Introduction
Breast cancer is the leading type of cancer in women
worldwide (1). Several therapeutic
strategies including hormone blocking therapy, chemotherapy, and
monoclonal antibodies, are used to treat breast cancer. Paclitaxel
exerts anti-tubulin activity, and is one of a number of
chemotherapeutic agents used in the treatment of patients with
breast cancer (2,3). Paclitaxel inhibits mitotic progression
by interfering with the homeostasis of microtubule assembly, and
induces apoptotic cell death. As has been demonstrated previously
with a number of other chemotherapeutic agents, the emergence of
cancer resistance to paclitaxel is an important issue in the clinic
(4). It has been suggested that
resistance to paclitaxel is associated with the overexpression of
multidrug transporters such as P-glycoprotein1 (also known as
multidrug resistant protein 1) (5),
and alterations in the tubulin/microtubule system (6). Cancer cells that are resistant to
paclitaxel can be established in vitro by culturing them in
the presence of increasing concentrations of paclitaxel for several
months. The final concentration at the end of the establishment
process of paclitaxel resistant cancer cells is far beyond its
GI50 concentration. A recent study has shown that
patients treated with 175 mg/m2 paclitaxel for 3 h had
plasma concentrations ranging from 80–280 nM, and intratumoral
concentrations of 1.1–9.0 µM at 20 h following administration of
the agent (7). These high
intratumoral concentrations are due to the intracellular
accumulation of paclitaxel. In addition, the study showed that
breast cancer cell lines treated with low nanomolar concentrations
of paclitaxel (5–50 nM for MDA-MB-231 cells and 10–50 nM for Cal51
cells), had intracellular concentrations of paclitaxel in the range
of 1–9 µM, which is a clinically relevant concentration range. This
suggests that low nanomolar concentrations of paclitaxel can mimic
intratumoral concentrations. The aim of the present study
therefore, was to examine whether nanomolar concentrations of
paclitaxel, which mimic intratumoral concentrations, are sufficient
to induce death of the TNBC cell line MDA-MB-231 in vitro;
and to isolate and characterize any surviving cells.
Materials and methods
Cell culture and cloning of
MDA-MB-231-JYJ cells
The MDA-MB-231 human breast adenocarcinoma cell line
was obtained from the American Type Culture Collection (Manassas,
VA, USA) and maintained in RPMI 1640 medium supplemented with 10%
heat-inactivated fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and 1% penicillin/streptomycin
(100 µg/ml). Cells were incubated at 37°C in a humidified
atmosphere containing 5% CO2. MDA-MB-231 cells were
treated with paclitaxel (Sigma-Aldrich, St. Louis, MO, USA) at a
concentration of 0.03 µM for 2 days. Any dead cells were removed by
gentle washing with RPMI 1640 medium, and this step was repeated
twice with the surviving cells. The remaining cells were briefly
incubated in the presence of 0.025% trypsin to ensure that only
dead, rounded cells detached from the bottom of the culture plate.
The surviving adherent cells were then passaged again to remove the
majority of cells with different morphologies. Finally, the
remaining round cells were diluted in maintenance culture medium
and seeded in 96-well culture plates. A clone was isolated and
named MDA-MB-231-JYJ.
Cell proliferation assay
MDA-MB-231 and MDA-MB-231-JYJ cells were seeded in
100-mm dishes at a density of 1×105 cells/ml. The cells
were trypsinized at 24, 48, and 72 h and stained with trypan blue.
The number of viable cells was counted using a hemocytometer.
Human tumor xenograft in nude
mice
6-week-old specific-pathogen-free BALB/c nude mice
(Charles River Development, Inc., Tokyo, Japan) were maintained as
previously described (8). The
protocols regarding the use of animals were reviewed by the Korea
University Institutional Animal Care & Use Committee (Seoul,
Korea). MDA-MB-231 cells, suspended in 200 µl of a 1:1 mixture of
phosphate-buffered saline (PBS) (pH 7.4) and BD Matrigel™ (BD
Biosciences, Franklin Lakes, NJ, USA), and MDA-MB-231-JYJ cells
suspended in 200 µl of PBS (pH 7.4), were injected subcutaneously
at a concentration of 6×106 cells/mouse. Tumor volumes
were measured three times a week for 3 weeks using a Vernier
caliper, and subsequently calculated using the formula 0.5 × height
× length × width.
Western blotting analysis
Protein extracts (20 µg) obtained from the cell
lysates were resolved on 8–10% sodium dodecyl sulphate
polyacrylamide gels and transferred to Immobilon-P transfer
membranes as described previously (9). The membranes were incubated in
Tris-buffered saline containing 0.2% Tween-20 and 5% nonfat dried
milk, and probed with rabbit monoclonal antibodies against Src
(36D10) (Cat No. 2110), E-cadherin (24E10) (Cat No. 3195), Notch 1
(C44H11) (Cat No. 3268), cleaved Notch 1 (V744) (D3B8) (Cat No.
4147), and c-Myc (D3N8F) (Cat No. 13987), and with rabbit
polyclonal antibodies against phospho-Src (Y416) (D49G4) (Cat No.
6943), Akt (11E7) (Cat No. 4685), phospho-Akt (S473) (D9E) (Cat No.
