Introduction
Langerhans' cell histiocytosis (LCH) is a rare
disease with a wide spectrum of clinical manifestations (1). The primary pathogenesis of this disease
is an abnormal proliferation of Langerhans' cells, which are
typically present only in the dermis (1). A total of three different terms, namely
eosinophilic granuloma, Hand-Schüller-Christian disease and
Letterer-Siwe disease, were used in the past to describe a group of
disorders characterized by increased numbers of histiocytes
(2). In 1953, Lichtenstein (3) summarized these disorders as
histiocytosis X, due to their similar histopathologies, and in
1987, the term LCH was recommended by The Writing Group of the
Histiocyte Society as the official definition for this syndrome
(4). The clinical appearance of LCH
is dependent on the location of the lesion. The abnormal
accumulation of histiocytes may occur in almost every organ,
including the central nervous system, skin, bone, bone marrow,
lung, liver, spleen and lymph nodes, and may cause associated signs
and symptoms (4). Therefore, LCH may
be classified as monosystemic or plurisystemic, according to the
number of organs involved (5). The
diagnosis of LCH is based on pathological examination. Typical
accumulation of histiocytes, electron microscopic observation of
Birbeck granules and the presence of Langerhans' cell-associated
markers, including cluster of differentiation (CD)1a and S-100
protein, are crucial for establishing a diagnosis of LCH (6). LCH is typically considered to be an
extremely rare disease of childhood, with an incidence rate of
