Introduction
Cancer is one of the most prevalent causes of
mortality in humans worldwide, with colorectal cancer being the
most common type of malignancy. The occurrence of colorectal cancer
in Asia is increasing, possibly due to dietary habits and lifestyle
factors (1,2). However, the diagnosis of progressive
colorectal cancer is inefficient, and the majority of the presently
offered therapeutic agents may be toxic, expensive and lacking in
efficacy. Cytotoxic chemotherapy has been applied, with limited
success, for treatment of patients with advanced colorectal cancer,
often causing severe side effects (3). Thus, identifying new and effective
chemotherapeutic agents is important for improving the treatment of
this disease. Recent evidence involving targeted therapy recommends
merging cancer prevention and cancer treatment, with inhibition of
cancer being desired over treatment (4,5).
Irinotecan (CPT-11) is a water-soluble,
semisynthetic derivative of camptothecin, which is converted in
vivo to its active metabolite, SN-38. CPT-11 is clinically
active in the treatment of colorectal cancer and exhibits no
cross-resistance (6). CPT-11
demonstrates antitumor activity against a variety of human tumor
xenografts when administered via intravenous, intraperitoneal
(i.p.) or oral routes (7,8). Clinical studies have also revealed that
CPT-11 has significant activity against a range of tumor types,
including colon cancer (9–11). Although CPT-11 has been shown to be
highly effective in the treatment of colon cancer, the dosage is
limited by toxicities, including diarrhea and neurotoxicity
(12,13). Therefore, developing new anticancer
drugs or establishing effective combinations of drugs would greatly
improve colon cancer therapy.
Adamantane and diamantane are closely analogous
polycyclic alkanes, which structurally comprise three and six fused
cyclohexane rings, respectively (14). Diamantane derivatives have been
extensively investigated by chemists; however, limited research
regarding the biological activity of diamantane derivatives has
been reported (15). A previous study
characterized the anticancer activities of diamantane derivatives
using 60 human cancer cell lines in the National Cancer Institute
(NCI) Anticancer Drug Screening, and evaluated the
structure-activity association.
1,6-Bis[4-(4-amino-3-hydroxyphenoxy)phenyl] diamantane (DPD)
demonstrated significant anticancer activity on the sub-panel of
cell lines (16). The
antiproliferative and differentiation-inducing effects of DPD were
observed in human colon cancer cells, and these effects were
irreversible following the removal of DPD (17). DPD has also been reported to induce
apoptosis in human leukemic cells via the elevation of reactive
oxygen species (18). However, the
molecular mechanisms of the combination of DPD and CPT-11, with
special reference to apoptotic signaling, still warrant study.
Therefore, the present study evaluated the effect of DPD and
CPT-11, alone or in combination, on apoptotic pathways and its
mechanisms in colon cancer cell lines. Its in vivo
anticancer effect in colon cancer xenograft mice was also
investigated.
Materials and methods
DPD assay and pharmacokinetic
evaluation
DPD was synthesized and provided by Professor
Yaw-Terng Chern (National Taiwan University of Science and
Technology, Taipei, Taiwan). DPD was weighed and dissolved in
dimethyl sulfoxide (DMSO) to produce 1 mg/ml stock solutions, which
were stored at −20°C when not in use. The standard solution was
prepared by dilution of the stock solution to 10 µg/ml, followed by
further serial dilutions with rat plasma obtained from Wistar rats.
A total of 8 standard solutions of DPD at 1,000, 500, 100, 50, 10,
5, 1 and 0.5 ng/ml were prepared. The internal standard (IS)
2,2-Bis (4-(4-amino-3-hydroxyphenoxy) phenyl) adamantane (DPA)
stock solution, obtained from Professor Yaw-Terng Chern (National
Taiwan University of Science and Technology, Taipei, Taiwan), was
prepared in DMSO at a concentration of 1 mg/ml and was stored at
4°C when not in use. The IS working solution was prepared by
dilution of the IS stock solution to 0.5 µg/ml with
acetonitrile.
The dynamic range of the calibration curve was
0.5–1,000 ng/ml. The calibration standards were freshly prepared
and run on the day when the samples were analyzed. Blank plasma
spiked with known amounts of DPD was prepared and analyzed along
with study samples and plasma standards on the day of analysis. The
quality control (QC) samples (800, 400, 80 and 4 ng/ml) were
prepared from 10, 0.8, 0.4 and 0.08 µg/ml standard solutions in
duplicate. The plasma sample (25 µl) was mixed with 50 µl of
acetonitrile containing the IS, and was then capped, vortexed and
centrifuged at 21,000 × g at ambient temperature for 20 min. The
supernatant portion was transferred to a clean autosampler vial
prior to injection (20 µl) into a liquid chromatography-mass
spectrometry (LC-MS) system. The high performance LC (HPLC) system
consisted of an Agilent 1100 Series HPLC System (Agilent
Technologies, Santa Clara, CA, USA) and a Waters C8 Column (5 µm;
3×150 mm; Waters, Elstree, UK), interfaced to the Agilent HPLC
System with ESI Positive Ion Spray (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Mobile phase consisted of 10 mM ammonium acetate
(Sigma-Aldrich, St. Louis, MO, USA) and acetonitrile
(Sigma-Aldrich) at a ratio of 30:70 (v/v). The flow rate was 0.6
ml/min (total running time, 10 min). The retention times of DPD and
DPA (IS) were 6.6 and 6.8 min, respectively. Nitrogen was used as
the nebulizing gas. The electrospray needle was maintained at 4.5
kV and heated-capillary temperature was set at 400°C. Data
acquisition was via multiple reactions monitoring. Ions
representing the [M+H]+ species for the analyte and IS
were selected in MS1 and collisionally dissociated with nitrogen
gas to form specific product ions, which were subsequently
monitored by MS2. The mass (M+1) for DPD (analyte) and DPA (IS)
were 587 and 536 amu, respectively. Plasma samples that had
concentrations above the upper limit of quantitation (1000 ng/ml)
were diluted proportionally with blank plasma prior to extraction
with acetonitrile. The calibration curve was calculated and plotted
based on the spiked drug concentration per sample. The plasma
calibration curve was generated from drug concentration vs. peak
area ratio, followed by a quadratic or linear regression. The
regression parameters of slope, intercept and correlation
coefficient were calculated by weight (1/x) linear regression in
Analyst® version 1.3 software (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The calibration curves require a
correlation coefficient r>0.999. The plasma concentrations in
the QC and unknown samples were calculated from the calibration
curve. The lower limit of quantitation was 0.5 ng/ml, calculated
from the lowest concentration of the calibration curve.