4060), Erk1/2 (137F5) (Cat No. 4695), phospho-Erk1/2 (T202/Y204)
(Cat No. 4370), c-Met (D1C2) (Cat No. 8198), phospho-c-Met
(Y1234/1235) (Cat No. 3077), Sox2 (D6D9) (Cat No. 3579), Nanog
(D73G4) (Cat No. 4903), Oct3/4A (C30A3) (Cat No. 2840) and β-actin
(Cat No. A5441). After washing, membranes were probed with a
horseradish peroxidase-conjugated secondary antibody. Detection was
performed using an enhanced chemiluminescent protein detection
system (Amersham Biosciences, Little Chalfont, United Kingdom) and
exposure was carried out using X-ray film. All antibodies were
obtained from Cell Signaling Technologies (Danvers, MA, USA) and
diluted 1:1,000 to 1:2,000 before use except the antibodies against
β-actin (1:10,000).
Cell cycle analysis
Cell cycle distribution was analyzed using a
modification of a previously described protocol (10). Briefly, cells were plated at a density
of 1×106 cells/100-mm dish. Cells were cultured for 48
h, collected, fixed in 70% ethanol, washed, and stained with
Krishan buffer (0.1% sodium citrate, 20 µg/ml RNase A, 0.3% Triton
X-100 and 50 µg/ml propidium iodide, pH 7.4). Samples were
centrifuged, resuspended in 1 ml PBS (pH 7.4) and applied to flow
cytometry using an LSRFortessa™ Cell Analyzer (BD Biosciences, San
Jose, CA, USA). Data were collected for 10,000 events and analyzed
with the FlowJo ver. 9.3.3 (Tree Star Inc., Ashland, OR, USA).
Cell growth inhibition assay
Inhibition of cell growth by anticancer agents was
determined according to the sulforhodamime B (SRB) assay (11). Cells were seeded at a density of
3×104 cells/well in a 96-well plate and incubated for 24
h. Cells were further incubated for 48 h in the presence and
absence of the compounds listed in Table
I, fixed, and stained with SRB. The absorbance was measured at
565 nm.
 | Table I.Comparison of the GI50
concentrations of anticancer agents against MDA-MB-231 and
MDA-MB-231-JYJ cells. |
Table I.
Comparison of the GI50
