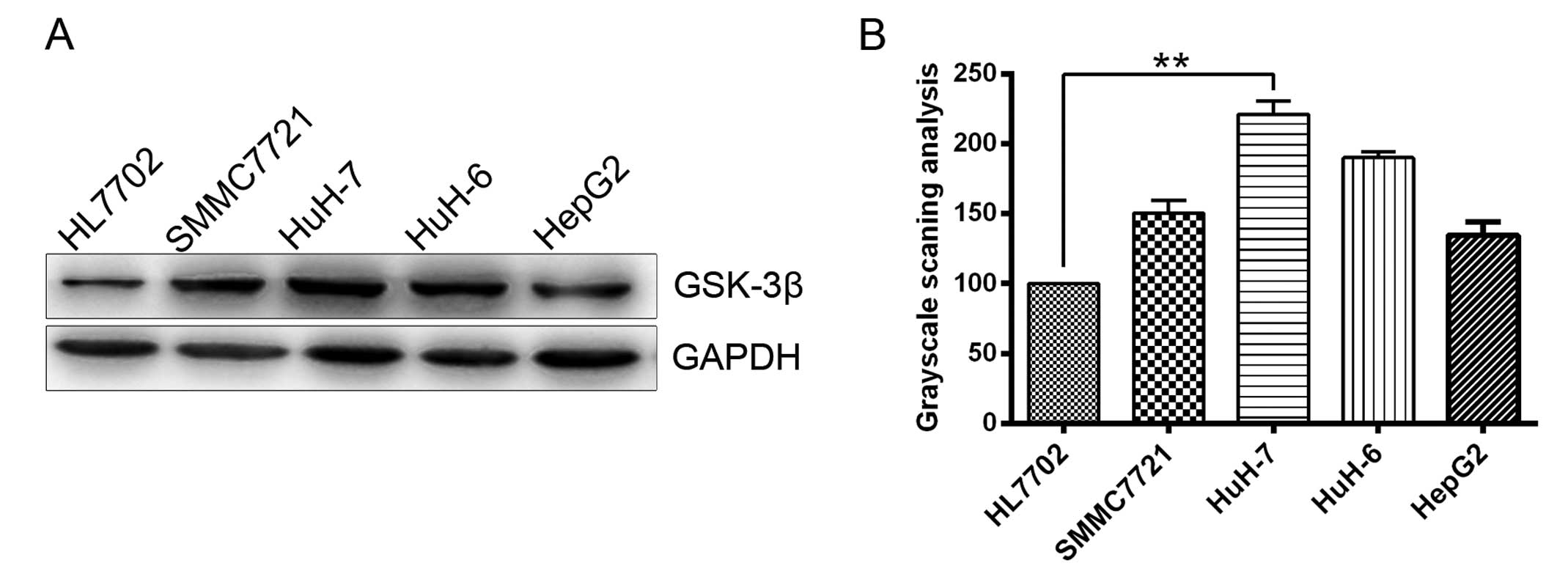
growth (19). GSK-3β is one of the
two GSK-3 isoforms and its expression was determined in five cell
lines using western blot analysis in the present study (Fig. 1). To examine the role of GSK-3β in HCC
cells, the effect of downregulation of GSK-3β protein expression on
HCC cell growth was investigated. HepG2 cells were divided into
three groups: Blank HepG2 cells, shGSK-3β-treated cells and
shRNA-treated control cells. Western blotting and RT-qPCR
identified that GSK-3β protein and mRNA expression levels,
respectively, were significantly downregulated by shGSK-3β in HepG2
cells (P<0.05; Fig. 2A–C).
Furthermore, shRNA-mediated downregulation of GSK-3β resulted in
growth inhibition, as revealed by the MTT assay. Compared with the
shRNA control cells, there was a significant growth delay in
shGSK-3β-treated cells by 72 h (P<0.05; Fig. 2D). These results demonstrate that
GSK-3β downregulation inhibited the proliferation of HCC cells.