Rats
In total, 8 male Wistar albino rats, weighing
250–350 g each (8–10 weeks old), were obtained from BioLASCO
(Taipei, Taiwan). The rats were housed individually and fed a
laboratory standard diet (Purina Laboratory Rodent Chow 5001;
Ralston Purina Co., St. Louis, MO, USA) ad libitum. Animals
were handled according to The Guide for the Care and Use of
Laboratory Animals (19). A single 2
mg/kg (intravenous to the tail vein) or 20 mg/kg (orally) dose of
DPD was separately administered to two groups (4 rats/group). At 0
(prior to dosing), 0.033, 0.083, 0.25, 0.67, 1, 1.5, 2, 4, 6, 9, 24
and 27 h following dosing, a blood sample (~150 µl) was collected
from each animal via the jugular-vein cannula and stored on ice
(0–4°C). Plasma was separated from the blood by centrifugation
(14,000 × g for 15 min at 4°C; Allegra® 6R; Beckman
Coulter, Inc., Brea, CA, USA) and stored in a freezer (−60°C). All
samples were analyzed for the parent compound by LC-MS. Data were
acquired via multiple reaction monitoring. Plasma concentration
data were analyzed using a standard non-compartmental method with
Phoenix WinNonLin version 3.1 software (Pharsight; Certara, L.P.,
Princeton, NJ, USA).
Cell culture and DPD treatment
The colon cancer cell line COLO 205 (CCL-222™) was
purchased from the American Type Culture Collection (Manassas, VA,
USA). COLO 205 cells were cultured in Hyclone RPMI-1640 with 10%
fetal bovine serum (GE Healthcare Life Sciences, Logan, UT, USA).
Cells were incubated in a humidified atmosphere of 5%
CO2 in air at 37°C. DPD was dissolved in DMSO at a stock
concentration of 10 mM and added to culture media at a final
concentration of 0.5–8 µM. Cells were seeded at 6×105
cells/60-mm dish in the growth medium. The following day, the cells
were replenished with a medium containing DPD. Cells were harvested
and counted by hemocytometer at 24, 48 and 72 h after treatment
with DPD and used for further analysis.
Microarray analysis
COLO 205 cells were treated with DPD (2 µM) for 24
h. Cells were harvested and 0.2 µg of total RNA was purified using
a Qiagen RNeasy Mini Kit (Qiagen, Inc., Valencia, CA, USA) and
labeled with Cy3 (GE Healthcare Life Sciences) during the in
vitro transcription process. Cy3-labeled cRNA (0.6 µg) was
fragmented to an average size of 50–100 nucleotides by incubation
with a fragmentation buffer (Asia BioInnovations Corporation,
Taipei, Taiwan) at 60°C for 30 min. Corresponding fragmented and
labeled cRNA was then pooled and hybridized to the ABC Human
UniversoChip 20K Microarray (Asia BioInnovations Corporation) at
65°C for 17 h. After washing and drying via nitrogen gun (Asia
BioInnovations Corporation), microarrays were scanned using a
GenePix 4000B Microarray Scanner (Molecular Devices, LLC,
Sunnyvale, CA, USA) at 535 nm for Cy3. Scanned images were analyzed
by GenePix Pro Microarray Acquisition & Analysis Software
v3.0.5.56 (Molecular Devices, LLC); the image analysis and
normalization software was used to quantify signal and background
intensities for each feature. Following the acquisition and initial
quantification of array images, raw array data were normalized per
chip and per gene and filtered based on raw signal intensity and
detection call. Genes with an expression fold change of ≥2 between
a treatment (cells treated with 2 µM DPD for 24 h) and a control
(cells treated with DMSO for 24 h) were considered to be
significant. To determine the potential mechanistic network,
transcripts with differential expression were studied using
Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA,
USA).
Chemosensitivity data
Pearson's correlation coefficients of DPD
(#NSC-706831) with Food and Drug Administration-approved
chemotherapy compounds whose mechanism of action is presumptively
known and published on the NCI Developmental Therapeutics Program
(DTP) (20) were used. These data
sets were downloaded from the NCI DTP's NCI-60 screen (discover.nci.nih.gov/cellminer/). Seven
compounds that were inactive in all cell lines were excluded. A
total of 99 drugs were analyzed. Drug sensitivity was measured by
the negative of log10 GI50 (50% growth inhibition).
Trypan blue dye exclusion method
Viability of cells was determined using an exclusion
assay based on trypan blue dye [0.4% in phosphate-buffered saline
(PBS)]. Cultured COLO 205 cells were washed with Hanks' Balanced
Salt Solution (HBSS; Gibco; Thermo Fisher Scientific, Inc.),
incubated with a solution of 0.125% trypsin, 0.05%
ethylenediaminetetraacetic acid (EDTA) and 0.05% glucose dissolved
in HBSS (pH 7.2) for 2 min, and then incubated with trypan blue
solution (1:1 dilution) for 5 min. Finally, cells were transferred
to a Bürker counting chamber and counted by microscopy
(Observer-A1; Carl Zeiss, Oberkochen, Germany). Dead cells were
defined as those stained with the dye. The percentage of living
cells was calculated as the number of viable cells out of the total
number of cells counted.
