Introduction
Non-small cell lung cancer (NSCLC) is the leading
cause of mortality due to cancer in the world (1). In locally advanced cancers, chemotherapy
and radiation therapy are always incorporated into the treatment
regimens of patients (1).
Platinum-based adjuvant chemotherapy is a standard treatment for
completely resected higher-stage NSCLC (2). Cisplatin is one of the most potent
platinum-based chemotherapeutic agents currently in use and is
effective as a single agent or in combination with other drugs for
the treatment of NSCLC. Cisplatin-based chemotherapy significantly
improves the prognosis of patients with NSCLC (3). However, a concerning clinical challenge
for cisplatin-based NSCLC chemotherapy is the intrinsic and
acquired chemoresistance to the drug (3). Therefore, the identification of factors
that contribute to cisplatin chemoresistance in NSCLC may be
pivotal for the development of novel therapeutic strategies for
this disease.
Deletion of sterile α motif domain-containing 9
(SAMD9) is commonly observed in cells from patients with myeloid
leukemia and myelodysplastic syndrome, suggesting that SAMD9 is an
inhibitor of tumor progression (4).
Ubiquitously expressed in human adult and fetal tissues, SAMD9 is
expressed at lower levels in tumors and has been reported to be a
potent tumor suppressor gene (5).
Overexpression of SAMD9 causes apoptosis and reduced proliferation
of malignant cells, whereas downregulation of SAMD9 is associated
with increased cellular proliferation and tumor growth in in
vivo and in vitro models (5). A recent study has revealed that SAMD9
suppresses tumorigenesis and progression of NSCLC (6).
MicroRNAs (miRNAs) have previously been implicated
in oncogenic cell processes, including chemoresistance (7). Lung cancer development is closely
correlated with miRNA expression (8).
Since miRNAs are small non-coding RNA molecules, they
post-transcriptionally regulate target gene expression by
incomplete base pairing with target mRNAs (9). miRNAs operate through RNA-induced
silencing complexes, targeting these complexes to mRNAs where
direct destructive cleavage or repression of translation takes
place (10). A previous study has
demonstrated marked alterations in the miRNA profile in NSCLC
compared to adjacent normal tissues (1).
The present study explored the role of miRNA/SAMD9
signaling in regulating cisplatin chemoresistance in NSCLC; to the
best of our knowledge this is the first study to do so.
Materials and methods
Cells lines, plasmid constructs and
reagents
The human NSCLC cell lines H358 (catalog no.,
CRL-5807) and H23 (catalog no., CRL-5800) were purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA). Human
SAMD9 3′-untranslated region (UTR) luciferase reporter (catalog
no., HmiT013716) and the LucPair Duo Luciferase Assay kit (catalog
no., LPFR-M010) were purchased from GeneCopoeia, Inc. (Rockville,
MD, USA). Human SAMD9 cDNA clone (catalog no., SC304503) was
purchased from OriGene Technologies China (Beijing, China) and
subcloned into the pcDNA 3.1 expression vector (catalog no.,
V790-20; Thermo Fisher Scientific, Inc., Waltham, MA, USA) to
generate a pCDNA3.1-SAMD9 expression vector. The 3′-UTR of human
SAMD9 was subcloned from the human SAMD9 3′-UTR luciferase reporter
and inserted downstream of human SAMD9 cDNA in the pcDNA3.1-SAMD9
expression vector to generate a pcDNA3.1-(SAMD9 cDNA plus 3′-UTR)
expression vector. Human SAMD9 short hairpin (sh)RNA lentiviral
particles (catalog no., sc-89746-V), control shRNA lentiviral
particles-A (catalog no., sc-108080) and goat anti-human polyclonal
GAPDH antibody (clone V-18; catalog no., sc-20357; 1:1,000) were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Rabbit anti-human polyclonal anti-SAMD9 antibody (catalog no.,
HPA021319; 100 µl), puromycin, G418, and cisplatin were purchased
from Sigma-Aldrich (St. Louis, MO, USA). Scrambled miR, miR mimics
and antagomirs were purchased from NeuroBiotech (Shanghai, China).
Lipofectamine® 2000 transfection reagent and TRIzol reagent were
purchased from Thermo Fisher Scientific, Inc. The TiterTACS in
situ apoptosis detection kit (catalog no., 4822-96-K) was
purchased from R&D Systems, Inc. (Minneapolis, MN, USA). The
MTT assay kit (catalog no., 30-1010K) was purchased from ATCC.
miRNAs that were potentially able to suppress SAMD9 gene (gene ID,
NM_017654) expression were selected using TargetScan prediction
software (available from http://www.targetscan.org) (10).
Tissue samples
Human NSCLC tumor and adjacent normal lung tissues
were obtained from the Tumor Tissue Bank of the Affiliated Cancer
Hospital of Xiangya School of Medicine, Central South University
(Changsha, Hunan, China). The tissues had been collected from 5
consecutive patients treated at the Affiliated Cancer Hospital of
Xiangya School of Medicine, Central South University in July 2005.
No patients received chemotherapy or radiotherapy prior to surgery.
All NSCLC and adjacent normal tissues (taken >5 cm from tumor)
were pathologically validated by a pathologist.
Luciferase assay
H358 and H23 cells were transfected with luciferase
reporter constructs using Lipofectamine 2000 transfection reagent.
Luciferase activity was measured 30 h subsequent to transfection
using the LucPair Duo Luciferase Assay kit following the
manufacturer's protocol. Each experiment was repeated three times
in duplicate.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was prepared from the cells using TRIzol
reagent. cDNA was synthesized using SuperScript II reverse
transcriptase (Thermo Fisher Scientific, Inc.). qPCR was performed
using the Applied Biosystems SYBR Green PCR master mix in an
Applied Biosystems 7300 real-time PCR System (Thermo Fisher
Scientific, Inc.). Applied Biosystems TaqMan microRNA assays
(Thermo Fisher Scientific, Inc.), which include RT primers and
TaqMan probes, were used to quantify the expression of miR-96. For
the measurement of SAMD9 mRNA, the following primers were used:
Human SAMD9 forward, 5′-GTGGCCTTTTGTGATCTCCT-3′ and reverse,
5′-CTTATGACTTTCTAACCACTGA-3′; and human GAPDH forward,
5′-GACTCATGACCACAGTCCATGC-3′ and reverse,
5′-AGAGGCAGGGATGATGTTCTG-3′. The quantification cycle
(Cq) for each PCR product was calculated with the
instrument's software and Cq values were normalized by
subtracting the Cq values of GAPDH. The resulting
ΔCq values were used to calculate the relative change in
mRNA expression as a ratio, according to the 2−ΔΔCq
method. Each experiment was repeated three times in duplicate.
Western blot analysis
Tissue homogenate or cultured cells were lysed with
a hypotonic buffer containing 2% Nonidet-P and a protease inhibitor
cocktail (Sigma-Aldrich) by sonication, which was performed three
times for 3 sec on ice. The supernatant obtained subsequent to
centrifugation at 2,000 × g for 15 min at 4°C, was used for protein
concentration determination by the Coomassie blue method and for
subsequent steps. Equal amounts of protein were used for each
sample and were separated by 8–15% SDS-polyacrylamide gel and
blotted onto a polyvinylidene difluoride microporous membrane
(Merck Millipore, Hong Kong, China). The membranes were incubated
for 1 h with a 1:1,000 dilution of primary antibody, and then
washed and revealed using bovine anti-goat (catalog no., sc-2350;
Santa Cruz Biotechnology, Inc.) or anti-rabbit (catalog no.,
sc-2370; Santa Cruz Biotechnology, Inc.) secondary antibodies with
horseradish peroxidase conjugate (dilution, 1:5,000; 1 h). The
peroxidase was revealed with an Amersham ECL Western Blotting
Detection kit (GE Healthcare Life Sciences, Shanghai, China). Three
independent experiments were performed for each western blot
analysis.