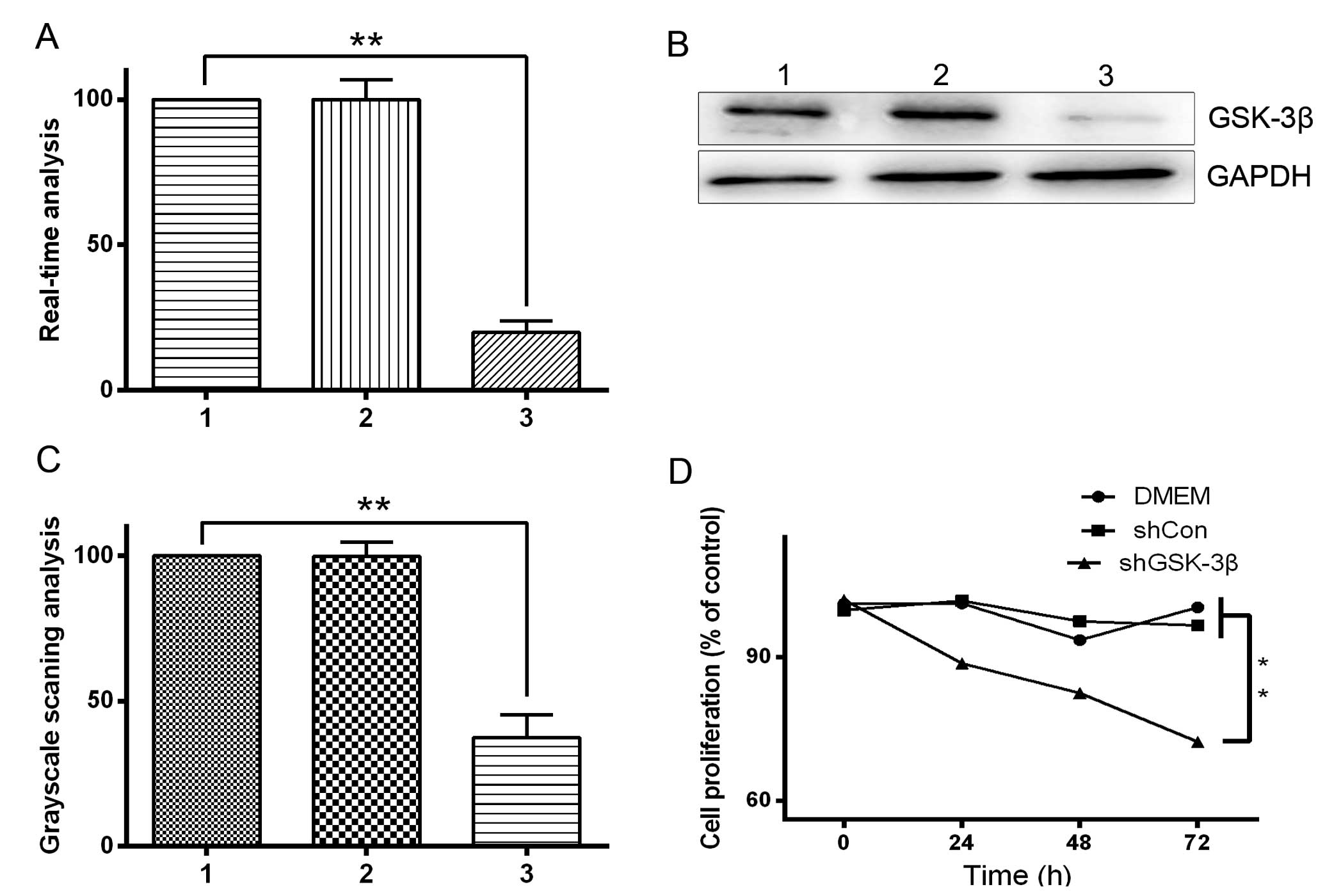 | Figure 2.Effect of GSK-3β knockdown by
shGSK-3β in HepG2 hepatocellular carcinoma cells. HepG2 cells were
transfected with pGenesil vector harboring shRNA targeting GSK-3β.
Lane 1, shRNA transfected into HepG2 cells; lane 2, blank HepG2
cells; lane 3, shGSK-3β transfected into HepG2 cells. (A) RT-qPCR
and (B) western blot analysis of GSK-3β mRNA and protein expression
level, respectively. (C) Grayscale scanning analysis revealing that
the expression of GSK-3β was reduced in HepG2 cells transfected
with shGSK-3β. (D) Effect of shGSK-3β on the viability of HepG2
cells. HepG2 cells were treated with shGSK-3β for 72 h and cell
viability was determined by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
**P<0.05. RT-qPCR, reverse transcription-quantitative polymerase
chain reaction; GSK-3β, glycogen synthase kinase 3 β; DMEM,
Dulbecco's modified Eagle's medium; sh, short hairpin; Con,
control. |
GSK-3β inhibitor sensitizes HCC cells
to TRAIL-induced apoptosis
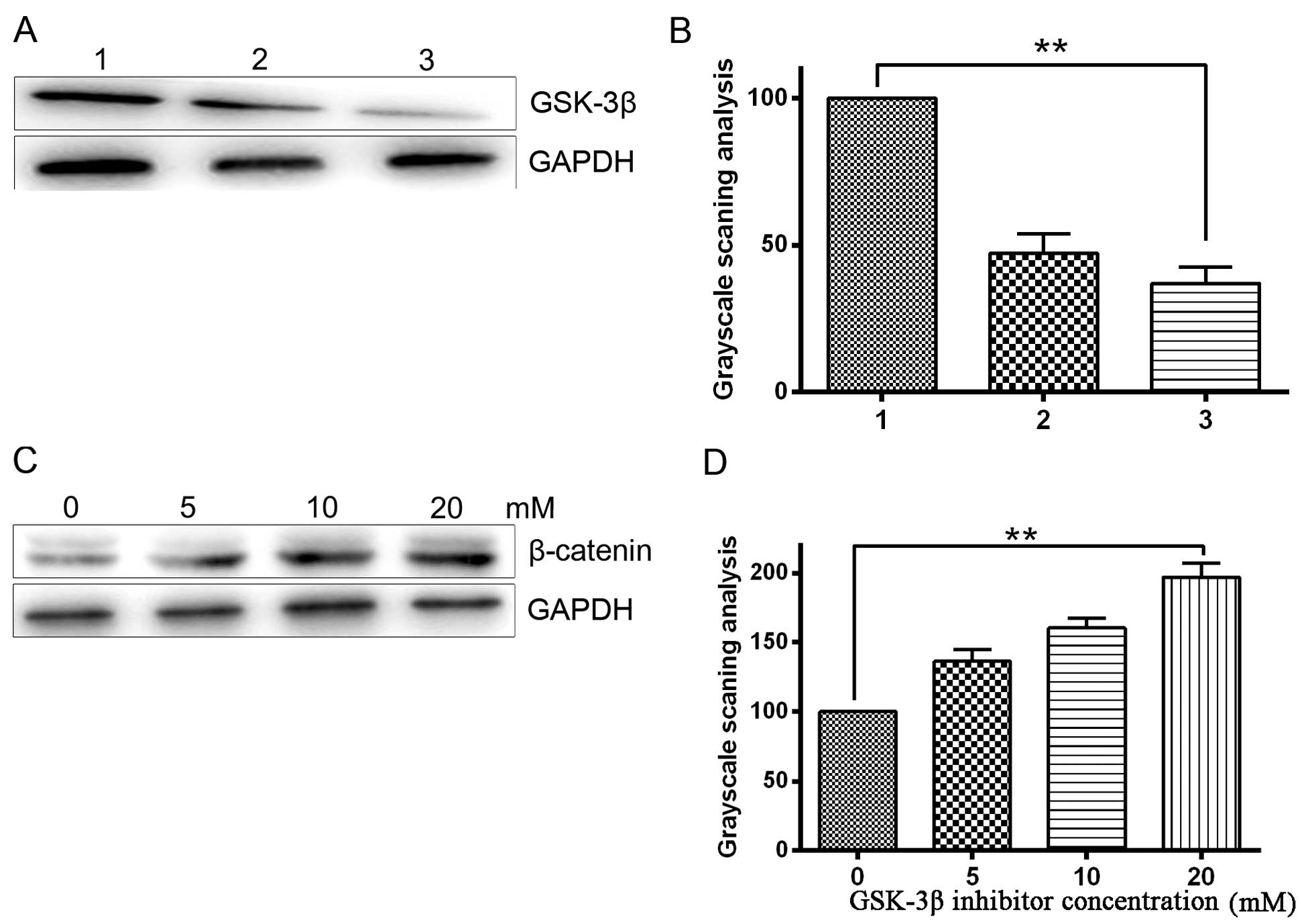