Western blot analysis
Cells were washed twice with ice-cold PBS and lysed
in a lysis buffer [0.5% proteinase inhibitors cocktail (Calbiochem;
EMD Millipore, Billerica, MA, USA) in 1 ml M-PER mammalian protein
extraction reagent (Thermo Fisher Scientific, Inc.)]. Cell lysates
were centrifuged at 12,000 × g for 30 min at 4°C, and supernatants
were separated. Protein concentration was measured using a
bicinchoninic acid protein assay kit (Thermo Fisher Scientific,
Inc.). After boiling for 5 min in the presence of 2-mercaptoethanol
(MP Biomedicals, LLC, Santa Ana, CA, USA), samples containing cell
lysate protein were electrophoretically separated on 10% or 7.5%
sodium dodecyl sulfate-polyacrylamide gels and then transferred
onto equilibrated nitrocellulose membranes. Following blocking in
skimmed-milk, the membranes were incubated with the following
primary antibodies: rabbit polyclonal anti-caspase-3 and
anti-procaspase-3 (catalog no. 14–264; 1:1,000) (Upstate™; EMD
Millipore); mouse monoclonal anti-poly ADP-ribose polymerase (PARP;
catalog no. P248; 1:1,000) and mouse monoclonal anti-β-actin
(catalog no. A3854; 1:2,500) (Sigma-Aldrich). The bound antibodies
were detected with horseradish peroxidase-labeled rabbit polyclonal
anti-mouse IgG secondary antibody (catalog no. A9044; 1:10,000;
Sigma-Aldrich) and an enhanced chemiluminescence detection kit (EMD
Millipore).
BALB/c-nu mouse tumor xenograft
model
All in vivo experiments were conducted with
ethics committee approval (Animal Research Committee of the
National Research Institute of Chinese Medicine; approval no.
A-100-1), and met the standards required by the UK Co-ordinating
Committee on Cancer Research guidelines (21). The 8-week-old male BALB/c-nu mice were
obtained from the National Laboratory Animal Center of National
Applied Research Laboratories (Taipei, Taiwan) and housed in a
laminar flow room under sterilized conditions with a temperature
maintained at 25°C and light controlled at a 12 h light and 12 h
dark cycle. COLO 205 cells were harvested and resuspended in
serum-free RPMI-1640 medium. Cells were adjusted to a concentration
of 1×107 cells/ml, and 0.1 ml was inoculated into each
mouse. Each experimental group included 6–7 mice bearing tumors.
DPD and CPT-11 were dissolved in DMSO and normal saline,
respectively. Treatment was initiated when tumor size reached 3–5
mm. DPD (18.75 mg/kg), CPT-11 (15 mg/kg) or a combination of DPD
(18.75 mg/kg) and CPT-11 (15 mg/kg) were administered via i.p.
injection twice per week (volume of injection, 0.1 ml/20 g body
weight). The control group received DMSO vehicle. Tumor size and
body weight were monitored twice per week throughout the
experiment. The tumor size was measured using a vernier caliper
twice per week. Tumor size (V) was calculated according to the
formula V (mm3) = 0.4 × A × B2, where A and B
are the longest diameter and the shortest diameter of the tumor,
respectively (22). At the end of the
experiment, all mice were sacrificed using CO2 gas. The
tumors, livers, kidneys and lungs were collected, fixed, embedded
and stained with hematoxylin and eosin for pathological
analysis.
Statistical analysis
All data are expressed as the mean ± standard error
(SE). The differences between the drug treatment groups and control
group were assessed using a Student's t-test with SigmaPlot version
12.5 software (Systat Software, Inc., San Jose, CA, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
DPD assay and pharmacokinetic
profile
Following intravenous administration of DPD (2
mg/kg) to rats, the plasma concentration-time profiles of DPD
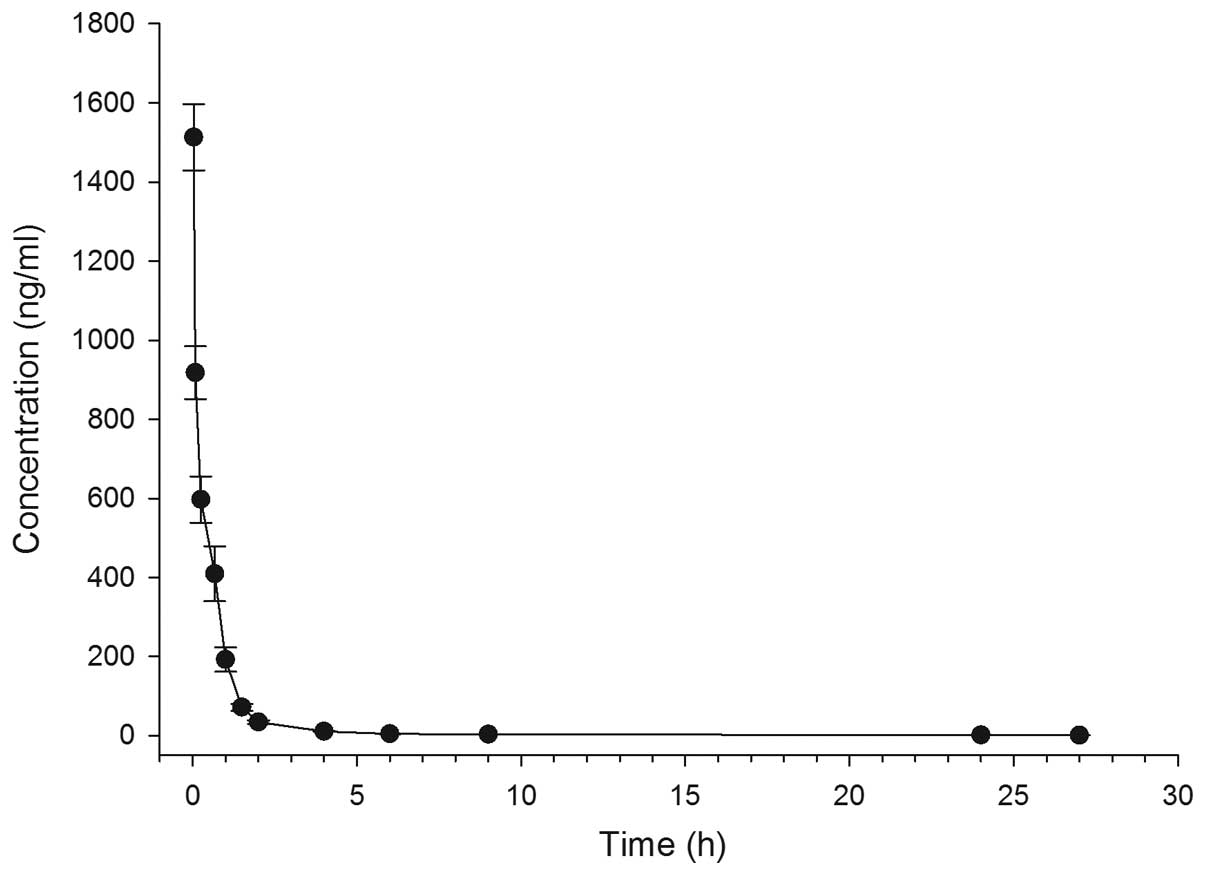
(Fig. 1) and the respective