concentrations of anticancer agents against MDA-MB-231 and
MDA-MB-231-JYJ cells.
|
| GI50
(µM) |
|---|
|
|
|
|---|
| Compounds | MDA-MB-231 | MDA-MB-231-JYJ |
|---|
| SN-38 | 0.034 | 0.018 |
| 5-FU | 2.202 | 2.339 |
| Docetaxel | 0.003 | 0.003 |
| Paclitaxel | 0.008 | 0.021 |
| Dasatinib | 0.014 | N/C |
| Doxorubicin | 0.447 | 0.103 |
Statistical analysis
Graphpad Prism 5.03 (GraphPad Software, Inc., La
Jolla, CA, USA) was used for statistical analysis. One-way ANOVA
and Dunnett's t-test were used for multiple comparisons. P<0.05
was considered to indicate a statistically significant
difference.
Results
Cloning and characterization of
MDA-MB-231-JYJ cells
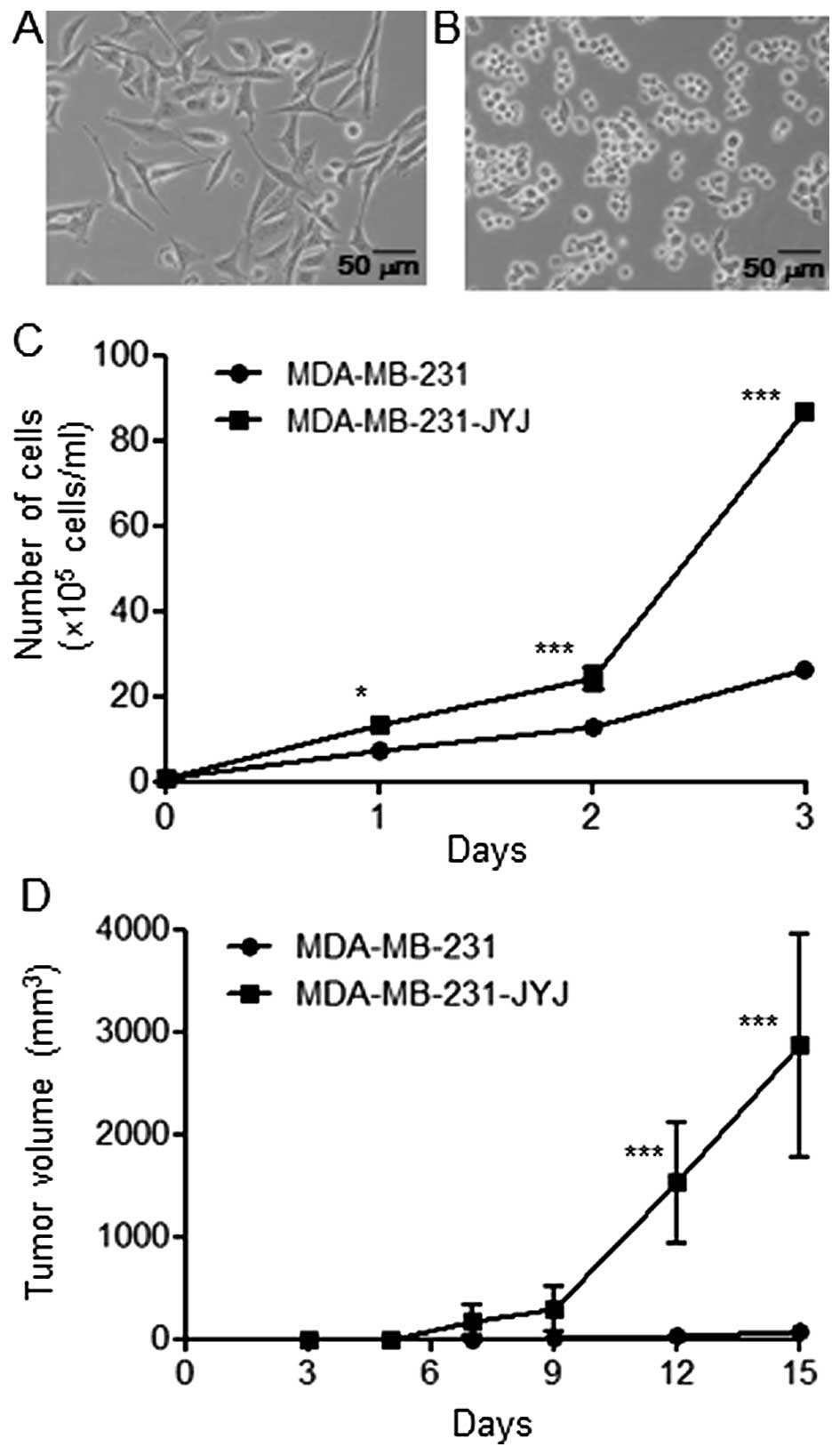
MDA-MB-231 cells cultured in culture medium
containing 10% fetal bovine serum had a flat and spindle shape
(Fig. 1A). After MDA-MB-231 cells
were treated with 0.03 µM paclitaxel (the GI50
concentration) for 4 days, the majority of cells were dead, with
the exception of a few cells with varying morphologies. Among the
surviving cells, the small, round cells were isolated, cloned, and
named MDA-MB-231-JYJ. These cells tended to grow together without
spreading out evenly (Fig. 1B).
Furthermore, MDA-MB-231-JYJ cells were highly proliferative
compared with MDA-MB-231 cells. After 3 days, the number of
MDA-MB-231 cells increased from 1×105 to 26.33±0.88,
while the number of MDA-MB-231-JYJ cells increased to 86.83±1.59
(Fig. 1C), a statistically
significant difference. MDA-MB-231 cells are tumorigenic when they
are transplanted subcutaneously into BALB/c nude mice, producing
small tumors (68.27±72.14 mm3) by day 15. However, the
growth rate of the tumors formed by MDA-MB-231-JYJ cells was
significantly higher, with a mean volume at day 15 of 2865.71±1091
mm3 (Fig. 1D).
Since the rates of proliferation and tumor growth of
MDA-MB-231-JYJ cells were significantly greater than those of
MDA-MB-231 cells (Fig. 1C and D), the
activation status of signal transduction molecules known to be