The protein expression level of GSK-3β was detected
by western blotting following treatment of HepG2 cells with
SB216763 (a specific of inhibitor of GSK-3β) and GSK-3β shRNA. As
GSK-3 phosphorylates β-catenin for proteasomal degradation, the
level of phosphorylated β-catenin reflects the activity of GSK-3.
It follows that pretreatment with a GSK-3 inhibitor completely
inhibits phosphorylation of β-catenin, thus, increasing the levels
of β-catenin (20). To determine the
effects of GSK-3β inhibitor on GSK-3β activity in HepG2 cells,
western blot was used to analyze the protein expression levels of
β-catenin. Western blotting confirmed that the GSK-3β inhibitor
significantly inhibited GSK-3β expression compared with the blank
control (P<0.05; Fig. 3A and B).
The results also demonstrated that GSK-3β inhibition significantly
enhanced β-catenin expression (P<0.05), indirectly indicating
effective inhibition of GSK-3β activity in HepG2 cells. This
enhancement of β-catenin expression occurred in a dose-dependent
manner (Fig. 3C and D). Our previous
study verified that infection of non-small cell lung cancer cells
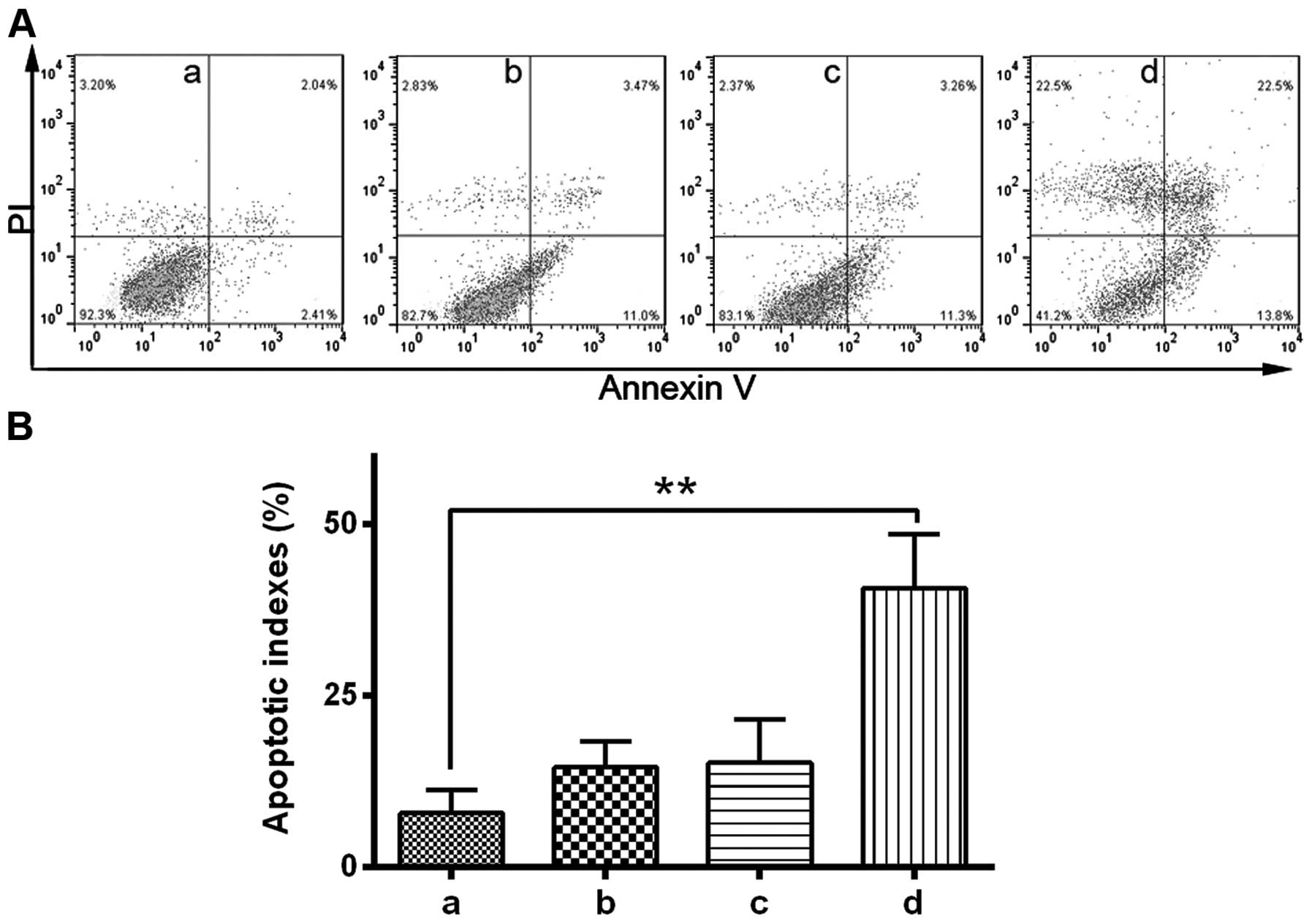
with Ad5.TRAIL resulted in significant cytotoxicity (21). To investigate the potential role of
GSK-3β in TRAIL-induced apoptosis, the effect of a GSK-3β inhibitor
on TRAIL-induced cytotoxicity was analyzed using
fluorescence-activated cell sorting. Due to the high expression of
GSK-3β compared with the other HCC cells investigated, HuH-7 cells
were used for this experiment. HuH-7 cells were treated with GSK-3β
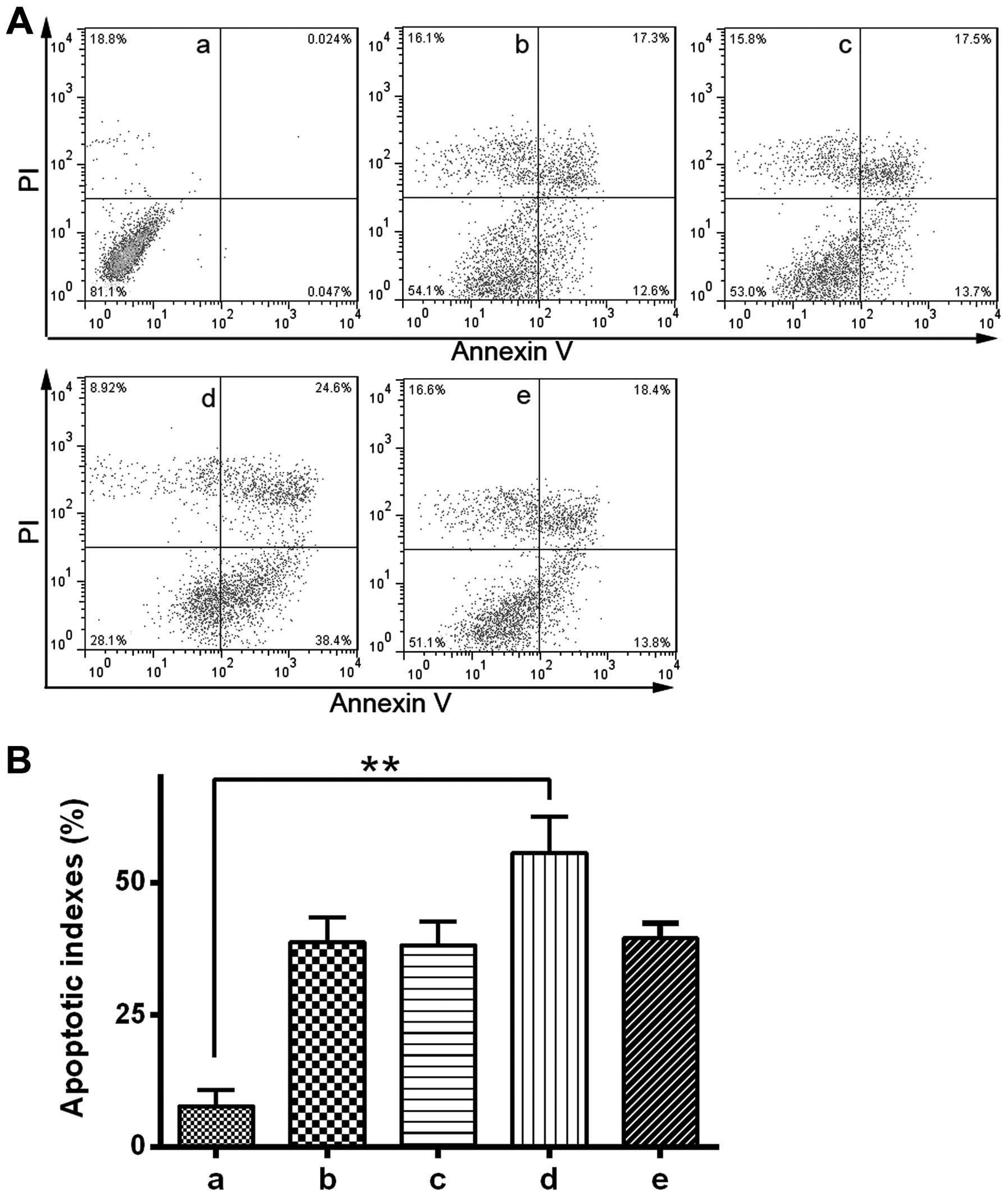
inhibitor and Ad5.TRAIL alone or in combination. The results
indicated that combined treatment with GSK-3β inhibitor and
Ad5.TRAIL led to a significant increase in apoptosis compared with
untreated cells (P<0.01). By contrast, GSK-3β
inhibitor/Ad5.TRAIL combined treatment resulted in a significant
induction of cell apoptosis (P<0.05). Thus, the present study
demonstrated that TRAIL-induced apoptosis is enhanced following
GSK-3β inhibition in HuH-7 cells (Fig. 4A