pharmacokinetic parameters were calculated (Table I). DPD exhibited a rapid distribution
phase and could not be detected at 27 h after injection. As shown
in Table I, the area under the curve
(AUC), elimination of half-life (T1/2),
maximum plasma concentration (Cmax), volume of distribution at
steady state (Vss) and clearance rate of DPD were 0.75±0.13 µg/ml ×
h, 8.0±1.8 h, 1.51±0.16 µg/ml, 4.51±1.28 l/kg, and 2.71±0.52
l/h/kg, respectively.
 | Table I.Plasma pharmacokinetic parameters
after intravenous administration of 2 mg/kg of DPD to rats
(n=4). |
Table I.
Plasma pharmacokinetic parameters
after intravenous administration of 2 mg/kg of DPD to rats
(n=4).
| Parameter | Mean ± standard
error |
|---|
| Clearance rate | 2.71±0.52
l/h/kg |
| Steady state volume
of distribution | 4.51±1.28 l/kg |
|
T1/2 | 8.0±1.8 h |
| Maximum plasma
concentration | 1.51±0.16 µg |
| Area under the
curve | 0.75±0.13 µg/ml ×
h |
Pathway analysis of gene expression
associated with DPD treatment
To identify gene expression signatures that are
associated with biological functions of DPD, microarray and pathway
enrichment analysis was conducted to compare expression patterns in
COLO 205 cells treated with DPD (2 µM) for 24 h. The top ten
pathways identified by the pathway enrichment analysis are
presented in Table II. Notably, the
most prominent transcriptional changes in COLO 205 cells treated
with DPD (2 µM) were enriched for cell cycle and apoptosis pathways
(7/10). The most common pathways associated with DPD treatment
include apoptosis and cell adhesion; other pathways are related to
cancer signaling. The results indicate that DPD may regulate cell
cycle progression and cancer cell apoptosis in COLO 205 cells.
| Table II.Top ten pathways identified by
pathway enrichment analysis of differentially expressed genes in
COLO 205 human colon cancer cells following treatment with DPD (2
µM) for 24 h. |
Table II.
Top ten pathways identified by
pathway enrichment analysis of differentially expressed genes in
COLO 205 human colon cancer cells following treatment with DPD (2
µM) for 24 h.
| Pathway name | P-value | Input nodes | Total nodes | Ratio |
|---|
| Cell cycle control
of chromosomal replication |
1.37×10−8 | 11 | 34 | 11/34 |
| Hereditary breast
cancer signaling |
1.84×10−7 | 20 | 134 | 20/134 |
|
Ataxia-telangiectasia mutated
signaling |
2.77×10−7 | 14 | 66 | 14/66 |
| Mitotic roles of
polo-like kinase |
1.17×10−6 | 14 | 74 | 14/74 |
| Cell cycle: G2/M
DNA damage checkpoint regulation |
2.54×10−6 | 11 | 49 | 11/49 |
| DNA damage-induced
14–3–3σ signaling |
1.06×10−5 | 7 | 22 | 7/22 |
| GADD45
signaling |
1.57×10−5 | 7 | 24 | 7/24 |
| Role of CHK
proteins in cell cycle checkpoint control |
2.53×10−5 | 11 | 59 | 11/59 |
| Molecular
mechanisms of cancer |
6.41×10−5 | 32 | 388 | 32/388 |
| Salvage pathways of
pyrimidine ribonucleotides |
4.11×10−4 | 14 | 96 | 14/96 |
Chemosensitivity data
The significant associations between the DPD
sensitivity profile and each of the 99 drug sensitivity profiles in
NCI-60 cell lines are listed in Table
III. There were 10 chemotherapy drugs that significantly
correlated with DPD sensitivity profiles in NCI-60 cell lines. A
significant negative correlation was found for capecitabine and
anastrozole, whilst a significant positive correlation was observed
for floxuridine, topotecan, nitrogen mustard, gemcitabine,
pemetrexed, megestrol acetate, hydroxyurea and methotrexate. From
this data, an analog of topotecan (a topoisomerase I inhibitor),
CPT-11, was selected to determine whether its combination with DPD
had a multiplied antitumor effect in colon cancer.
| Table III.Correlation between the invasion
profile and each of the 99 drug sensitivity profiles of NCI-60 cell
lines. |
Table III.