Transfection and lentiviral
transduction
Plasmids, miR mimics and antagomirs were transfected
into cells using Lipofectamine 2000 transfection reagent, following
the manufacturer's protocol. For stable transfections, pools of
stable transfectants of pcDNA3.1-(SAMD9 cDNA plus 3′-UTR) were
generated via selection with 700 µg/ml G418, according to the
manufacturer's protocol. Lentiviral transduction of SAMD9-shRNA was
performed and pools of stable transductants were generated via
selection with 4.5 µg/ml puromycin.
Cisplatin chemoresistance assay
The cells were plated in duplicate in 96-well plates
at a density of 5×103 cells per well. Subsequent to 24-h
incubation, Dulbecco's modified Eagle's medium (Thermo Fisher
Scientific, Inc.) was replaced with fresh medium, with or without
various concentrations of cisplatin (0.1, 0.25, 0.5, 1.0, 1.5, 3.0,
6.0, 15.0, 30.0 and 55.0 mM). Cell viability was assayed 96 h later
using a MTT assay kit following the manufacturer's protocols. The
half maximal inhibitory concentration (IC50) was defined
as the concentration resulting in a 50% reduction in growth of
cells compared to control cells.
Cell apoptosis assay
The cells were cultured at 8×104 cells
per well in 96-well tissue culture plates and incubated at 37°C for
24 or 48 h with cisplatin (1 µM). Cell apoptosis was measured at 24
and 48 h with a microplate reader-based TiterTACS in situ
apoptosis detection kit, according to the manufacturer's protocol
(11). The cell apoptosis rate at 24
h and 48 h was identified as the percentage of apoptotic cells
relative to 100% cell apoptosis induced by nuclease treatment. Each
experiment was repeated for three times in duplicate.
Statistical analysis
Statistical analyses were performed using SPSS for
Windows 10.0 (SPSS, Inc., Chicago, IL, USA). All continuous
variable values were expressed as the mean ± standard deviation.
Comparison between the means of two groups was performed using
Student's t-test. Comparison between the means of multiple
groups was performed using one-way analysis of variance followed by
post hoc pairwise comparisons using Tukey's test. A two-tailed
P<0.05 was considered statistically significant.
Results
miR-96 targets SAMD9 in NSCLC
cells
TargetScan prediction software analyzed the 3′-UTR
of human SAMD9 gene and cross-referenced the results with a
previous study that identified miRNAs that are differentially
expressed between NSCLC and adjacent normal lung tissues (1). The previous study revealed that 4 miRNAs
were upregulated in NSCLC, consisting of miR-96, miR-374b, miR-182
and miR-1229 (1), and the TargetScan
analysis in the present study demonstrated that these 4 miRNAs
could potentially target SAMD9. The 4 candidate miRNAs were
co-transfected with a SAMD9 3′-UTR luciferase reporter into H358
human NSCLC cells. In total, 7 other miRNAs that demonstrated a
highly favorable context+ score (≤-0.4) (12) on TargetScan and were not upregulated
in NSCLC tumors (1), consisting of
miR-298, miR-432, miR-483-3p, miR-626, miR-4628, miR-133b and
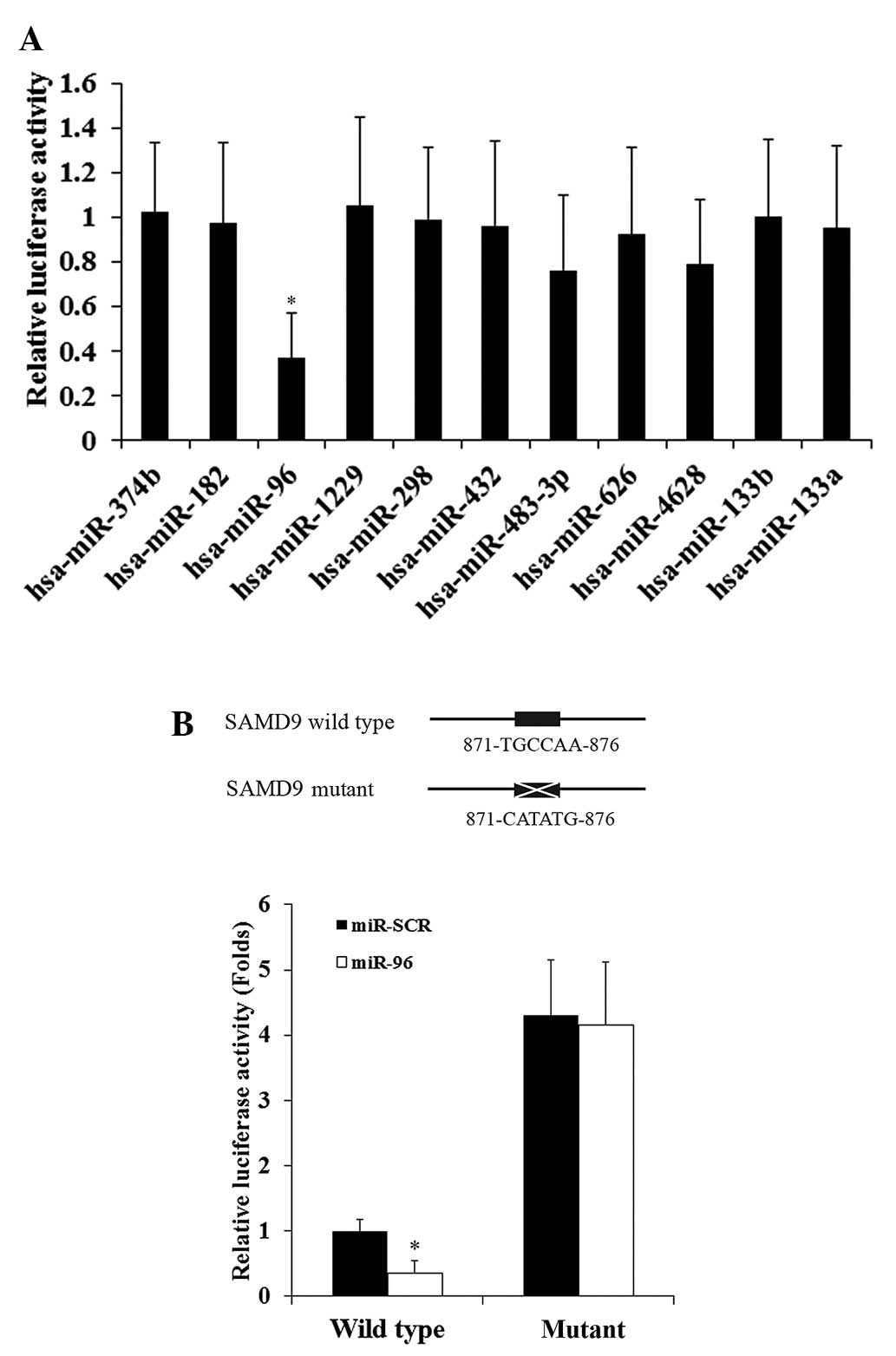
miR-133a, were also included in the luciferase assays (Fig. 1A). The luciferase activity was
measured and normalized to that in cells co-transfected with
scramble miR (miR-SCR) and the SAMD9 3′-UTR luciferase reporter 30
h subsequent to transfection. As demonstrated by Fig. 1A, among the miRNAs tested, only miR-96
significantly decreased the luciferase activity (cut off value,
1.0), suggesting that miR-96 targeted SAMD9.