~2-5/1,000,000 children/year (7).
There are no specific signs and symptoms associated with LCH
involving the skull, and the most common presentation is a painful
and immobile scalp mass, which may be palpable in certain cases
(5,8).
Epistaxis or otorrhagia may be exhibited when the lesion invades
the paranasal sinuses or external ear canal (5,8).
The present study reports the case of an 8-year-old
male patient with an LCH lesion in his left temporal fossa, and
aims to provide clinical experience in the diagnosis and treatment
of intracranial LCH.
Case report
An 8-year-old male patient presented at the
Department of Neurosurgery of the First Affiliated Hospital of
Xi'an Jiaotong University (Xi'an, China) in July 2014 with a
1-month history of left temporal pain and headache. Physical
examination revealed slight exophthalmos and conjunctival
hemorrhage in the left eye, with no other positive signs.
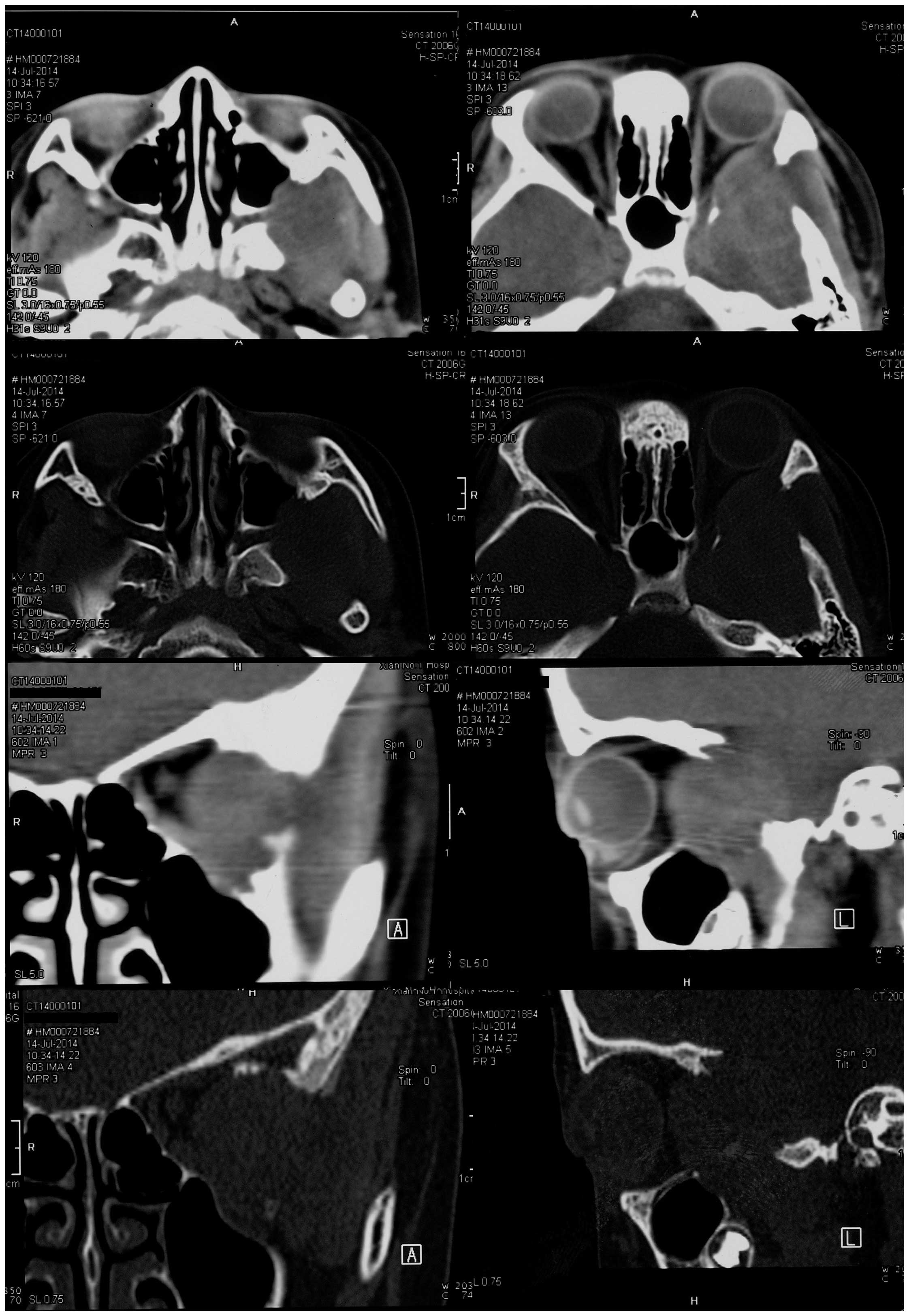
Non-contrast computed tomography (CT; Brilliance 64; Philips
Medical Systems, Inc., Bothell, WA, USA) imaging of the head
revealed a soft tissue mass with unclear margins located in the
left temporal fossa. The mass extended into the patient's left
orbit and maxillary sinus. Bone window CT imaging revealed a wide
bony defect, including part of the greater wing of the left
sphenoid bone, the left lateral orbit and the posterior wall of the
left maxillary sinus (Fig. 1).
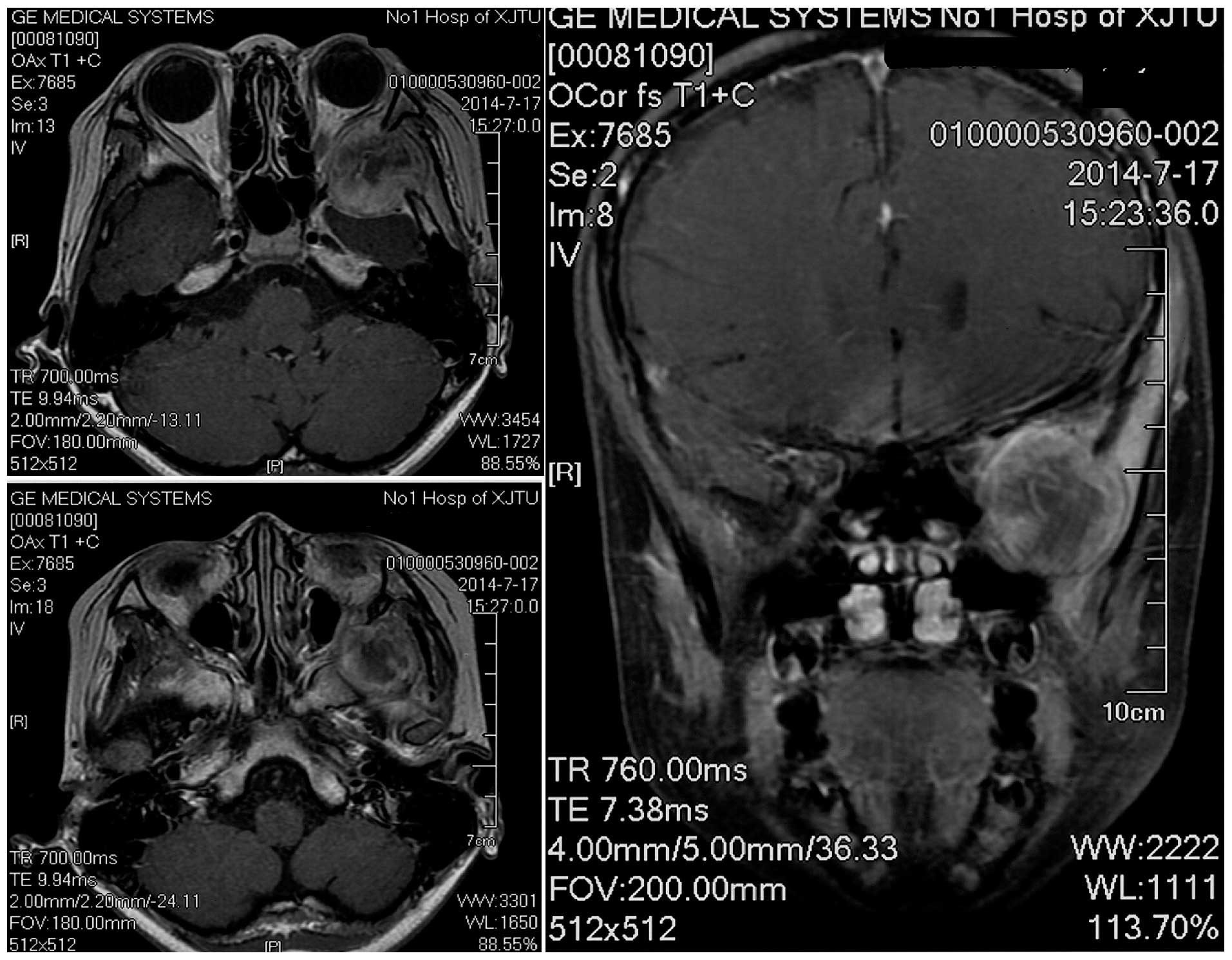
Magnetic resonance imaging (MRI; Signa HDxt 3.0T; GE Healthcare
Bio-Sciences, Pittsburgh, PA, USA) revealed a heterogeneously