involved in the regulation of cell survival, proliferation, and
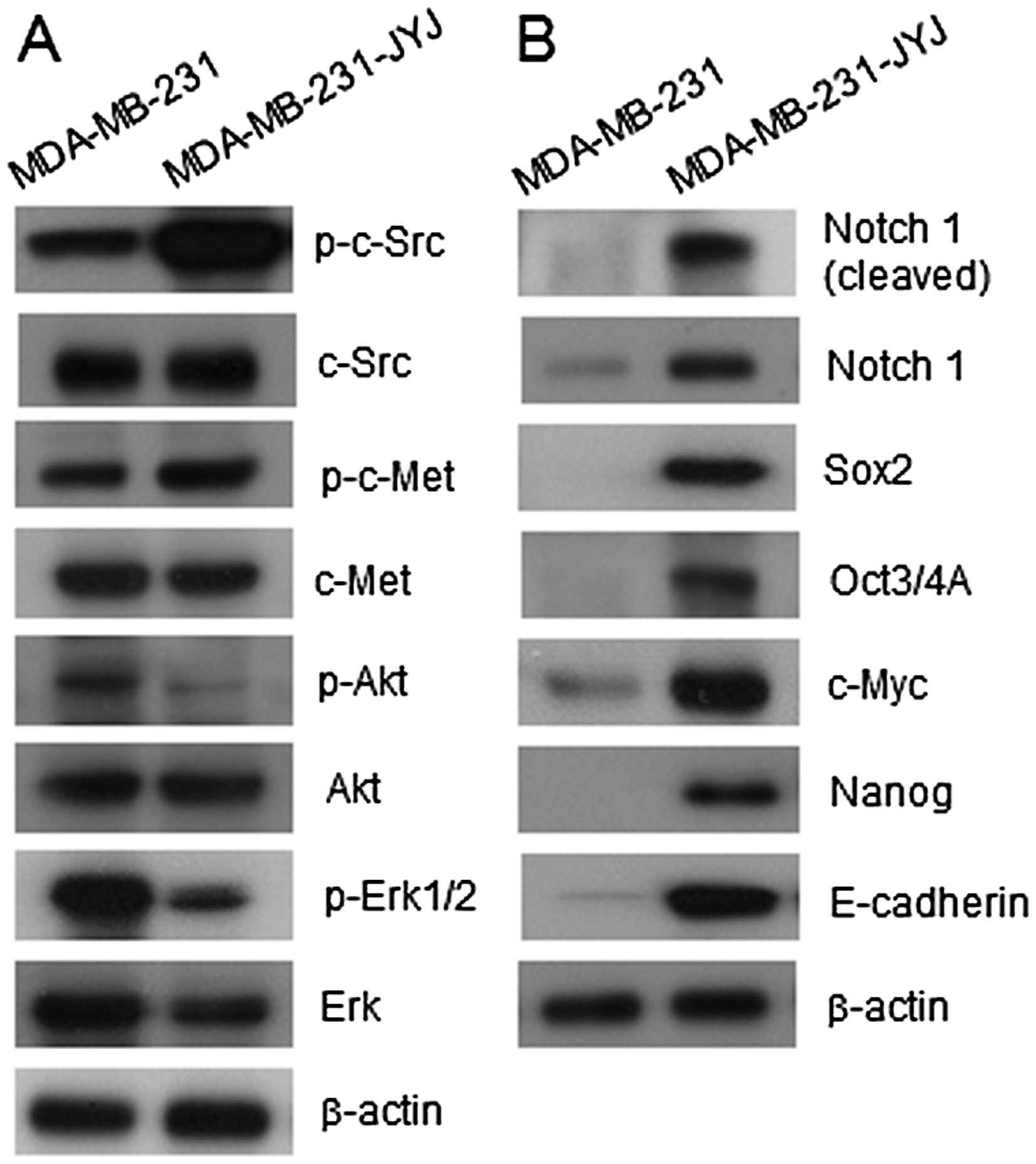
apoptosis was compared between the two cell types (Fig. 2B). Levels of phosphorylated c-Src and
c-Met (also known as hepatocyte growth factor receptor) in
MDA-MB-231-JYJ cells, which are involved in the invasive growth of
cancer, were elevated compared to MDA-MB-231 cells. However, levels
of Akt and phosphor-Erk1/2, which are involved in the regulation of
cell survival, were lower in MDA-MB-231-JYJ cells than in
MDA-MB-231 cells. The activation and expression of signal
transduction molecules that increase the malignancy or stemness of
cancer cells were also compared (Fig.
2B). While the expression and cleavage of Notch 1 was either
barely detected or not detected at all in MDA-MB-231 cells, they
were greatly increased in MDA-MB-231-JYJ cells. Similarly,
expression of Sox2, Oct3/4, c-Myc, Nanog, and E-cadherin was absent
or barely detectable in MDA-MB-231 cells, but the expression of
these molecules was highly increased in MDA-MB-231-JYJ cells.
Selective resistance of MDA-MB-231-JYJ
cells to dasatinib
To examine whether the MDA-MB-231-JYJ cells could
develop resistance to a number of anticancer agents, they were
treated with SN-38 (an active metabolite of irinotecan),
5-fluorouracil (5-FU), docetaxel, paclitaxel, dasatinib, and
doxorubicin, and their GI50 concentrations were
calculated for both MDA-MB-231 and MDA-MB-231-JYJ cells (Table I). Although MDA-MB-231-JYJ cells were
isolated from cells treated with paclitaxel, the GI50
concentrations of paclitaxel in these cells only slightly increased
from 0.008 to 0.021 µM, showing they maintained susceptibility to
the drug. By contrast, MDA-MB-231-JYJ cells were resistant only to
dasatinib of all the anticancer agents tested. While the
GI50 concentration of dasatinib for MDA-MB-231 cells was
0.014 mM, the concentration for MDA-MB-231-JYJ cells was >10 µM,
indicating that these cells had become resistant.