Correlation between the invasion
profile and each of the 99 drug sensitivity profiles of NCI-60 cell
lines.
| NSC# | Name | Mechanism | Correlation | P-value |
|---|
| 27640 | Floxuridine |
Antimetabolite/nucleoside | 0.321165 | 0.013130 |
| 609699 | Topotecan |
Anthracycline/topoisomerase poison | 0.288541 | 0.026673 |
| 762 | Nitrogen
mustard | DNA damaging
agent | 0.28534 | 0.028481 |
| 613327 | Gemcitabine |
Antimetabolite/nucleoside | 0.295385 | 0.028566 |
| 712807 | Capecitabine |
Antimetabolite/nucleoside | −0.2889 | 0.032425 |
| 698037 | Pemetrexed |
Antimetabolite/nucleoside | 0.279781 | 0.035049 |
| 71423 | Megestrol
acetate | Hormonal agent | 0.273379 | 0.039627 |
| 32065 | Hydroxyurea |
Antimetabolite/nucleoside | 0.267059 | 0.040882 |
| 740 | Methotrexate |
Antimetabolite/nucleoside | 0.266899 | 0.041007 |
| 719344 | Anastrozole | Hormonal agent | −0.27687 | 0.042682 |
DPD enhances in vitro antitumor
effects of CPT-11 in human colon cancer
The in vitro antitumor effects of DPD and
CPT-11 were tested by trypan blue dye exclusion assay. COLO 205
cells were treated with vehicle control, DPD (1 µM, 2 µM), CPT-11
(6.25 µg/ml) or DPD in combination with CPT-11 for 24, 48 and 72 h.
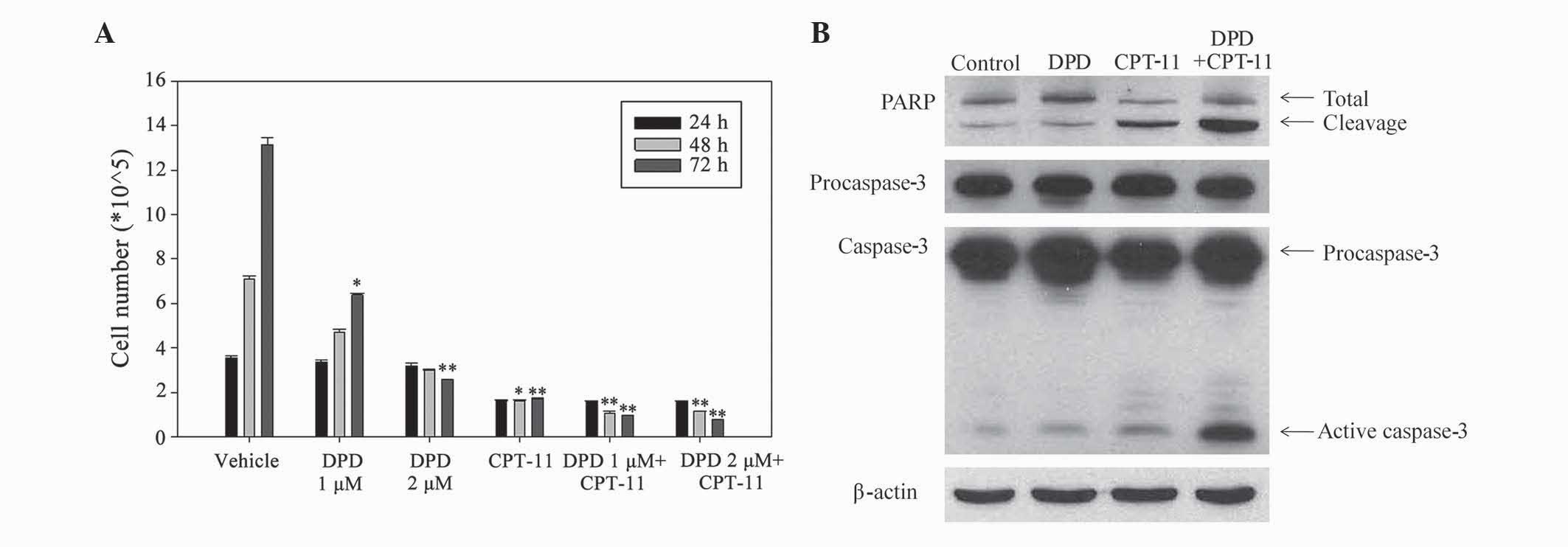
As shown in Fig. 2A, DPD alone had a
positive antitumor effect (1 µM, P=0.04 at 72 hr; 2 µM, P=0.009 at
72 hr), and combination with CPT-11 had a markedly greater
antitumor effect than that of CPT-11 alone. Western blotting showed
that DPD alone induced the active caspase-3 expression. In
addition, DPD combined with CPT-11 increased the active caspase-3
expression significantly (Fig. 2B).
The result suggest that caspase-3 activity is a possible mechanism
of this antitumor effect. The results indicated that PARP and
caspase-3 may contribute to these antitumor effects.
DPD enhances in vivo antitumor effects
of CPT-11 in human colon cancer xenografts
To further investigate whether DPD could enhance the
antitumoral activity of the chemotherapeutic agent CPT-11 in
vivo, COLO 205 cells were transplanted into BALB/c-nu mice.