and B).
GSK-3β modulates a specific set of
NF-κB target genes
NF-κB is a critical factor in protecting hepatocytes
from apoptosis (22). TRAIL has been
reported to activate the transcription factor NF-κB, which
transactivates genes encoding the key anti-apoptotic proteins
Bcl-xL and IAPs, both of which have been implicated in TRAIL
resistance (10,11). The current findings demonstrated that
GSK-3β inhibitor sensitizes HCC cells to TRAIL-induced apoptosis,
suggesting that GSK-3β may be involved in the regulation of NF-κB
activation. To prove this hypothesis, the effect of GSK-3β on NF-κB
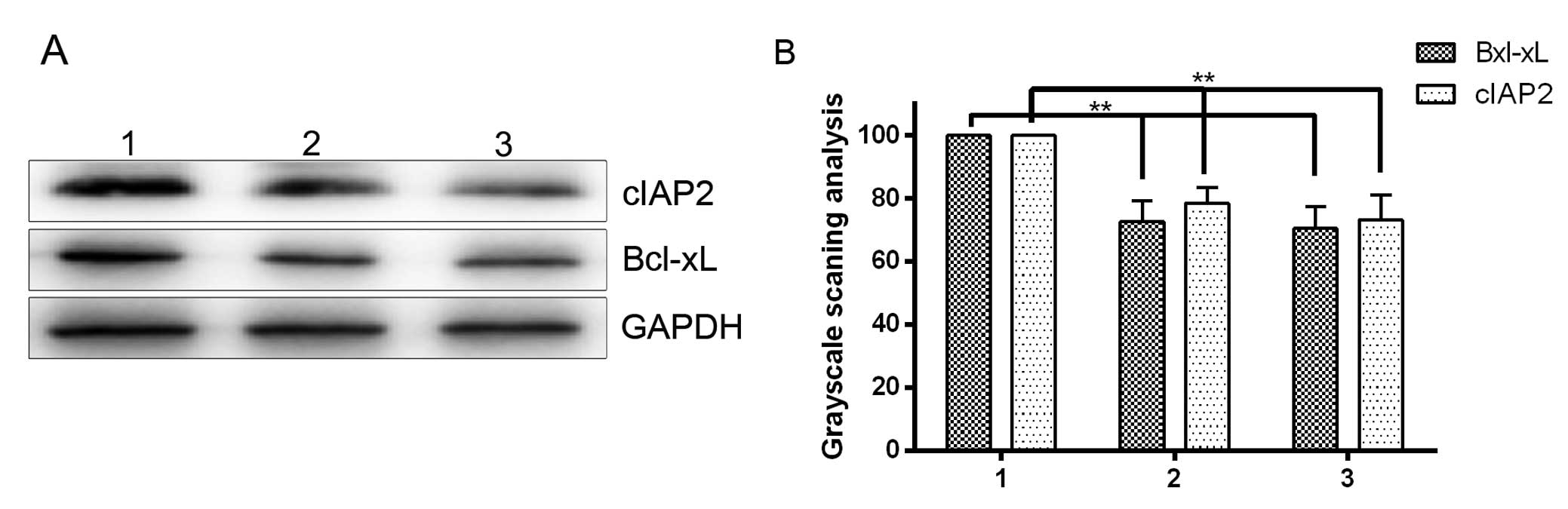
target genes, which encode the key anti-apoptotic proteins Bcl-xL
and cIAP2, was evaluated. Western blotting demonstrated that
downregulation of GSK-3β by shGSK-3β or GSK-3β inhibitor in HepG2
HCC cells significantly decreased the expression of Bcl-xL and
cIAP2 (P<0.05; Fig. 5A and B). The
results indicated that GSK-3β has the potential to regulate NF-κB
target genes involved in cell survival.