contrast-enhancing mass close to the patient's left temporal pole,
eroding into the left orbit and maxillary sinus (Fig. 2). There was a clear plane between the
mass and the left temporal pole. The imaging findings suggested a
diagnosis of meningioma.
The patient underwent resection of the tumor of the
middle cranial fossa. A pterional craniotomy was performed. The
bony defect of the sphenoid bone was observed, and the tumor
extended outside the temporal fossa through this defect. Following
removal of the bone flap, the tumor was revealed to be completely
extradural, but adhered to the dura. Although the patient's left
lateral orbit and the posterior wall of his left maxillary sinus
were eroded by the tumor, the orbital fascia and the mucous
membrane of the maxillary sinus remained complete, and did not
adhere to the tumor. The tumor was removed from around the normal
anatomical structures, and was completely resected under a surgical
microscope (OPMI Pentero; Zeiss AG, Oberkochen, Germany).
Tissues were fixed with 10% formalin (Xi'an Chemical
Reagent Factory, Xi'an, China) for 24 h at room temperature. The
fixed tissues were trimmed and placed in embedding cassettes with
paraffin (Shanghai Huayong Paraffin Wax Co., Ltd, Shanghai, China).
The paraffin-embedded, trimmed blocks were cut into 4-µm sections
and adhered to microscope slides. The paraffin sections were
deparaffinized by xylene (Xi'an Chemical Reagent Factory) and
hydrated routinely, then hematoxylin and eosin staining (ZSGB-BIO,
Beijing, China) was performed.