When the tumors were palpable (3–5 mm), the mice were treated with
vehicle control, DPD (18.75 mg/kg, i.p., twice per week), CPT-11
(15 mg/kg, i.p., twice per week), or DPD in combination with
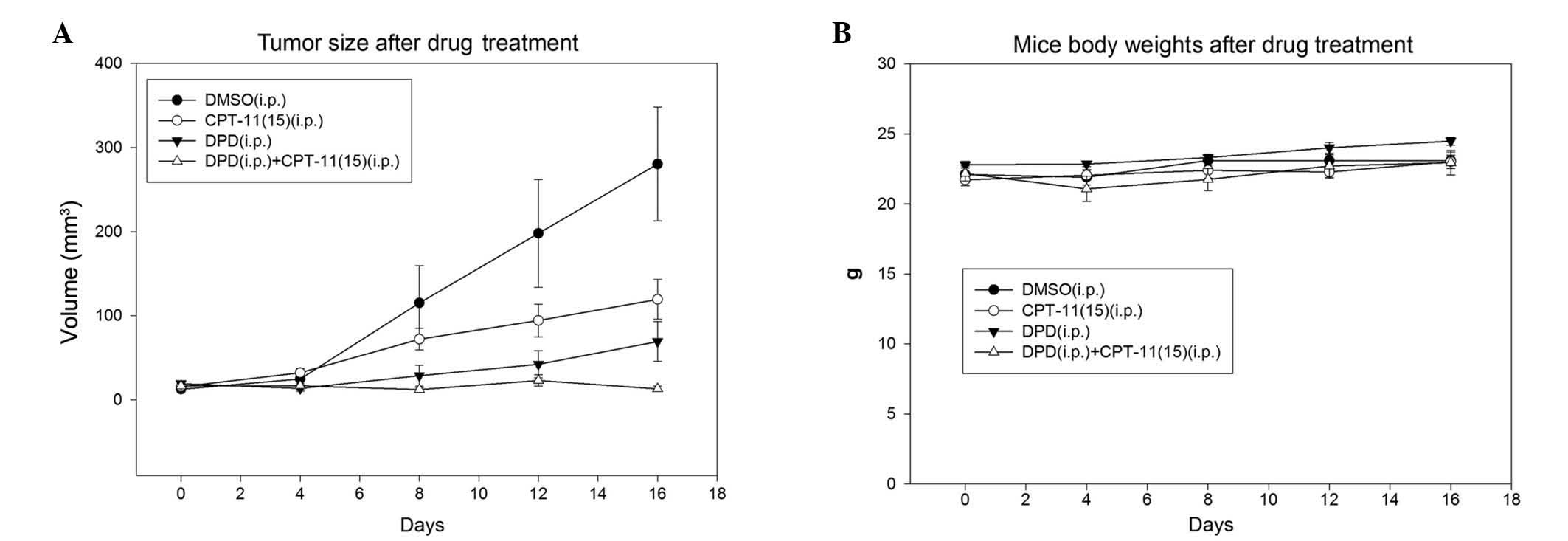
CPT-11. As shown in Fig. 3A, the mean
(±SE) tumor size in the control animals was 280.4±67.6
mm3 at the end of the study. By contrast, the mean tumor
size in the DPD plus CPT-11 combination treatment group was
12.9±3.1 mm3 (P=0.028). The mean tumor sizes of DPD and
CPT-11 single-treatment animals were 69.2±23.6 (P=0.036)and
119.4±23.6 mm3 (P=0.080), respectively. The antitumoral
activity of DPD in combination with CPT-11 showed 5-fold and 9-fold
increases as compared with DPD and CPT-11 alone, respectively.
These results clearly demonstrated that DPD enhances the
antitumoral activity of CPT-11.
Treatment with DPD (18.75 mg/kg, i.p., twice per
week) or DPD in combination with CPT-11 in nude mice produced no
obvious acute toxicity. No significant reduction in body weight was
observed in DPD-treated mice (Fig.
3B). In addition, no tissue damage was observed in the liver,
lungs or kidneys after examination of the hematoxylin and
eosin-stained tissue slices (data not shown).
Discussion
The current study demonstrated, for the first time,
that DPD potentiates the anticancer effects of CPT-11 by the
stimulation of caspase-3 and PARP signaling in vitro, thus
reducing the tumor size of colon cancer xenografts implanted in
mice. A previous study showed that co-treatment with DPA and CPT-11
increased the p53-independent induction of p21/Cip1 and p27/Kip1 in
the tumor tissue of nude mice, and that DPA induced the elevation
of p21/Cip1 and p27/Kip1 in COLO 205 cells in vitro
(23). However, to the best of our
knowledge, no studies have reported a chemotherapeutic effect of
DPD or its apoptosis-related mechanisms. Thus, the present findings
provide novel insight into the potential role and mechanisms of DPD
in the treatment of colon cancer.
Apoptosis is a common and intricate cell suicide
pathway, and is an effective way for the body to eliminate damaged
cells (24). Studies have
demonstrated that components of apoptotic signaling pathways may be
promising targets for the development of novel anticancer agents
(25–28). A number of plant-derived, bio-active
substances have been shown to act as chemopreventive agents via the
induction of apoptosis in various experimental models of
carcinogenesis (29). At present, it
is generally accepted that agents able to induce apoptosis in
cancer cells may have applications in the development of
mechanism-based preventions and treatments for cancer (30). Thus, further elucidation of the
mechanism of action of DPD and CPT-11 may be of significance for
the development for cancer prevention and/or therapy.
Knowledge of the mechanisms of caspase regulation
may aid in the manipulation of apoptosis for therapeutic
applications in human cancer. Caspases, which are constitutively
expressed as latent proenzymes in living cells, affect apoptosis in
a manner dependent upon the specific tissue or cell type, or the
presence of a particular death signal (31). A previous study demonstrated that
cells in which caspase-3 was disrupted, and which were highly
resistant to apoptosis induced by ultraviolet irradiation or
osmotic shock, remained sensitive to apoptosis induced by
γ-irradiation or heat shock (32),
suggesting that caspase-3 is important in cell apoptosis. Caspase-3
is considered to be the major effector caspase among the known
execution caspases, which include caspases-3, −6 and −7 (33). In a previous study using
caspase-3-defective MCF-7 human breast cancer cells, induction of
apoptosis was accompanied by cleavage of PARP without the
corresponding appearance of characteristic DNA internucleosomal
degradation and typical morphological changes associated with
apoptosis (34). Therefore,
additional investigation into the cell morphological changes
following DPD treatment are required. The current findings indicate
that pro- and active caspase-3 are major contributors to apoptosis
in COLO 205 cells. In addition, the results demonstrated that
stimulation of caspase-3 and procaspase activities by DPD and
CPT-11 strongly activated induction of apoptosis with concomitant
stimulation of PARP cleavage.