Knockdown of Bcl-xL enhances the
effect of GSK-3β inhibitor on TRAIL-induced apoptosis
A recent study revealed that Bcl-xL downregulation
contributes to GSK-3β inhibitor-induced sensitization to TRAIL in
pancreatic ductal adenocarcinoma cells (11). To evaluate the role of Bcl-xL and
cIAP2 on TRAIL-induced apoptosis in HCC cells, shRNA was used to
generate HuH-7 cells with stable knockdown of each gene.
Subsequently, the response of shBcl-xL- and shcIAP2-treated HuH-7
cells to TRAIL-induced apoptosis was determined. The results
indicated that Bcl-xL and cIAP2 knockdown each markedly enhanced
TRAIL-induced apoptosis (Fig. 6). To
further examine whether Bcl-xL actively participates in GSK-3β
inhibitor-mediated TRAIL sensitization, apoptosis was measured
following treatment with GSK-3β inhibitor (Fig. 6). The results indicated that knockdown
of Bcl-xL significantly enhances the effect of GSK-3β inhibitor on
TRAIL-induced apoptosis (P<0.05; Fig.
6).
Discussion
GSK-3β modulates cell survival and apoptosis through
multiple intracellular signaling pathways (23,24). In
the present study, downregulation of GSK-3β resulted in growth
inhibition in HCC cells, which is consistent with previous reports
(12). The aim of the current study
was to investigate whether inhibition of GSK-3β could induce
TRAIL-reduced apoptosis in HCC cells. The results herein
demonstrated that chemical inhibition or genetic silencing of
GSK-3β sensitizes HCC cells to TRAIL-induced apoptosis.
Furthermore, it was found that GSK-3β inhibitor-induced TRAIL
sensitization depends upon the activity of NF-κB. These results
provide a novel mechanism for GSK-3β modulation of TRAIL
sensitivity in HCC cells.
TRAIL resistance has been observed in a variety of
cell types (25,26) and is caused by the differential
expression of TRAIL receptors, as well as by intracellular
molecules, such as Akt and Bcl-2, which modulate the downstream
effect of TRAIL signaling (27,28).
GSK-3β has been implicated in apoptosis under a variety conditions,
including DNA damage, endoplasmic reticulum stress and hypoxia
(13,29,30).
GSK-3β inhibitors are used in the treatment of Alzheimer's disease,
diabetes and inflammatory disorders (31,32). In an
oncology setting, previous studies have shown that GSK-3β
inhibitors sensitize prostate cancer cells to TRAIL-induced
apoptosis (33). The present study
demonstrates the mechanism by which GSK-3β inhibits TRAIL-induced
apoptosis of sensitized HCC cells. The rationale was based on the
premise that GSK-3β inhibition in HCC cells downregulates the
expression of key anti-apoptotic proteins that mediate TRAIL
resistance. For example, previous studies have demonstrated that
TRAIL stimulates NF-κB activation, while inhibition of NF-κB
sensitizes cells to TRAIL-mediated apoptosis in various cell lines
(10,34). Since the first report that genetic
ablation of the murine GSK-3β gene perturbs NF-κB activation
(35), the role of GSK-3β in NF-κB
signaling has been investigated in a number of cell types. However,
conflicting results have been reported regarding the role of GSK-3β
in promoting NF-κB activity, ranging from major defects in
TNFα-induced inhibitor of κBα (IκBα) phosphorylation to minor
effects on cytosolic signaling pathways and p65 nuclear
translocation (16,36). In addition, Bcl-2 was reported to
inhibit TRAIL-mediated cell death (27). IAPs constitute a protein family that
regulates programmed cell death (37), and a recent study demonstrated that
GSK-3β induces Bcl-xL and cIAP2 expression in pancreatic cancer
cells (11). Consistent with this,
the results of the present study indicate that GSK-3β may also
regulate the expression of the BCL2L1 and BIRC3 genes, which encode
anti-apoptosis proteins Bcl-xL and cIAP2 in HCC cells,
respectively, and are known downstream targets of NF-κB. Notably,
suppression of Bcl-xL and cIAP2 sensitized HCC cells to
TRAIL-induced apoptosis. Furthermore, knockdown of Bcl-xL signaling
enhanced the effect of GSK-3β inhibitor on TRAIL-induced apoptosis.
These results are consistent with a previous study that
demonstrated a role for Bcl-xL in mediating TRAIL resistance
(25). A limitation of the current
study is that only two NF-κB target genes were examined. Thus, it
is unknown whether the NF-κB inhibitor IκBα and a number of other
NF-κB target genes are not involved in mediating TRAIL resistance.