Change in the response of signal
transduction molecules to dasatinib
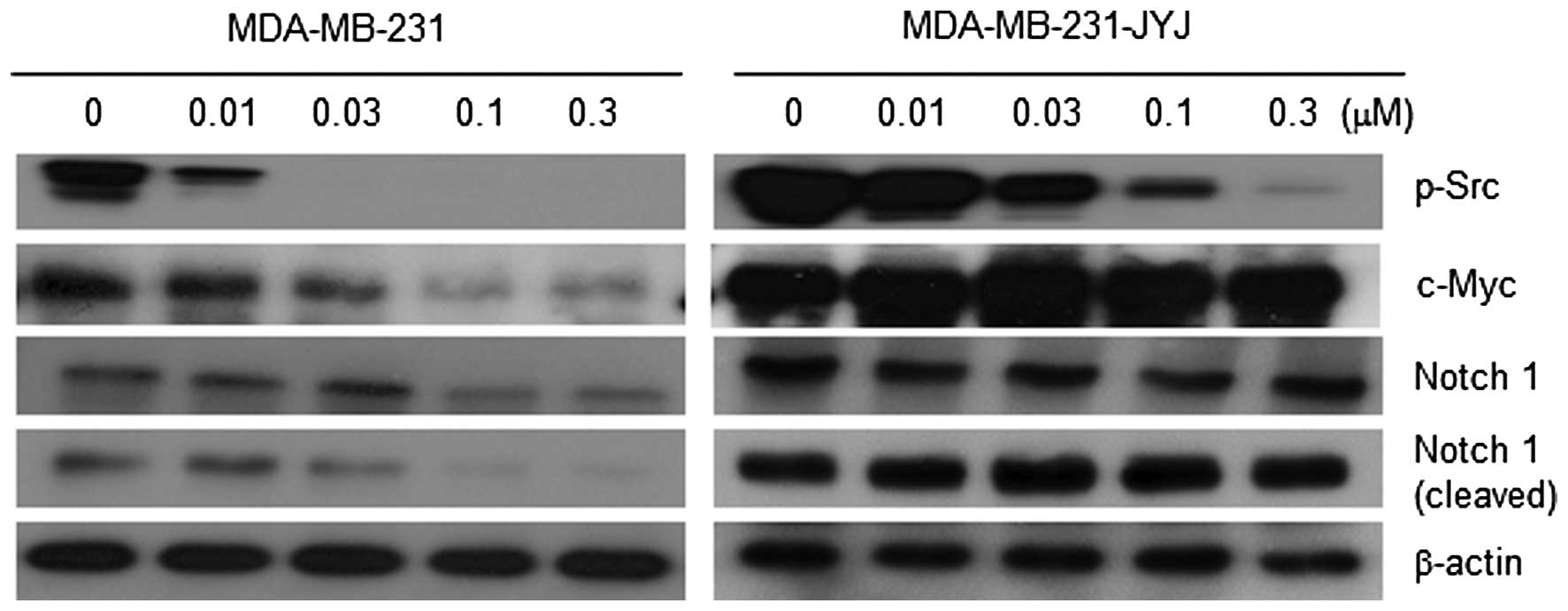
To understand the mechanism by which dasatinib
caused the growth inhibition of MDA-MB-231 cells, the expression
and/or activation status of signal transduction molecules known to
regulate cell survival or proliferation were measured in the
presence and absence of dasatinib. Phosphorylation of c-Src (p-Src)
in MDA-MB-231 cells was almost completely inhibited by 0.01 and
0.03 µM dasatinib. However in MDA-MB-231-JYJ cells, phosphorylation
of c-Src was only slightly inhibited at these concentrations,
meaning the signal transduction pathway regulated by c-Src was
activated at higher concentrations of dasatinib in MDA-MB-231-JYJ
cells (Fig. 3). While the expression
of c-Myc, a representative signal transduction molecule involved in
cell proliferation, gradually decreased with increasing
concentrations of dasatinib in MDA-MB-231 cells, it did not
decrease at all in MDA-MB-231-JYJ cells, even at the highest
concentration of dasatinib. Both the expression and cleavage of
Notch 1, which has been reported to be activated in malignant
tumors (12), were inhibited with
increasing concentrations of dasatinib in MDA-MB-231 cells, but
were unchanged in MDA-MB-231-JYJ cells (Fig. 3).
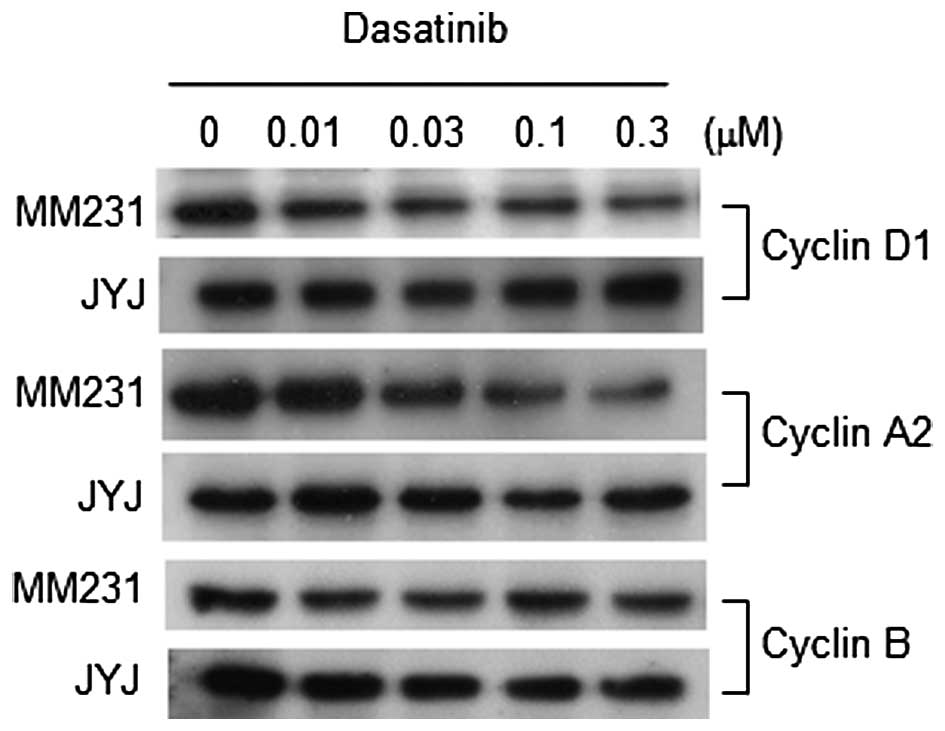
In order to examine whether these changes in signal
transduction regulatory proteins affected cell cycle progression,
levels of cyclins D1, A2 and B were measured in both cell
populations following treatment with dasatinib. As shown in
Fig. 4, dasatinib gradually inhibited
the expression of Cyclin D1 and A2 with increasing concentrations
in MDA-MB-231 cells. It is possible that this inhibition
contributed, at least in part, to the growth inhibition of
MDA-MB-231 cells by dasatinib. However, dasatinib did not inhibit
the expression of Cyclin D1, A2, or B in MDA-MB-231-JYJ cells. Flow
cytometry analysis supports the results obtained with cyclins
(Table II). While dasatinib
decreased the proportion of MDA-MB-231 cells in S-phase and
increased those in the G0/G1 phase, it did
not significantly affect the proportion of MDA-MB-231-JYJ cells in
any phase of the cell cycle.