The observed involvement of the caspase cascade in
DPD- and CPT-11-treated COLO 205 cells provides important insights
to further examine the mechanism. Circumvention of apoptosis is one
of the hallmarks of cancer, and induction of apoptosis during tumor
development is a critical step in chemoprevention (35). In addition to inducing apoptosis in
colon cancer cells, the present study revealed that DPD potentiates
the effects of CPT-11 when administered in combination via
preventing colonic tumor growth in a xenograft model; this suggests
that it has the potential for utilization as a chemotherapeutic
agent for colon cancer.
The current study also identified that combination
treatment with DPD and CPT-11 suppressed tumor growth more
prominently than treatments with either of these drugs alone.
Additionally, pharmacokinetic studies have revealed that CPT-11 was
eliminated faster (T1/2, 1.87±0.43 h)
(36) than DPD
(T1/2, 8.0±1.8 h). By contrast, DPD exhibited
a markedly smaller distribution volume (Vss, 4.51±1.28 l/kg) than
CPT-11 (Vss, 12.50±1.46 l/kg) (36).
This finding suggests that DPD may have a higher protein binding
rate in the circulation, or may exhibit specific accumulation in
vivo. The Cmax (1.51±0.16 µg/ml/ml × h) of DPD was comparable
with that of CPT-11 (Cmax, 1.22±0.13 µg/ml × h) identified by a
previous study (36). However, the
AUC of DPD (0.75±0.13 µg/ml × h) was ~1.77-fold lower than that of
CPT-11 (2.19±0.55 µg/ml × h). This could be explained by the fact
that a 5-fold lower dose of DPD (2 mg/kg) was administered than
that of CPT-11 (10 mg/kg) (36).
These results indicate that DPD may cause fewer side effects than
CPT-11 whilst achieving a comparable Cmax with that of CPT-11, by
applying a relatively lower administration dose.
In conclusion, the current study demonstrates that
DPD potentiates the anticancer effects of CPT-11 by inducing
apoptosis in colon cancer cells. Results from a xenograft model
suggest that the anticancer effects of DPD may arise from induction
of apoptosis. Taken together, these results indicate that DPD is a
potential therapeutic agent for the treatment of colorectal
cancer.
Acknowledgements
The authors would like to thank Professor Yaw-Terng
Chern (National Taiwan University of Science and Technology,
Taipei, Taiwan), who provided the DPD compound used in these
experiments, Professor Chin-Wen Chi (Department of Medical
Research, Taipei Veterans General Hospital, Taipei, Taiwan) and Dr
Shen Yuh-Chiang (National Research Institute of Chinese Medicine,
Taipei, Taiwan) for arranging the animal study.
References
|
1
|
Byeon JS, Yang SK, Kim TI, Kim WH, Lau JY,
Leung WK, Fujita R, Makharia GK, Abdullah M, Hilmi I, et al:
Colorectal neoplasm in asymptomatic Asians: A prospective
multinational multicenter colonoscopy survey. Gastrointest Endosc.
65:1015–1022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pham NM, Mizoue T, Tanaka K, Tsuji I,
Tamakoshi A, Matsuo K, Wakai K, Nagata C, Inoue M, Tsugane S, et
al: Meat consumption and colorectal cancer risk: An evaluation
based on a systematic review of epidemiologic evidence among the
Japanese population. Jpn J Clin Oncol. 44:641–650. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wasserman E, Myara A, Lokiec F, Goldwasser
F, Trivin F, Mahjoubi M, Misset JL and Cvitkovic E: Severe CPT-11
toxicity in patients with Gilbert's syndrome: Two case reports. Ann
Oncol. 8:1049–1051. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Saltz LB, Clarke S, Díaz-Rubio E,
Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS,
Rivera F, et al: Bevacizumab in combination with oxaliplatin-based
chemotherapy as first-line therapy in metastatic colorectal cancer:
A randomized phase III study. J Clin Oncol. 26:2013–2019. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aggarwal BB, Takada Y and Oommen OV: From
chemoprevention to chemotherapy: Common targets and common goals.
Expert Opin Investig Drugs. 13:1327–1338. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Robert J and Rivory L: Pharmacology of
irinotecan. Drugs Today (Barc). 34:777–803. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kawato Y, Furuta T, Aonuma M, Yasuoka M,
Yokokura T and Matsumoto K: Antitumor activity of a camptothecin
derivative, CPT-11, against human tumor xenografts in nude mice.
Cancer Chemother Pharmacol. 28:192–198. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Houghton PJ, Cheshire PJ, Hallman JD II,
Lutz L, Friedman HS, Danks MK and Houghton JA: Efficacy of
topoisomerase I inhibitors, topotecan and irinotecan, administered
at low dose levels in protracted schedules to mice bearing
xenografts of human tumors. Cancer Chemother Pharmacol. 36:393–403.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Karato A, Sasaki Y, Shinkai T, Eguchi K,
Tamura T, Ohe Y, Oshita F, Nishio M, Kunikane H, Arioka H, et al:
Phase I study of CPT-11 and etoposide in patients with refractory
solid tumors. J Clin Oncol. 11:2030–2035. 1993.PubMed/NCBI
|
|
10
|
Armand JP, Terret C, Couteau C and Rixe O:
CPT-11. The European experience. Ann NY Acad Sci. 803:282–291.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rothenberg ML: Irinotecan (CPT-11): Recent
developments and future directions - colorectal cancer and beyond.