Histological section of the tumor tissue stained
with hematoxylin and eosin revealed the presence of histocytes,
with moderate amounts possessing eosinophilic cytoplasms and pale
staining nuclei (Fig. 3).
Immunohistochemistry of the tumor tissue revealed positivity for
S-100 (monoclonal mouse anti-human; catalog no., ZM-0224; ZSGB-BIO,
Beijing, China), CD1a (monoclonal rabbit anti-human; catalog no.,
ZA-0544; ZSGB-BIO), vimentin (monoclonal rabbit anti-human; catalog
no., ZA-0511; ZSGB-BIO) and cytokeratin (monoclonal mouse
anti-human; catalog no., ZM-0067; ZSGB-BIO), and negativity for
epithelial membrane antigen (monoclonal mouse anti-human; catalog
no., ZM-0095; ZSGB-BIO), desmin (monoclonal rabbit anti-human;
catalog no., ZA-0610; ZSGB-BIO), CD68 (monoclonal mouse anti-human;
catalog no., ZM-0060; ZSGB-BIO), glial fibrillary acidic protein
(monoclonal rabbit anti-human; catalog no., ZA-0529; ZSGB-BIO),
synaptophysin (monoclonal rabbit anti-human; catalog no., ZA-0236;
ZSGB-BIO) and chromogranin A (monoclonal rabbit anti-human; catalog
no., ZA-0507; ZSGB-BIO). The final pathological diagnosis of the
tumor was LCH.
The patient experienced a good recovery with no
neurological deficits following surgery, and was treated with
chemotherapy (vinblastine 6 mg/m2 intravenously administered once a
week for 6 weeks following surgery; prednisone 40 mg/m2 once a day
administrated orally for 4 weeks following surgery and then
gradually at a reduced dosage over 2 weeks) and radiation treatment
(three dimensional-conformal radiotherapy; total dose, 15 Gy in 1.5
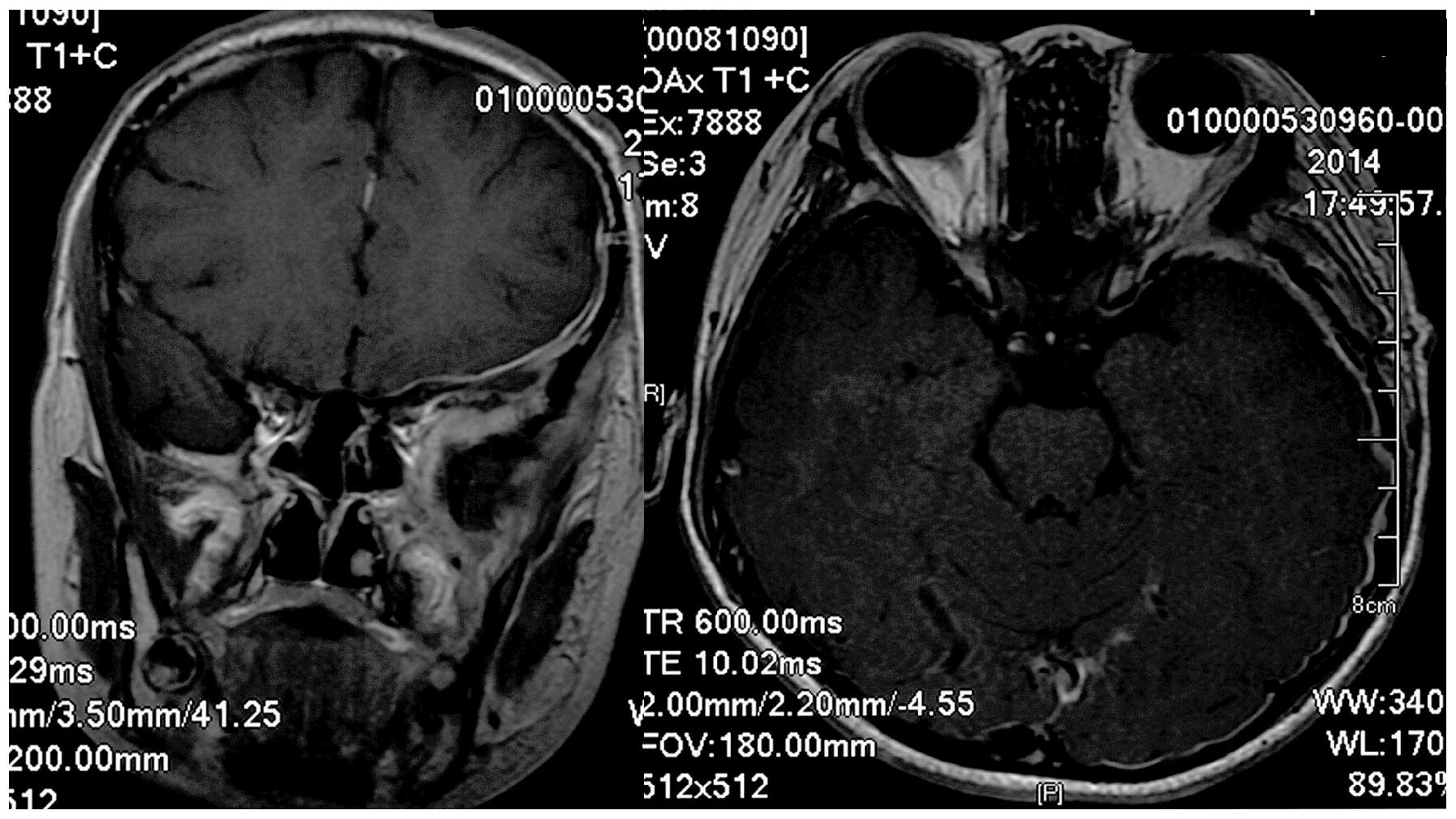
Gy increments). Follow-up MRI revealed no residual tumor matter or
recurrence (Fig. 4). Written informed
consent was obtained from the family of the patient for the
publication of the present study.
Discussion
As mentioned previously, there are no specific signs
and symptoms for LCH involving the skull, and the most common
presentation is a painful and immobile scalp mass, which may be
palpable in certain cases (5,8). In the present study, the patient