| Table II.Proportion of MDA-MB-231 and
MDA-MB-231-JYJ breast cancer cells at each phase of the cell cycle
following treatment with dasatinib. |
Table II.
Proportion of MDA-MB-231 and
MDA-MB-231-JYJ breast cancer cells at each phase of the cell cycle
following treatment with dasatinib.
|
| MDA-MB-231 | MDA-MB-231-JYJ |
|---|
|
|
|
|
|---|
| Dasatinib (µM) |
G0/G1 | S | G2/M |
G0/G1 | S |
G2/M |
|---|
| 0 | 64.2 | 7.3 | 22.8 | 49.0 | 10.3 | 23.1 |
| 0.03 | 71.5 | 3.7 | 19.3 | 46.6 |
9.7 | 24.6 |
| 0.1 | 68.3 | 3.1 | 20.8 | 44.3 | 10.3 | 25.3 |
| 0.3 | 74.4 | 2.8 | 19.5 | 40.1 | 10.2 | 27.5 |
Discussion
It has been reported previously that a tumor
contains a variety of cancer cells that are phenotypically,
genetically, and functionally different from each other (13,14).
Cancer cells in a cell line in vitro are more homogeneous
compared with those in a tumor, but they still maintain a certain
level of heterogeneity (15,16). This is one reason why a tumor and
cancer cells in culture respond differently to an anticancer agent.
Since breast cancer also contains many types of stromal and cancer
cells, it is difficult to understand the underlying mechanism in a
tumor treated with an anticancer agent. A breast cancer cell line
may be an easy and simple model to investigate what happens in
cancer cells when they are treated with an anticancer agent.
Although paclitaxel has long been used to treat
patients with breast cancer, its use is limited due to several
significant toxicities such as bone marrow suppression, peripheral
neuropathy, and cardiac disturbances (17). Paclitaxel is usually administered by
intravenous injection to inhibit a sudden increase in its plasma
concentration and to maintain the plasma concentration at an
appropriate range for a certain period of time. Since a tumor
contains a heterogeneous population of cancer cells, there is a
possibility that the tumors in patients treated with paclitaxel
include partially resistant or resistant cancer cells. When
MDA-MB-231 cells were treated with 30 nM paclitaxel for 6 days, the
majority of the cells died, however, a few cells survived that had
different morphologies to the parental cells. Among these, the
majority were round cells and appeared highly proliferative
(Fig. 1B). A small number of large
and flat cells were also observed, but their proliferation rate
seemed very slow. It is believed that this kind of event, evoked by
paclitaxel, may occur more frequently in vivo due to the
fact that a tumor is formed with a complex structure of different
types of cells and tissues, which provides cancer cells with a
protective environment. Although the concentration of paclitaxel
treated in vitro was much lower than that of its plasma
concentration in patients (80–280 nM), it is thought that the
intracellular concentration of paclitaxel accumulated in MDA-MB-231
cells in vitro and in a tumor are similar, in view of a
study conducted by Zasadil et al (7).
Since MDA-MB-231-JYJ cells, the small round cells
that survived treatment with paclitaxel, were much more
proliferative and tumorigenic than MDA-MB-231 cells (Fig. 1C and D), the possibility of that these
cells had acquired resistance to paclitaxel was tested. In
addition, there were great differences in signal transduction
pathways between MDA-MB-231 and MDA-MB-231-JYJ cells (Fig. 2A and B). Signal transduction molecules
involved in the proliferation, survival, angiogenesis, and
metastasis of cancer, such as c-Src and c-Myc (18,19) were
phosphorylated at greater levels in MDA-MB-231-JYJ cells compared
to MDA-MB-231 cells. While the expression and cleavage of Notch 1,
which are involved in the malignancy or stemness of cancer cells
(12,20), were either absent or undetectable in
MDA-MB-231 cells, their expression was elevated and they were
activated in MDA-MB-231-JYJ cells. In addition to Notch 1, other
intracellular molecules that participate in and regulate the
stemness of cells, such as Sox2, Oct3/4, c-Myc, Nanog, and
E-cadherin were highly expressed in MDA-MB-231-JYJ cells. These
results suggest that MDA-MB-231-JYJ cells are more malignant than
their parental cell line and appear to possess some characteristics
of cancer stem/precursor cells. In contrast to the response of the
signal transduction molecules described above, the level of
phosphorylation of Akt and Erk1/2, which are known to promote cell
survival and proliferation (21,22), were
low in MDA-MB-231-JYJ cells compared with MDA-MB-231 cells. Further
study is required, however over-activation of other signal
transduction molecules involved in cell survival and proliferation
such as c-Src, c-Myc, and Notch 1, may circumvent the effect of the
lower level of activation of Akt and Erk1/2.