Oncologist. 6:66–80. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gamelin E, Gamelin L, Bossi L and
Quasthoff S: Clinical aspects and molecular basis of oxaliplatin
neurotoxicity: Current management and development of preventive
measures. Semin Oncol. 29(5 Suppl 15): 21–33. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hecht JR: Gastrointestinal toxicity or
irinotecan. Oncology (Williston Park). 12(8 Suppl 6): 72–78.
1998.PubMed/NCBI
|
|
14
|
Chen CSH, Shen DM and Wentzek SE:
Preparation of adamantane and diamantane derivatives as antivirals.
Patent WO 9428885 Al. 1994.
|
|
15
|
Chen CSHSD and Wentzek SE: Preparation of
adamantane and diamantane derivatives as antivirals. WO 9428885 Al
(patent). 1994.
|
|
16
|
Wang JJ, Huang KT and Chern YT: Induction
of growth inhibition and G1 arrest in human cancer cell lines by
relatively low-toxic diamantane derivatives. Anticancer Drugs.
15:277–286. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang JJ, Chang YF, Chern YT and Chi CW:
Study of in vitro and in vivo effects of 1,6-Bis
[4-(4-amino-3-hydroxyphenoxy)phenyl] diamantane (DPD), a novel
cytostatic and differentiation inducing agent, on human colon
cancer cells. Br J Cancer. 89:1995–2003. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chang YF, Chi CW, Chern YT and Wang JJ:
Effects of 1,6-Bis [4-(4-amino-3-hydroxyphenoxy) phenyl] diamantane
(DPD), a reactive oxygen species and apoptosis inducing agent, on
human leukemia cells in vitro and in vivo. Toxicol
Appl Pharmacol. 202:1–12. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
National Research Council: Guide for the
Care and Use of Laboratory Animals (8th). The National Academies
Press. Washington, DC: 2011.
|
|
20
|
Holbeck SL, Collins JM and Doroshow JH:
Analysis of food and drug administration-approved anticancer agents
in the NCI60 panel of human tumor cell lines. Mol Cancer Ther.
9:1451–1460. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
United Kingdom Co-ordinating Committee on
Cancer Research (UKCCCR) Guidelines for the Welfare of Animals in
Experimental Neoplasia (Second Edition). Br J Cancer. 77:1–10.
1998. View Article : Google Scholar
|
|
22
|
Attia MA and Weiss DW: Immunology of
spontaneous mammary carcinomas in mice. V. Acquired tumor
resistance and enhancement in strain A mice infected with mammary
tumor virus. Cancer Res. 26:1787–1800. 1966.PubMed/NCBI
|
|
23
|
Wang JJ, Lee JY, Chen YC, Chern YT and Chi
CW: The antitumor effect of a novel differentiation inducer,
2,2-Bis (4-(4-amino-3-hydroxyphenoxy) phenyl) adamantane (DPA), in
combinatory therapy on human colon cancer. Int J Oncol.
28:1003–1012. 2006.PubMed/NCBI
|
|
24
|
Singh RP, Dhanalakshmi S and Agarwal R:
Phytochemicals as cell cycle modulators-a less toxic approach in
halting human cancers. Cell Cycle. 1:156–161. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaufmann SH and Earnshaw WC: Induction of
apoptosis by cancer chemotherapy. Exp Cell Res. 256:42–49. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Makin G and Hickman JA: Apoptosis and
cancer chemotherapy. Cell Tissue Res. 301:143–152. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cotter TG: Apoptosis and cancer: The
genesis of a research field. Nat Rev Cancer. 9:501–507. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang R, Ma L, Weng D, Yao J, Liu X and Jin
F: Gallic acid induces apoptosis and enhances the anticancer
effects of cisplatin in human small cell lung cancer H446 cell line
via the ROS-dependent mitochondrial apoptotic pathway. Oncol Rep.
Mar 17–2016.(Epub ahead of print). View Article : Google Scholar
|
|
29
|
Khuda-Bukhsh AR, Das S and Saha SK:
Molecular approaches toward targeted cancer prevention with some
food plants and their products: Inflammatory and other signal
pathways. Nutr Cancer. 66:194–205. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang L and Yu J: Role of apoptosis in
colon cancer biology, therapy, and prevention. Curr Colorectal
Cancer Rep. 9:2013. View Article : Google Scholar
|
|
31
|
Lavrik IN, Golks A and Krammer PH:
Caspases: Pharmacological manipulation of cell death. J Clin
Invest. 115:2665–2672. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Woo M, Hakem R, Soengas MS, Duncan GS,
Shahinian A, Kägi D, Hakem A, McCurrach M, Khoo W, Kaufman SA, et
al: Essential contribution of caspase 3/CPP32 to apoptosis and its
associated nuclear changes. Genes Dev. 12:806–819. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Slee EA, Adrain C and Martin SJ:
Executioner caspase-3, −6, and −7 perform distinct, non-redundant
roles during the demolition phase of apoptosis. J Biol Chem.
276:7320–7326. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Looi CY, Arya A, Cheah FK, et al:
Induction of apoptosis in human breast cancer cells via caspase
pathway by vernodalin isolated from Centratherum
anthelminticum (L.) seeds. PloS One. 8:e566432013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin Y, Bai L, Chen W and Xu S: The
NF-kappaB activation pathways, emerging molecular targets for
cancer prevention and therapy. Expert Opin Ther Targets. 14:45–55.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou S, Li N, Wang X, et al: In
vitro cytotoxicity, pharmacokinetics and tissue distribution in
rats of MXN-004, a novel conjugate of polyethylene glycol and SN38.
Xenobiotica. 44:562–569. 2014. View Article : Google Scholar : PubMed/NCBI
|