To demonstrate a direct interaction between miR-96
and SAMD9, the potential binding sequence for miR-96 in the 3′-UTR
of the SAMD9 gene, as predicted by TargetScan, was mutated to
generate a mutant SAMD9 3′-UTR luciferase reporter (Fig. 1B). H358 cells were co-transfected with
miR-96 or miR-SCR and the wild-type or mutant SAMD9 3′-UTR
luciferase reporter. Fig. 1B reveals
that miR-96 decreased the luciferase activity of the wild-type
SAMD9 3′-UTR luciferase reporter by ~65% compared with miR-SCR.
However, there was no significant difference between the effects of
miR-96 and miR-SCR on the mutant SAMD9 3′-UTR luciferase reporter.
The results suggest that miR-96 may directly target SAMD9 in NSCLC
cells. In addition, the mutant SAMD9 3′-UTR luciferase reporter
showed markedly higher luciferase activity compared to the
wild-type, possibly due to mutation of the miR-96 binding sequence
eliminating the effect of constitutively expressed miR-96 on the
luciferase reporter.
miR-96 inhibits the expression of
SAMD9 in NSCLC cells
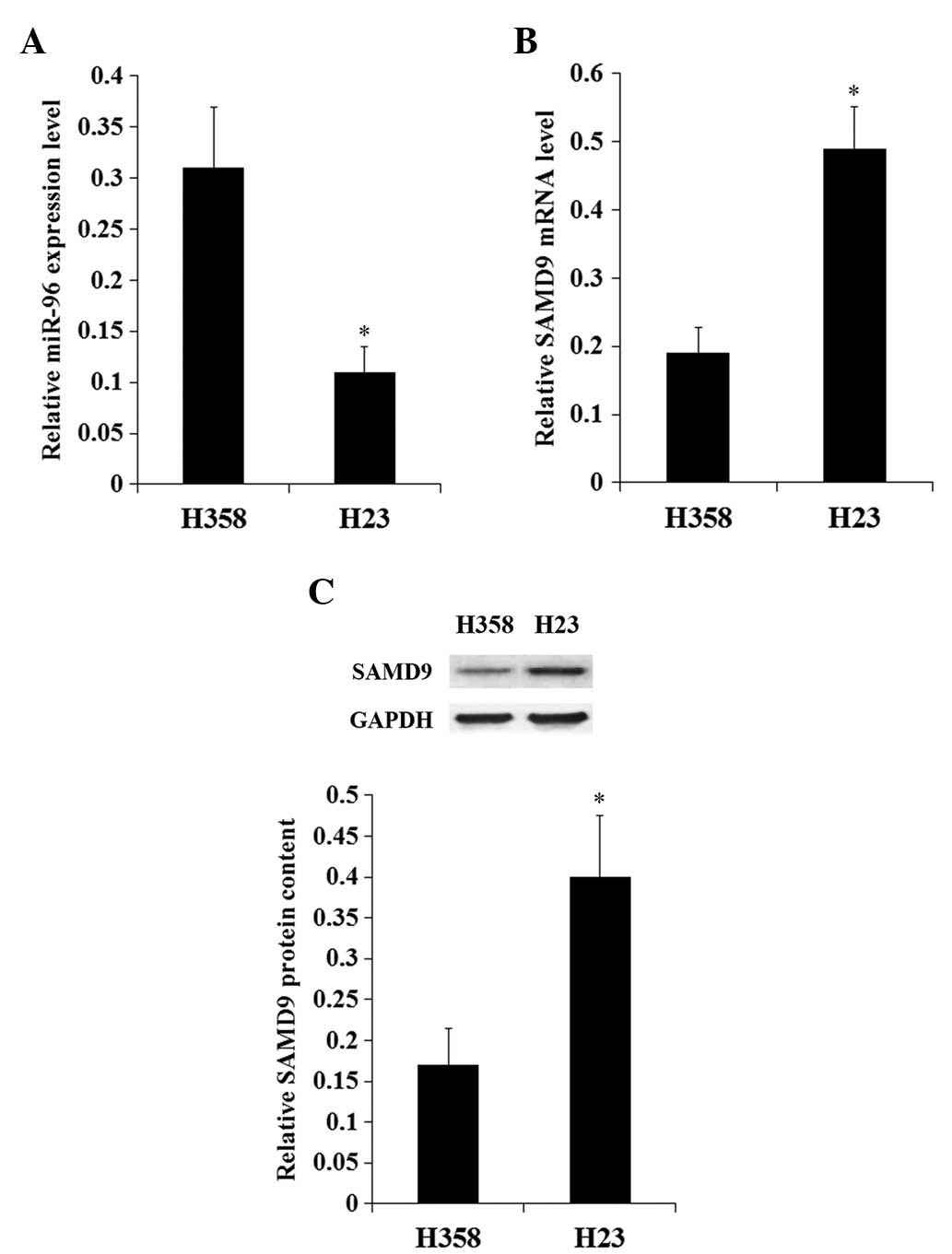
The constitutive expression of miR-96 and SAMD9 in
NSCLC cell lines was investigated. The constitutive expression
level of miR-96 in H358 cells was increased ~3-fold compared with
H23 cells (Fig. 2A). By contrast, the
expression of SAMD9 at the mRNA and protein levels in H358 cells
was <50% that in H23 cells (Fig.
2B). The results suggest a negative association between miR-96
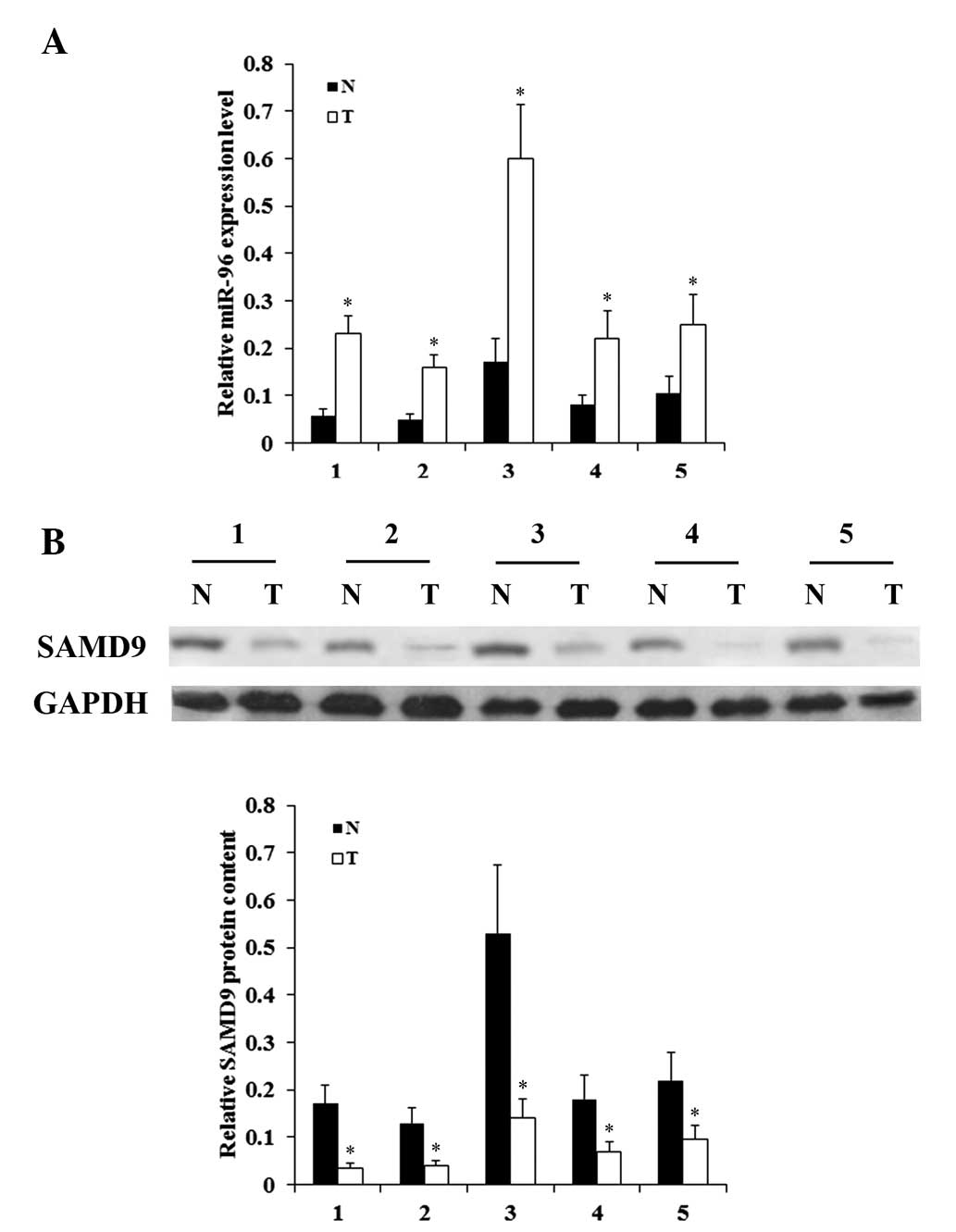
and SAMD9 in NSCLC cells. In addition, the expression levels of
miR-96 and SAMD9 were determined in NSCLC and adjacent normal lung
tissues in 5 consecutive patients, who received no chemotherapy or
radiotherapy prior to surgery. As demonstrated in Fig. 3, while NSCLC tumor samples exhibited
significantly increased expression levels of miR-96 compared with
adjacent normal tissues, the expression levels of SAMD9 were
significantly decreased compared to those in adjacent normal
tissues. The in vitro and in vivo results suggest
that miR-96 is negatively associated with SAMD9 in NSCLC.