The detailed mechanisms underlying GSK-3β regulation of the NF-κB
signaling pathway have yet to be fully elucidated.
GSK-3β inhibition can overcome TRAIL resistance
through the extrinsic apoptotic pathway by promoting the initial
step of death-inducing signaling complex formation, leading to
caspase-8 and caspase-3 activation in breast cancer cells (38). The results of the current study
revealed that GSK-3β can regulate the TRAIL-dependent inhibition of
apoptosis at the mRNA and protein levels in HCC cells. Thus, the
NF-κB signaling pathways are promising targets for cancer therapy
(39). The canonical and noncanonical
pathways can be inhibited to modulate NF-κB activity, and possibly
treat various types of disease. The present study indicates that
GSK-3β inhibitor enhances TRAIL-induced apoptosis in HCC cells.
Furthermore, GSK-3β contributes to TRAIL resistance by modulating
the expression of a set of specific anti-apoptotic NF-κB target
genes. The subcellular location (nucleus or cytoplasm) in which
GSK-3β regulates NF-κB function was not explored in the present
study. Future studies may determine whether GSK-3β contributes to
the expression of Bcl-xL and cIAP2 via nucleic or cytoplasmic
targeting and, thus, indicate whether the signaling cascades
differ.
In conclusion, the present study demonstrates that
GSK-3β suppression sensitizes HCC cells to TRAIL-induced apoptosis,
and is dependent on the NF-κB signaling pathway. An understanding
of the role of GSK-3β in TRAIL resistance may offer improved
therapeutic strategies for patients with HCC.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81372632).
References
|
1
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheng CF, Lu IH, Tseng HW, Sun CY, Lin LT,
Kuo ZK, Pan IH and Ko CH: Antitumor Effect of Periplocin in
TRAIL-Resistant Human Hepatocellular Carcinoma Cells through
Downregulation of IAPs. Evid Based Complement Alternat Med.
2013:Article ID 958025. 2013.
|
|
4
|
Chiao PJ, Na R, Niu J, Sclabas GM, Dong Q
and Curley SA: Role of Rel/NF-kappaB transcription factors in
apoptosis of human hepatocellular carcinoma cells. Cancer.
95:1696–1705. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wiley SR, Schooley K, Smolak PJ, et al:
Identification and characterization of a new member of the TNF
family that induces apoptosis. Immunity. 3:673–682. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wajant H, Pfizenmaier K and Scheurich P:
TNF-related apoptosis inducing ligand (TRAIL) and its receptors in
tumor surveillance and cancer therapy. Apoptosis. 7:449–459. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Srivastava RK: TRAIL/Apo-2L: Mechanisms
and clinical applications in cancer. Neoplasia. 3:535–546. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Spierings DC, de Vries EG, Vellenga E, et
al: Tissue distribution of the death ligand TRAIL and its
receptors. J Histochem Cytochem. 52:821–831. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smyth MJ, Takeda K, Hayakawa Y, et al:
Nature's TRAIL - on a path to cancer immunotherapy. Immunity.
18:1–6. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Omar HA, Arafa SA, Maghrabi IA and Weng
JR: Sensitization of hepatocellular carcinoma cells to Apo2L/TRAIL
by a novel Akt/NF-κB signalling inhibitor. Basic Clin Pharmacol
Toxicol. 114:464–471. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang JS, Herreros-Villanueva M, Koenig A,
et al: Differential activity of GSK-3 isoforms regulates NF-κB and
TRAIL- or TNFα induced apoptosis in pancreatic cancer cells. Cell
Death Dis. 5:e11422014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Forde JE and Dale TC: Glycogen synthase
kinase 3: A key regulator of cellular fate. Cell Mol Life Sci.
64:1930–1944. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song L, De Sarno P and Jope RS: Central
role of glycogen synthase kinase-3beta in endoplasmic reticulum
stress-induced caspase-3 activation. J Biol Chem. 277:44701–44708.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pak C and Miyamoto S: A new alpha in line
between KRAS and NF-κB activation? Cancer Discov. 3:613–615. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yao HB, Shaw PC, Wong CC and Wan DCC:
Expression of glycogen synthase kinase-3 isoforms in mouse
tissuesand their transcription in the brain. J Chem Neuroanat.
23:291–297. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schwabe RF and Brenner DA: Role of
glycogen synthase kinase-3 in TNF-alpha-induced NF-kappaB
activation and apoptosis in hepatocytes. Am J Physiol Gastrointest
Liver Physiol. 283:G204–G211. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Coghlan MP, Culbert AA, Cross DA, et al:
Selective small molecule inhibitors of glycogen synthase kinase-3
modulate glycogen metabolism and gene transcription. Chem Biol.
7:793–803. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu J, Jiang G, Liu S, et al:
Lentivirus-delivered short hairpin RNA targeting SNAIL inhibits
HepG2 cell growth. Oncol Rep. 30:1483–1487. 2013.PubMed/NCBI
|
|
19
|
Kotliarova S, Pastorino S, Kovell LC,
Kotliarov Y, Song H, Zhang W, Bailey R, Maric D, Zenklusen JC, Lee
J, et al: Glycogen synthase kinase-3 inhibition induces glioma cell
death through c-MYC, nuclear factor-kappaB, and glucose regulation.