The GI50 concentrations of several
anticancer agents including paclitaxel and dasatinib were
unexpected (Table I). Although
MDA-MB-231-JYJ cells were isolated and cloned from MDA-MB-231 cells
that survived paclitaxel treatment, the GI50
concentration (0.021 µM) for paclitaxel was only slightly increased
compared with that of the MDA-MB-231 cell line (0.008 µM). This may
be because MDA-MB-231-JYJ cells are a subpopulation of MDA-MB-231
cells that were only partially resistant to paclitaxel. Recent
studies regarding cancer stem cells have shown that cancer contains
several subpopulations of cells, which express different surface
and intracellular markers (23–25). Many
cancer patients, if not all, eventually succumb to pan-resistant
cancer (26). However, partial
resistance is also a problem in the treatment of primary cancer, as
a proportion of cancer cells that are slightly resistant to an
anticancer agent may escape the activity of anticancer agents,
survive, and proliferate in the body. There is a possibility that
cells similar to MDA-MB-231-JYJ may arise from the treatment of
paclitaxel, owing to their partial resistance. This type of
phenomenon has previously been observed with other cancer cell
lines. When LNCaP, a human prostatic adenocarcinoma cell line, was
exposed to paclitaxel, round cells appeared together with large
cells and multinucleated cells (27).
The round cells were observed with optical and electron microscopy,
but were not cloned and studied further for changes in transduction
pathways.
Dasatinib is an oral dual Bcr-Abl and Src family
inhibitor approved for use in patients with chronic myelogeneous
leukemia and Philadelphia chromosome-positive acute lymphoblastic
leukemia (28,29). MDA-MB-231-JYJ cells only exhibited
resistance to dasatinib among the six anticancer agents tested in
the present study. These results raised concern regarding the
combined use of dasatinib and paclitaxel, or similar drugs, in
patients with breast or other types of cancer. Since many clinical
trials using dasatinib are currently being conducted or planned,
elucidation of the mechanism by which MDA-MB-231-JYJ cells become
resistant to dasatinib will help clinical trial researchers to
design their studies to minimize the chances of resistance arising.
As shown in Fig. 3, phosphorylation
of c-Src in MDA-MB-231 cells was effectively inhibited at low
concentrations of dasatinib, however, inhibition of phosphorylation
of c-Src in MDA-MB-231-JYJ cells occurred at higher concentrations
of dasatinib. The cleavage of Notch 1 and the expression of c-Myc
did not decrease in the treated concentration range of dasatinib.
This may explain, at least in part, why MDA-MB-231-JYJ cells were
resistant to dasatinib.
In addition to the effects of dasatinib on the
signal transduction molecules involved in the proliferation,
survival, and stemness of cancer cells, the effect of dasatinib on
the cell cycle itself or on the cellular molecules related to the
cell cycle was also different between MDA-MB-231 and MDA-MB-231-JYJ
cells (Table II and Fig. 4). MDA-MB-231-JYJ cells had a higher
proportion of cells in the S- and G2/M-phases, meaning that they
were more proliferative, and were not affected by dasatinib
treatment, with the cell cycle continuously and actively proceeding
even in its presence. The results regarding the cell cycle may, at
least partly, be explained by the fact that the levels of cyclin
D1, A2, and B in MDA-MB-231-JYJ cells were not affected by the
presence of dasatinib.
In conclusion, MDA-MB-231-JYJ cells were round,
highly proliferative, and tumorigenic. They were resistant to
dasatinib but remained susceptible to paclitaxel. The resistance of
certain signaling molecules and cyclins in MDA-MB-231-JYJ cells to
the inhibitory activity of dasatinib may be the reason why these
cells are resistant to dasatinib. Signal transduction molecules
related to the malignancy or stemness of cancer cells were also
more actively expressed and activated. Further study is required in
order to elucidate whether MDA-MB-231-JYJ cells are genetically or
epigenetically different from their parental line, and whether the
changes in the response of signal transduction molecules are
somehow manageable. However, cancer cells may become resistant to
dasatinib during the process of paclitaxel therapy, and this must
be taken into consideration by researchers and medical
practitioners.
Acknowledgements
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea,
funded by the Ministry of Education (grant no. 2014R1A1A2059237)
and by a grant from the Korea Research Institute of Bioscience and
Biology research initiative program.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crown J, O'Leary M and Ooi WS: Docetaxel
and paclitaxel in the treatment of breast cancer: A review of
clinical experience. Oncologist. 9(Suppl 2): S24–S32. 2004.
View Article : Google Scholar
|
|
3
|
Gradishar WJ: Taxanes for the treatment of
metastatic breast cancer. Breast Cancer (Auckl). 6:159–171.