To determine the regulatory effects of miR-96 on
SAMD9 expression in NSCLC cells, miR-96 was overexpressed and
knocked down in H358 and H23 cells. Overexpression of miR-96
decreased the constitutive mRNA level of SAMD9 by ~60 and 55% in
H358 and H23 cells, respectively (Fig.
4). By contrast, knocking down miR-96 with antagomir-96
increased the constitutive mRNA level of SAMD9 by ~3- and 2-fold in
H358 and H23 cells, respectively (Fig.
4). Compared with the controls, stable overexpression of SAMD9
(SAMD9 cDNA + 3′-UTR) in H358 cells increased the mRNA level of
SAMD9 by >4.5 fold, which was reversed by overexpression of
miR-96 (Fig. 4A). Stable transduction
of H23 cells with lentiviral SAMD9-shRNA knocked down the SAMD9
mRNA level by ~80%, which was only partially reversed by
antagomir-96 (Fig. 4B). Similar data
trends were observed at the protein level of SAMD9 in H358 and H23
cells (Fig. 5).
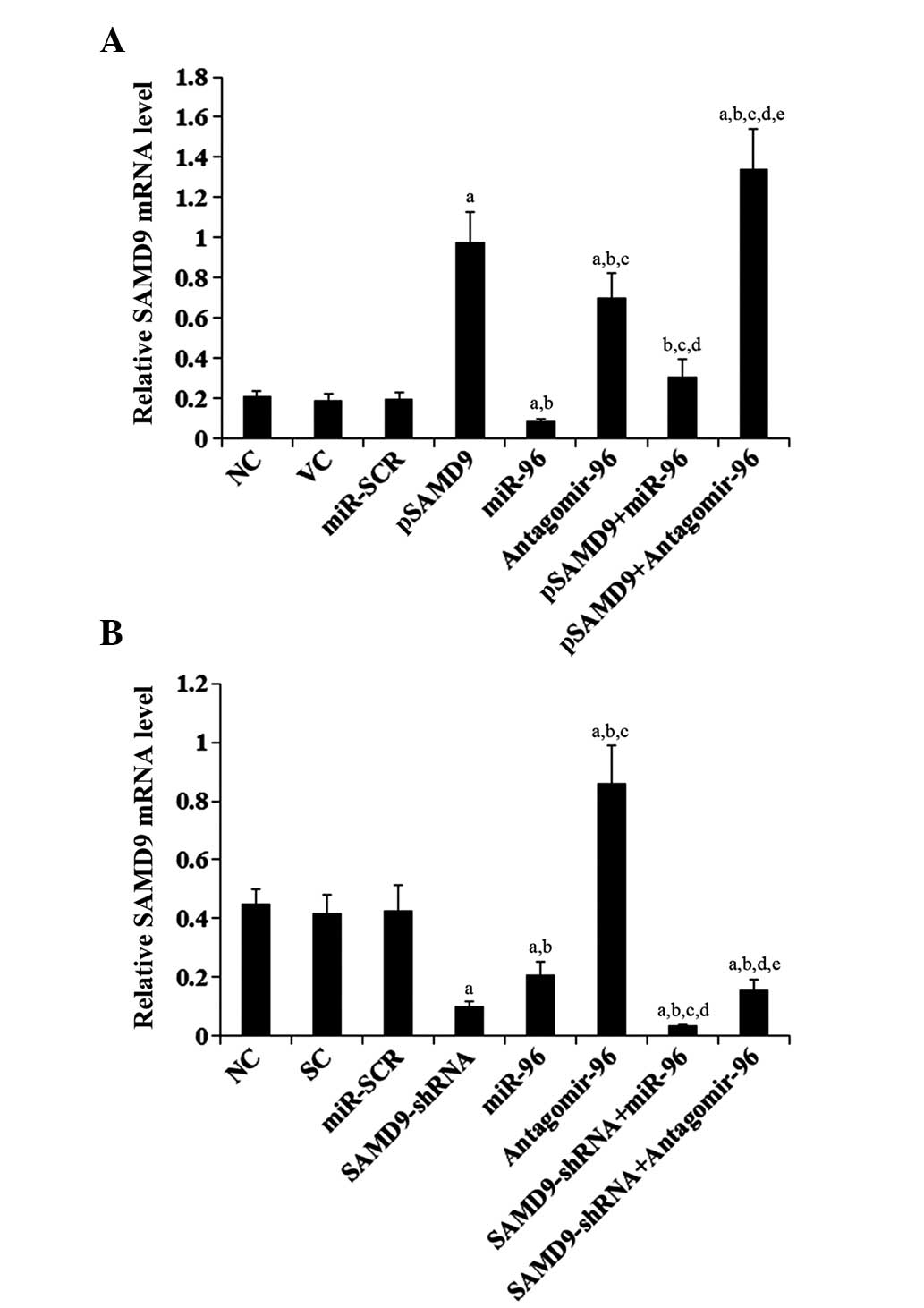 | Figure 4.Effect of miR-96 on SAMD9 mRNA levels
in non-small cell lung cancer cells (A) In H358 cells, the SAMD9
mRNA level was determined with RT-qPCR in NC, cells stably
transfected with VC, transfected with miR-SCR, stably transfected
with pSAMD9, transfected with miR-96 mimics, transfected with
antagomir-96, stably transfected with pSAMD9 + miR-96, and stably
transfected with pSAMD9 + antagomir-96. (B) In H23 cells, the SAMD9
mRNA level was determined with RT-qPCR in NC cells and cells stably
transduced with SC, transfected with miR-SCR, stably transduced
with SAMD9-shRNA, transfected with miR-96 mimics, transfected with
antagomir-96, stably transduced with SAMD9-shRNA and transiently
transfected with miR-96 mimics, and stably transduced with
SAMD9-shRNA and transiently transfected with antagomir-96. In (A)
H358 cells, aP<0.05 vs. NC, VC and miR-SCR;
bP<0.05 vs. pSAMD9; cP<0.05 vs. miR-96;
dP<0.05 vs. antagomir-96; eP<0.05 vs.