experienced left temporal pain and headache, however, there was no
obvious palpable mass. Therefore, imaging examinations of the
cranium were essential in order to locate the lesion.
LCH lesions may develop in the diploic space and are
round or oval-shaped with well-defined margins, which is described
as a ‘punched-out’ image on an X-ray (6). CT scanning is useful for identifying
bony defects that may be eroded by the lesion, while MRI may be
used to identify the intracranial lesion (9). Due to the absence of LCH diagnostic
criteria for imaging examination, meningioma, rhabdomyosarcoma and
Ewing's sarcoma should be considered during differential diagnosis
(9). In the present case, the lesion
was located in the patient's left temporal fossa, and a wide bony
defect was detected. As 60–80% of LCH cases have skull involvement
(8), it was appropriate to
hypothesize that the lesion originated from the skull. In recent
years, positron emission tomography/CT has proven to be the most
sensitive test available for the identification of LCH lesions and
in the evaluation of patient responses to therapy (10,11).
Once the diagnosis of LCH has been confirmed, it is
important to assess the involvement of other organs, as LCH is
typically a systemic disease. According to the current guidelines,
full blood count, liver function, electrolytes, erythrocyte
sedimentation rate, abdominal ultrasound, coagulation studies,
chest radiograph and skeletal radiographic survey should been
performed during this phase (9). In
the present case, all these tests were performed, which confirmed
that no other organs were involved (data not shown).
Due to the potential for development of sequelae,
according to the current guidelines, systemic therapy is
recommended for the treatment of patients with lesions involving
the skull base, orbits and temporal bone (9). Therefore, chemotherapy and radiation
treatment were performed following surgery in the present case. The
standard chemotherapy treatment is based on vinblastine and
steroids, and the clinical response should be evaluated following
the first 6 weeks of treatment (9).