Cancer Res. 68:6643–6651. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim SJ, Lim JY, Lee JN, Choe SK, Kim YI,
Song SR, Cho M, So HS and Park R: Activation of β-catenin by
inhibitors of glycogen synthase kinase-3 ameliorates
cisplatin-induced cytotoxicity and pro-inflammatory cytokine
expression in HEI-OC1 cells. Toxicology. 320:74–82. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang H, Sui A, Wang Z, Liu S and Yao R:
Adenovirus-mediated TRAIL expression and downregulation of Bcl-2
expression suppresses non-small cell lung cancer growth in vitro
and in vivo. Int J Mol Med. 30:358–364. 2012.PubMed/NCBI
|
|
22
|
Kim HS, Loughran PA, Rao J, Billiar TR and
Zuckerbraun BS: Carbon monoxide activates NF-kappaB via ROS
generation and Akt pathways to protect against cell death of
hepatocytes. Am J Physiol Gastrointest Liver Physiol.
295:G146–G152. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jope RS and Johnson GV: The glamour and
gloom of glycogen synthase kinase-3. Trends Biochem Sci. 29:95–102.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Beurel E and Jope RS: The paradoxical pro-
and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic
apoptosis signaling pathways. Prog Neurobiol. 79:173–189. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen KF, Yeh PY, Hsu C, Hsu CH, Lu YS,
Hsieh HP, Chen PJ and Cheng AL: Bortezomib overcomes tumor necrosis
factor-related apoptosis-inducing ligand resistance in
hepatocellular carcinoma cells in part through the inhibition of
the phosphatidylinositol 3-kinase/Akt pathway. J Biol Chem.
284:11121–11133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Newsom-Davis T, Prieske S and Walczak H:
Is TRAIL the holy grail of cancer therapy? Apoptosis. 14:607–623.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Munshi A, Pappas G, Honda T, McDonnell TJ,
Younes A, Li Y and Meyn RE: TRAIL (APO-2L) induces apoptosis in
human prostate cancer cells that is inhibitable by Bcl-2. Oncogene.
20:3757–3765. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen X, Thakkar H, Tyan F, Gim S, Robinson
H, Lee C, Pandey SK, Nwokorie C, Onwudiwe N and Srivastava RK:
Constitutively active Akt is an important regulator of TRAIL
sensitivity in prostate cancer. Oncogene. 20:6073–6083. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Loberg RD, Vesely E and Brosius FC III:
Enhanced glycogen synthase kinase-3beta activity mediates
hypoxia-induced apoptosis of vascular smooth muscle cells and is
prevented by glucose transport and metabolism. J Biol Chem.
277:41667–41673. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Watcharasit P, Bijur GN, Zmijewski JW,
Song L, Zmijewska A, Chen X, Johnson GV and Jope RS: Direct,
activating interaction between glycogen synthase kinase-3beta and
p53 after DNA damage. Proc Natl Acad Sci USA. 99:7951–7955. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Klamer G, Song E, Ko KH, O'Brien TA and
Dolnikov A: Using small molecule GSK3β inhibitors to treat
inflammation. Curr Med Chem. 17:2873–2881. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Medina M and Avila J: Glycogen synthase
kinase-3 (GSK-3) inhibitors for the treatment of Alzheimer's
disease. Curr Pharm Des. 16:2790–2798. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liao X, Zhang L, Thrasher JB, Du J and Li
B: Glycogen synthase kinase-3β suppression eliminates tumor
necrosis factor-related apoptosis-inducing ligand resistance in
prostate cancer. Mol Cancer Ther. 2:1215–1222. 2003.PubMed/NCBI
|
|
34
|
Huerta-Yepez S, Vega M, Jazirehi A, Garban
H, Hongo F, Cheng G and Bonavida B: Nitric oxide sensitizes
prostate carcinoma cell lines to TRAIL-mediated apoptosis via
inactivation of NF-kappa B and inhibition of Bcl-xl expression.
Oncogene. 23:4993–5003. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hoeflich KP: Requirement for glycogen
synthase kinase-3β cell survival and NF-κB activation. Nature.
406:86–90. 2001. View
Article : Google Scholar
|
|
36
|
Wilson W III and Baldwin AS: Maintenance
of constitutive IkappaB kinase activity by glycogen synthase
kinase-3alpha/beta in pancreatic cancer. Cancer Res. 68:8156–8163.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Deveraux QL and Reed JC: IAP family
proteins - suppressors of apoptosis. Genes Dev. 13:239–252. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun M, Zhou T, Jonasch E and Jope RS: DDX3
regulates DNA damage-induced apoptosis and p53 stabilization.
Biochim Biophys Acta. 1833:1489–1497. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Carbone C and Melisi D: NF-kB as a target
for pancreatic cancer therapy. Expert Opin Ther Targets. 16:1–10.
2012. View Article : Google Scholar : PubMed/NCBI
|