pSAMD9 + miR-96. In (B) H23 cells, aP<0.05 vs. NC, SC
and miR-SCR; bP<0.05 vs. SAMD9-shRNA;
cP<0.05 vs. miR-96; dP<0.05 vs.
antagomir-96; eP<0.05 vs. SAMD9-shRNA + miR-96. miR,
microRNA; SAMD9, sterile α motif domain-containing 9; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction; NC,
normal control cells; VC, pcDNA3.1 plasmid; miR-SCR, scramble miR;
UTR, untranslated region; pSAMD9, pcDNA3.1-(SAMD9 cDNA plus UTR)
plasmid; pSAMD9 + miR-96, pcDNA3.1-(SAMD9 cDNA plus UTR) plasmid
and transiently transfected with miR-96 mimics; pSAMD9 +
antagomir-96, pcDNA3.1-(SAMD9 cDNA plus UTR) plasmid and
transiently transfected with antagomir-96; SC, scramble control
short hairpin RNA. |
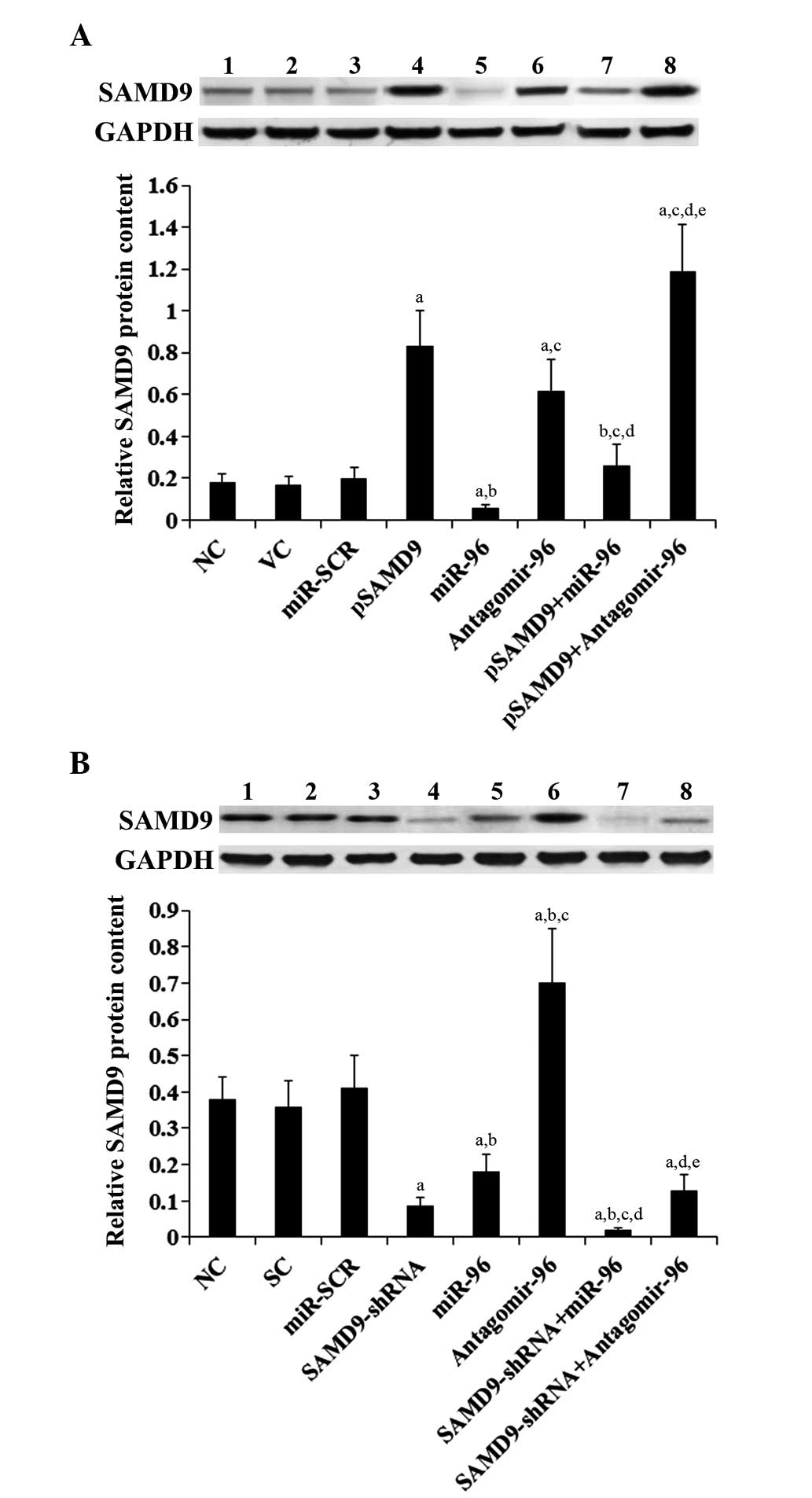 | Figure 5.Effect of miR-96 on SAMD9 protein
levels in non-small cell lung cancer cells. (A) In H358 cells, the
SAMD9 protein level was determined with western blot analysis in NC
(lane 1), cells stably transfected with VC (lane 2), transfected
with miR-SCR (lane 3), stably transfected with pSAMD9 (lane 4),
transfected with miR-96 mimics (lane 5), transfected with
antagomir-96 (lane 6), stably transfected with pSAMD9 + miR-96
(lane 7), and stably transfected with pSAMD9 + antagomir-96 (lane
8). (B) In H23 cells, the SAMD9 protein level was determined with
western blot analysis in NC cells (lane 1), cells stably transduced
with SC (lane 2), transfected with miR-SCR (lane 3), stably
transduced with SAMD9-shRNA (lane 4), transfected with miR-96
mimics (lane 5), transfected with antagomir-96 (lane 6), stably
transduced with SAMD9-shRNA and transiently transfected with miR-96
mimics (lane 7), and stably transduced with SAMD9-shRNA and
transiently transfected with antagomir-96 (lane 8). Density of the
SAMD9 blot was normalized against that of GAPDH to obtain a
relative blot density to represent relative SAMD9 protein content.
In (A) H358 cells, aP<0.05 vs. NC, VC and miR-SCR;
bP<0.05 vs. pSAMD9; cP<0.05 vs. miR-96;
dP<0.05 vs. antagomir-96; eP<0.05 vs.
pSAMD9 + miR-96. In (B) H23 cells, aP<0.05 vs. NC, SC
and miR-SCR; bP<0.05 vs. SAMD9-shRNA;
cP<0.05 vs. miR-96; dP<0.05 vs.
antagomir-96; eP<0.05 vs. SAMD9-shRNA + miR-96. miR,
microRNA; SAMD9, sterile α motif domain-containing 9; NC, normal
control cells; VC, pcDNA3.1 plasmid; miR-SCR, scramble miR; UTR,
untranslated region; pSAMD9, pcDNA3.1-(SAMD9 cDNA plus UTR)
plasmid; pSAMD9 + miR-96, pcDNA3.1-(SAMD9 cDNA plus UTR) plasmid
and transiently transfected with miR-96 mimics; pSAMD9 +
antagomir-96, pcDNA3.1-(SAMD9 cDNA plus UTR) plasmid and
transiently transfected with antagomir-96; SC, scramble control
short hairpin RNA. |
Effect of miR-96/SAMD9 signaling on
cisplatin chemoresistance in NSCLC cells
To explore the individual effect and interaction
between miR-96 and SAMD9 on NSCLC chemoresistance, the cisplatin
IC50 in NSCLC cells was investigated. An increased
IC50 value was considered to correspond with clinical
chemoresistance to cisplatin (13).