There remains controversy regarding the use of
radiotherapy to treat LCH patients. Certain experts no longer
recommend radiotherapy due to the risk of long-term sequelae
(12). However, other experts
consider radiotherapy to be an effective and safe treatment option
for LCH patients (13,14). Thus, the overall safety of
radiotherapy to treat LCH patients requires additional
investigation in future studies.
In conclusion, the rare case described in the
present report highlights the importance of neurosurgeons to be
familiar with LCH, as this disease frequently involves head tissues
and organs. LCH should be considered during differential diagnosis
in children with imaging examination results suggestive of an
intracranial lesion associated with a bony defect.
References
|
1
|
Saliba I, Sidani K, El Fata F, Arcand P,
Quintal MC and Abela A: Langerhans' cell histiocytosis of the
temporal bone in children. Int J Pediatr Otorhinolaryngol.
72:775–786. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kleinjung T, Woenckhaus M, Bachthaler M,
Wolff JE and Wolf SR: Langerhans' cell histiocytosis with bilateral
temporal bone involvement. Am J Otolaryngol. 24:265–270. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lichtenstein L: Histiocytosis X;
integration of eosinophilic granuloma of bone, Letterer-Siwe
disease, and Schüller-Christian disease as related manifestations
of a single nosologic entity. AMA Arch Pathol. 56:84–102.
1953.PubMed/NCBI
|
|
4
|
The Writing Group of the Histiocyte
Society: Histiocytosis syndromes in children. Lancet. 1:208–209.
1987.PubMed/NCBI
|
|
5
|
Martini A, Aimoni C, Trevisani M and
Marangoni P: Langerhans' cell histiocytosis: Report of a case with
temporal localization. Int J Pediatr Otorhinolaryngol. 55:51–56.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Azouz EM, Saigal G, Rodriguez MM and Podda
A: Langerhans' cell histiocytosis: Pathology, imaging and treatment
of skeletal involvement. Pediatr Radiol. 35:103–115. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Badalian-Very G, Vergilio JA, Degar BA,
Rodriguez-Galindo C and Rollins BJ: Recent advances in the
understanding of Langerhans cell histiocytosis. Br J Haematol.
156:163–172. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Binning MJ and Brockmeyer DL: Novel
multidisciplinary approach for treatment of langerhans cell
histiocytosis of the skull base. Skull Base. 18:53–58. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haupt R, Minkov M, Astigarraga I, Schäfer
E, Nanduri V, Jubran R, Egeler RM, Janka G, Micic D,
Rodriguez-Galindo C, et al: Euro Histio Network: Langerhans
cell histiocytosis (LCH): Guidelines for diagnosis, clinical
work-up, and treatment for patients till the age of 18 years.
Pediatr Blood Cancer. 60:175–184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou W, Wu H, Han Y, Wang S, Dong Y and
Wang Q: Preliminary study on the evaluation of Langerhans cell
histiocytosis using F-18-fluoro-deoxy-glucose PET/CT. Chin Med J
(Engl). 127:2458–2462. 2014.PubMed/NCBI
|
|
11
|
Yamaki T, Kokubo Y, Saito Y, Matsuda K,
Funiu H, Sakurada K, Sato S and Kayama T: A case of Langerhans cell
histiocytosis of the skull in which preoperative methionine
positron emission tomography was useful in comprehending the
spreading of the lesion. Surg Neurol Int. 5:272014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
The French Langerhans' Cell Histiocytosis
Study Group: A multicentre retrospective survey of Langerhans' cell
histiocytosis: 348 cases observed between 1983 and 1993. Arch Dis
Child. 75:17–24. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meyer A, Stark M, Karstens JH,
Christiansen H and Bruns F: Langerhans cell histiocytosis of the
cranial base: Is low-dose radiotherapy effective? Case Rep Oncol
Med. 2012:7896402012.PubMed/NCBI
|
|
14
|
Kriz J, Eich HT, Bruns F, Heyd R, Schäfer
U, Haverkamp U, Büntzel J, Seegenschmiedt H and Micke O:
Radiotherapy in langerhans cell histiocytosis - a rare indication
in a rare disease. Radiat Oncol. 8:2332013. View Article : Google Scholar : PubMed/NCBI
|