As demonstrated in Fig. 6A,
subsequent to 96 h of cisplatin treatment, the cisplatin
IC50 for H358 cells was 1.31 µM. Overexpression of
miR-96 increased the IC50 to 3.78 µM, which was
eliminated by overexpression of SAMD9 (Fig. 6A). By contrast, antagomir-96 decreased
the IC50 to 0.42 µM, which was enhanced by
overexpression of SAMD9 (Fig. 6A). In
H23 cells, the IC50 for cisplatin was 0.74 µM (Fig. 6B). Overexpression of miR-96 increased
the IC50 to 1.52 µM, which was enhanced by knockdown of
SAMD9 (Fig. 6B). By contrast,
antagomir-96 decreased the IC50 to 0.43 µM, which was
eliminated by knockdown of SAMD9 (Fig.
6B). In addition, overexpression of SAMD9 decreased the
IC50 for cisplatin to 0.35 µM in H358 cells (Fig. 6A), and knockdown of SAMD9 increased
the IC50 to 3.05 µM in H23 cells (Fig. 6B).
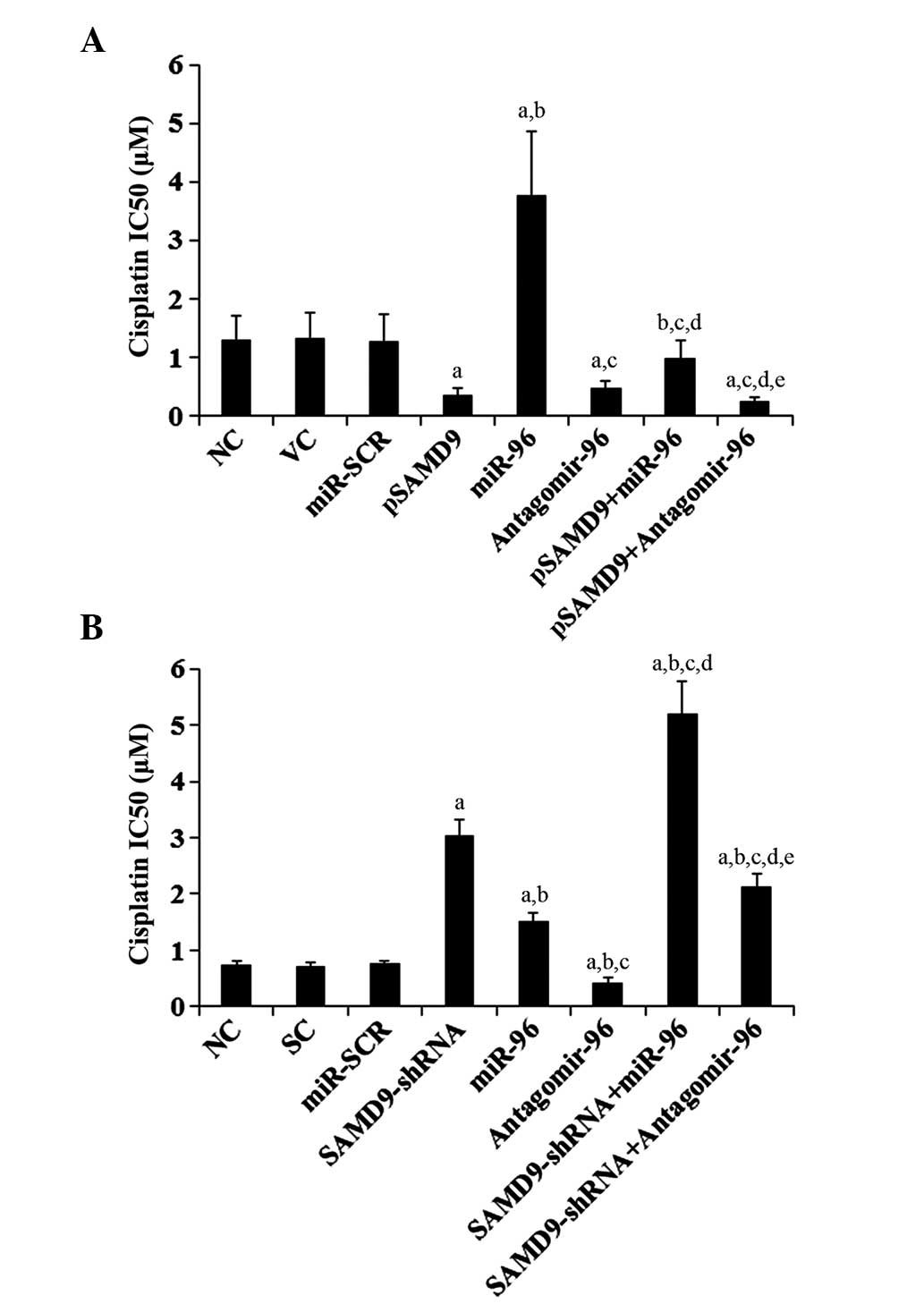 | Figure 6.Effect of miR-96/SAMD9 signaling on
cisplatin chemoresistance in non-small cell lung cancer cells. H358
and H23 cells were treated with or without various concentrations
of cisplatin (0.1, 0.25, 0.5, 1.0, 1.5, 3.0, 6.0, 15.0, 30.0 and
55.0 mM) for 96 h. (A) In H358 cells, IC50 was
determined in NC, cells stably transfected with VC, transfected
with miR-SCR, stably transfected with pSAMD9, transfected with
miR-96 mimics, transfected with antagomir-96, stably transfected
with pSAMD9 + miR-96, and stably transfected with pSAMD9 +
antagomir-96. (B) In H23 cells, IC50 was determined in
NC, cells stably transduced with SC, transfected with miR-SCR,
stably transduced with SAMD9-shRNA, transfected with miR-96 mimics,
transfected with antagomir-96, stably transduced with SAMD9-shRNA
and transiently transfected with miR-96 mimics, and stably
transduced with SAMD9-shRNA and transiently transfected with
antagomir-96. In (A) H358 cells, aP<0.05 vs. NC, VC
and miR-SCR; bP<0.05 vs. pSAMD9;
cP<0.05 vs. miR-96; dP<0.05 vs.
antagomir-96; eP<0.05 vs. pSAMD9 + miR-96. In (B) H23
cells, aP<0.05 vs. NC, SC and miR-SCR;
bP<0.05 vs. SAMD9-shRNA; cP<0.05 vs.
miR-96; dP<0.05 vs. antagomir-96;
eP<0.05 vs. SAMD9-shRNA + miR-96. miR, microRNA;
SAMD9, sterile α motif domain-containing 9; IC50, the
half maximal inhibitory concentration; NC, normal control cells;
VC, pcDNA3.1 plasmid; miR-SCR, scramble miR; UTR, untranslated
region; pSAMD9, pcDNA3.1-(SAMD9 cDNA plus UTR) plasmid; pSAMD9 +
miR-96, pcDNA3.1-(SAMD9 cDNA plus UTR) plasmid and transiently
transfected with miR-96 mimics; pSAMD9 + antagomir-96,
pcDNA3.1-(SAMD9 cDNA plus UTR) plasmid and transiently transfected
with antagomir-96; SC, scramble control short hairpin RNA. |
Effect of miR-96/SAMD9 signaling on
cisplatin-induced apoptosis in NSCLC cells
The individual effect and interaction between miR-96
and SAMD9 on cisplatin-induced apoptosis in NSCLC cells was
investigated. As revealed in Fig. 7,
in untreated H358 and H23 cells at 0 h, overexpression and
knockdown of SAMD9 and/or miR-96 demonstrated no significant effect
on NSCLC cell apoptosis. Subsequent to 48 h of cisplatin (1 µM)
treatment, the percentage of apoptotic cells in H358 cells
increased to ~21.5% (Fig. 7A).
Overexpression of miR-96 decreased cell apoptosis to 15.6%, which
was eliminated by overexpression of SAMD9 (Fig. 7A). Antagomir-96 increased cell
apoptosis to 30.7%, which was enhanced by overexpression of SAMD9
(Fig. 7A). In H23 cells, the cell
apoptosis rate subsequent to 48 h of treatment with 1µM cisplatin
was ~28% (Fig. 7B). Overexpression of
miR-96 decreased cell apoptosis to 23.8%, which was enhanced by the
knockdown of SAMD9 (Fig. 6B). By
contrast, antagomir-96 increased the cell apoptosis rate to 35.7%,
which was eliminated by knockdown of SAMD9 (Fig. 7B). In addition, overexpression of
SAMD9 increased the cell apoptosis rate to 34.9% in H358 cells
(Fig. 7A), and knockdown of SAMD9
decreased the cell apoptosis rate to 17.5% in H23 cells (Fig. 7B).
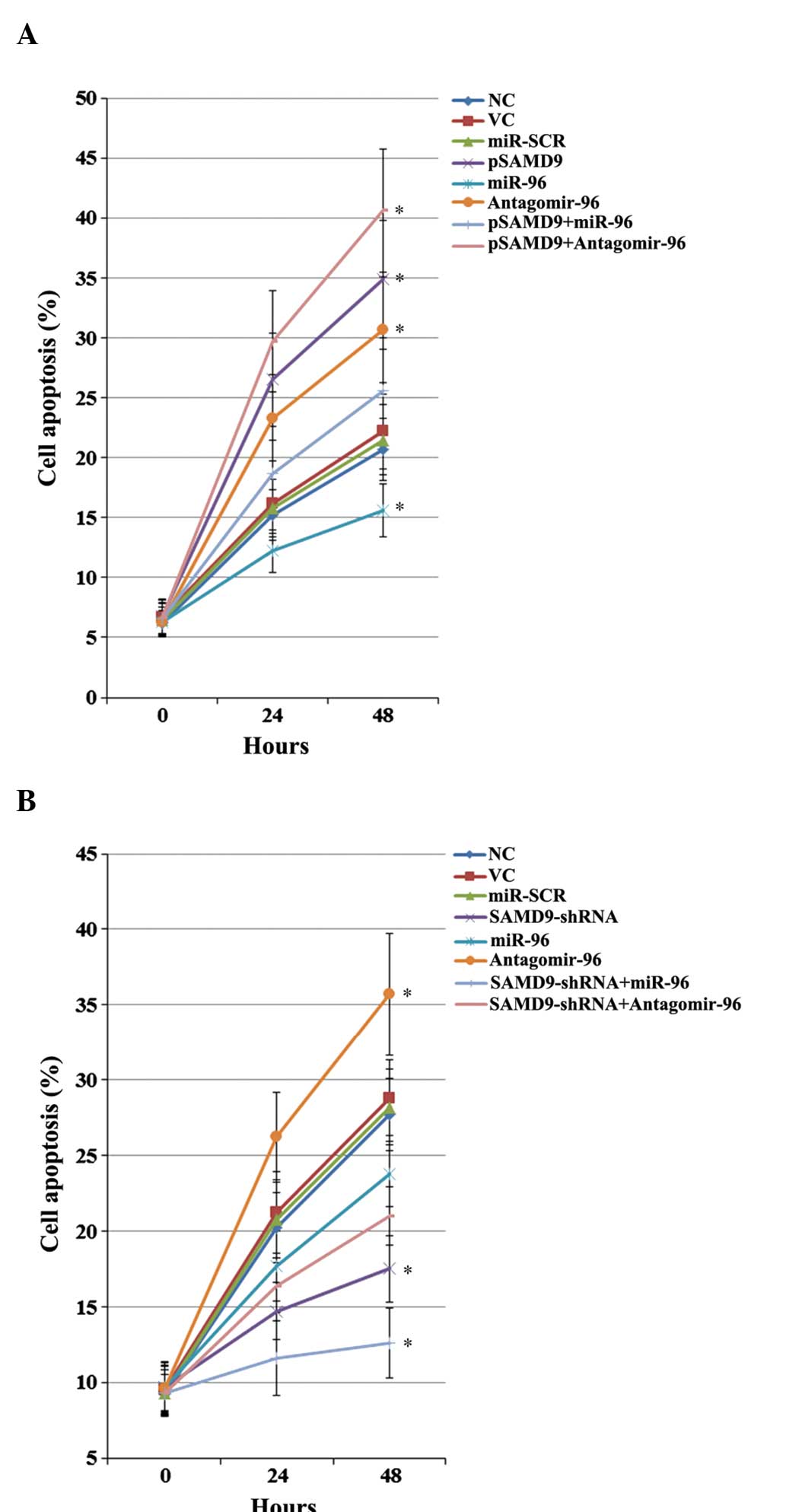 | Figure 7.Effect of miR-96/SAMD9 signaling on
cisplatin-induced apoptosis in non-small cell lung cancer (NSCLC)
cells. H358 and H23 cells were treated with cisplatin (1 µM) for 24
h and 48 h. Apoptosis was measured with a microplate reader-based
TiterTACS in situ apoptosis detection kit. (A) In H358
cells, apoptosis was determined in NC, cells stably transfected
with VC, transfected with miR-SCR, stably transfected with pSAMD9,
transfected with miR-96 mimics, transfected with antagomir-96,
stably transfected with pSAMD9 + miR-96, and stably transfected
with pSAMD9 + antagomir-96. (B) In H23 cells, apoptosis was
determined in NC, cells stably transduced with SC, transfected with
miR-SCR, stably transduced with SAMD9-shRNA, transfected with
miR-96 mimics, transfected with antagomir-96, stably transduced
with SAMD9-shRNA and transiently transfected with miR-96 mimics,
and stably transduced with SAMD9-shRNA and transiently transfected
with antagomir-96. The cell apoptosis rates at 24 h and 48 h were
shown as the percentage of apoptotic cells (as compared to 100%
cell apoptosis induced by nuclease treatment). In (A) H358 cells,
*P<0.05 vs. NC, VC and miR-SCR. In (B) H23 cells, *P<0.05 vs.
NC, SC and miR-SCR. miR microRNA; SAMD9, sterile α motif
domain-containing 9; IC50, the half maximal inhibitory
concentration; NC, normal control cells; VC, pcDNA3.1 plasmid;
miR-SCR, scramble miR; UTR, untranslated region; pSAMD9,
pcDNA3.1-(SAMD9 cDNA plus UTR) plasmid; pSAMD9 + miR-96,
pcDNA3.1-(SAMD9 cDNA plus UTR) plasmid and transiently transfected
with miR-96 mimics; pSAMD9 + antagomir-96, pcDNA3.1-(SAMD9 cDNA
plus UTR) plasmid and transiently transfected with antagomir-96;
SC, scramble control shRNA. |
Discussion
Platinum-based therapy is the mainstay of
chemotherapy for NSCLC (3). Although
platinum-based adjuvant chemotherapy significantly increases the
overall 5-year survival rate of NSCLC patients, the treatment
regimen fails in ~50% of patients, due to intrinsic and acquired
cisplatin resistance (2). SAMD9 is
reportedly a potent tumor suppressor gene (5) that has been demonstrated to inhibit
tumorigenesis and progression of NSCLC (6). The present study reports, to the best of
our knowledge, the first evidence that SAMD9 increases
cisplatin-induced apoptosis and decreases cisplatin chemoresistance
in NSCLC cells.
miRNAs have been identified to play important roles
in the regulation of cancer chemoresistance (7). The aim of the current study was to
identify miRNAs that regulate SAMD9 expression, and the results
revealed that miR-96 directly targets and downregulates SAMD9 in
NSCLC. The results are supported by the following in vivo
and in vitro findings: A negative association between miR-96
and SAMD9 expression in NSCLC and adjacent normal lung tissues;
target-sequence-specific inhibition of the SAMD9 3′-UTR luciferase
reporter by miR-96 in NSCLC cells; and alteration of SAMD9
expression by overexpression and inhibition of miR-96 in NSCLC
cells.
The cisplatin IC50 was employed as a
measure of cisplatin chemoresistance in NSCLC cells. A higher
IC50 value was considered to be associated with clinical
chemoresistance to cisplatin, which is one of the most potent
platinum-based chemotherapeutic agents currently in use (3). miR-96/SAMD9 signaling significantly
altered cisplatin chemoresistance in NSCLC cells. In the presence
of cisplatin, antagomir-96 significantly enhanced cisplatin-induced
apoptosis and decreased cisplatin chemoresistance in NSCLC cells,
suggesting that inhibition of miR-96 may be a potential novel
strategy to enhance chemotherapy for NSCLC. The effects of
antagomir-96 were reversed by knockdown of SAMD9 and enhanced by
overexpression of SAMD9, indicating that miR-96 promotes NSCLC cell
resistance to cisplatin mainly by downregulating SAMD9, or
antagomir-96 suppresses cisplatin chemoresistance by upregulating
SAMD9.
miR-96 has been shown to promote proliferation and
chemoresistance by downregulating reversion-inducing-cysteine-rich
protein with Kazal motifs (RECK) in esophageal cancer (14). A recent study has reported that miR-96
inhibits NSCLC cell apoptosis by targeting forkhead box O3 (FOXO3)
(15). In addition, another recent
study has suggested that miR-96 acts as a tumor suppressor in
pancreatic cancer, and therefore, may act as a useful therapeutic
target for the development of novel anticancer therapies (16). In the present study, miR-96 has been
revealed to inhibit cisplatin-induced apoptosis and induce
cisplatin chemoresistance in NSCLC cells by inhibiting the
expression of SAMD9. Overall, the results suggest that miR-96 plays
a dual role in cancer malignancy and chemoresistance, depending on
tissue specificity. The enhancing effect of miR-96 on NSCLC
chemoresistance, through downregulation of SAMD9 expression, is a
novel function of this miR and miR-96/SAMD9 signaling may be a
novel mechanism involved in the development of NSCLC
chemoresistance. How and whether SAMD9, FOXO3 and possibly RECK
interact with each other to affect cisplatin chemoresistance in
NSCLC cells may be a notable topic for future studies.
Cisplatin elicits DNA repair mechanisms by
crosslinking DNA that in turn, activates apoptosis when DNA repair
is impossible (17). In the present
study, only the effect of miR-96/SAMD9 signaling on cisplatin
chemoresistance in NSCLC cells was investigated; therefore, it is
unclear whether miR-96/SAMD9 is involved in chemoresistance to
other types of chemotherapy agents for NSCLC. Additional studies
with various types of chemotherapy agents and NSCLC cell lines may
resolve this issue. Furthermore, since SAMD9 has been associated
with aggressive fibromatosis and breast and colon cancers (6), it is worth defining the role of
miR-96/SAMD9 signaling in other cancers.
In conclusion, the present study has demonstrated
that miR-96 targets and downregulates SAMD9 in NSCLC, which
decreases cisplatin-induced apoptosis and induces cisplatin
chemoresistance in NSCLC cells. The current study provides novel
insights into the functions of miR-96 and SAMD9 in cancer and the
molecular mechanisms underlying NSCLC chemoresistance.
References
|
1
|
Ma L, Huang Y, Zhu W, et al: An integrated
analysis of miRNA and mRNA expressions in non-small cell lung
cancers. PLoS One. 6:e265022011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Merk J, Rolff J, Dorn C, Leschber G and
Fichtner I: Chemoresistance in non-small-cell lung cancer, Can
multidrug resistance markers predict the response of xenograft lung
cancer models to chemotherapy? Eur J Cardiothorac Surg. 40:e29–e33.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang A: Chemotherapy chemoresistance and
the changing treatment landscape for NSCLC. Lung Cancer. 71:3–10.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Asou H, Matsui H, Ozaki Y, et al:
Identification of a common microdeletion cluster in 7q21.3 subband
among patients with myeloid leukemia and myelodysplastic syndrome.
Biochem Biophys Res Commun. 383:245–251. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li CF, MacDonald JR, Wei RY, et al: Human
sterile alpha motif domain 9, a novel gene identified as
down-regulated in aggressive fibromatosis, is absent in the mouse.
BMC Genomics. 8:922007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ma Q, Yu T, Ren YY, Gong T and Zhong DS:
Overexpression of SAMD9 suppresses tumorigenesis and progression
during non small cell lung cancer. Biochem Biophys Res Commun.
454:157–161. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Feng B, Wang R and Chen LB: Review of
miR-200b and cancer chemosensitivity. Biomed Pharmacother.
66:397–402. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Van Den Broeck A, Ozenne P, Eymin B and
Gazzeri S: Lung cancer, A modified epigenome. Cell Adh Migr.
4:107–113. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartel DP: MicroRNAs: Genomics biogenesis,
mechanism, and function. Cell. 116:281–297. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Filipowicz W, Jaskiewicz L, Kolb FA and
Pillai RS: Post-transcriptional gene silencing by siRNAs and
miRNAs. Curr Opin Struct Biol. 15:331–341. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Byun K, Bayarsaikhan E, Kim CY, Mook-Jung
I, Paek SH, Kim SU, Yamamoto T, Won MH, Song BJ, et al: Induction
of neuronal death by microglial AGE-albumin: Implications for
Alzheimer's disease. PLoS ONE. 7:e379172012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garcia DM, Baek D, Shin C, Bell GW,
Grimson A and Bartel DP: Weak seed-pairing stability and high
target-site abundance decrease the proficiency of lsy-6 and other
microRNAs. Nat Struct Mol Biol. 18:1139–1146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu M, Wang J, Huang H, Hou J, Zhang B and
Wang A: miR-181a-Twist1 pathway in the chemoresistance of tongue
squamous cell carcinoma. Biochem Biophys Res Commun. 441:364–370.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xia H, Chen S, Chen K, Huang H and Ma H:
MiR-96 promotes proliferation and chemo- or radioresistance by
down-regulating RECK in esophageal cancer. Biomed Pharmacother.
68:951–958. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Li P, Chen T, Gao G, Chen X, Du Y,
Zhang R, Yang R, Zhao W, Dun S, Gao F, et al: Expression of
microRNA-96 and its potential functions by targeting FOXO3 in
non-small cell lung cancer. Tumour Biol. 36:685–692. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Feng J, Yu J, Pan X, Li Z, Chen Z, Zhang
W, Wang B, Yang L, Xu H, Zhang G and Xu Z: HERG1 functions as an
oncogene in pancreatic cancer and is downregulated by miR-96.
Oncotarget. 5:5832–5844. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rosenberg B, VanCamp L, Trosko JE and
Mansour VH: Platinum compounds, A new class of potent antitumour
agents. Nature. 222:385–386. 1969. View
Article : Google Scholar : PubMed/NCBI
|