2012.PubMed/NCBI
|
|
4
|
Galletti E, Magnani M, Renzulli ML and
Botta M: Paclitaxel and docetaxel resistance: Molecular mechanisms
and development of new generation taxanes. ChemMedChem. 2:920–942.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Krishna R and Mayer LD: Multidrug
resistance (MDR) in cancer. Mechanisms, reversal using modulators
of MDR and the role of MDR modulators in influencing the
pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 11:265–283.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Orr GA, Verdier-Pinard P, McDaid H and
Horwitz SB: Mechanisms of Taxol resistance related to microtubules.
Oncogene. 22:7280–7295. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zasadil LM, Andersen KA, Yeum D, Rocque
GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME and Weaver BA:
Cytotoxicity of paclitaxel in breast cancer is due to chromosome
missegregation on multipolar spindles. Sci Transl Med.
6:229ra432014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim HM, Lim J, Park SK, Kang JS, Lee K,
Lee CW, Lee KH, Yun MJ, Yang KH, Han G, et al: Antitumor activity
of cytokine-induced killer cells against human lung cancer. Int
Immunopharmacol. 7:1802–1807. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kang MR, Park SK, Lee CW, Cho IJ, Jo YN,
Yang JW, Kim JA, Yun J, Lee KH, Kwon HJ, et al: Widdrol induces
apoptosis via activation of AMP-activated protein kinase in colon
cancer cells. Oncol Rep. 27:1407–1412. 2012.PubMed/NCBI
|
|
10
|
Krishan A: Rapid flow cytofluorometric
analysis of mammalian cell cycle by propidium iodide staining. J
Cell Biol. 66:188–193. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kang MR, Kang JS, Yang JW, Kim BG, Kim JA,
Jo YN, Lee K, Lee CW, Lee KH, Yun J, et al: Gene expression
profiling of KBH-A42, a novel histone deacetylase inhibitor, in
human leukemia and bladder cancer cell lines. Oncol Lett.
3:113–118. 2012.PubMed/NCBI
|
|
12
|
Li L, Zhao F, Lu J, Li T, Yang H, Wu C and
Liu Y: Notch-1 signaling promotes the malignant features of human
breast cancer through NF-kB activation. PLoS One. 9:e959122014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meacham CE and Morrison SJ: Tumour
heterogeneity and cancer cell plasticity. Nature. 501:328–337.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Burrell RA, McGranahan N, Bartek J and
Swanton C: The causes and consequences of genetic heterogeneity in
cancer evolution. Nature. 501:338–345. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Croce MV, Colussi AG, Price MR and
Segal-Eiras A: Identification and characterization of different
subpopulations in a human lung adenocarcinoma cell line (A549).
Pathol Oncol Res. 5:197–204. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stockholm D, Benchaouir R, Picot J, Rameau
P, Neildez TM, Landini G, Laplace-Builhé C and Paldi A: The origin
of phenotypic heterogeneity in a clonal cell population in vitro.
PLoS One. 2:e3942007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marupudi NI, Han JE, Li KW, Renard VM,
Tyler BM and Brem H: Paclitaxel: A review of adverse toxicities and
novel delivery strategies. Expert Opin Drug Saf. 6:609–621. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gherardi E, Birchmeier W, Birchmeier C and
Vande Woude G: Targeting MET in cancer: Rationale and progress. Nat
Rev Cancer. 12:89–103. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang S and Yu D: Targeting Src family
kinases in anti-cancer therapies: Turning promise into triumph.
Trends Pharmacol Sci. 33:122–128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li D, Masiero M, Banham AH and Harris AL:
The notch ligand JAGGED1 as a target for anti-tumor therapy. Front
Oncol. 4:2542014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu Z and Xu S: ERK1/2 MAP kinases in cell
survival and apoptosis. IUBMB Life. 58:621–631. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu N, Lao Y, Zhang Y and Gillespie DA:
Akt: A double-edged sword in cell proliferation and genome
stability. J Oncol. 2012:9517242012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tirino V, Desiderio V, Paino F, De Rosa A,
Papaccio F, La Noce M, Laino L, De Francesco F and Papaccio G:
Cancer stem cells in solid tumors: An overview and new approaches
for their isolation and characterization. FASEB J. 27:13–24. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ling GQ, Chen DB, Wang BQ and Zhang LS:
Expression of the pluripotency markers Oct3/4, Nanog and Sox2 in
human breast cancer cell lines. Oncol Lett. 4:1264–1268.
2012.PubMed/NCBI
|
|
25
|
Klonisch T, Wiechec E, Hombach-Klonisch S,
Ande SR, Wesselborg S, Schulze-Osthoff K and Los M: Cancer stem
cell markers in common cancers-therapeutic implications. Trends Mol
Med. 14:450–460. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Borst P: Cancer drug pan-resistance:
Pumps, cancer stem cells, quiescence, epithelial to mesenchymal
transition, blocked cell death pathways, persisters or what? Open
Biol. 2:1200662012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Röyttä M, Laine KM and Härkönen P:
Morphological studies on the effect of taxol on cultured human
prostatic cancer cells. Prostate. 11:95–106. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Talpaz M, Shah NP, Kantarjian H, Donato N,
Nicoll J, Paquette R, Cortes J, O'Brien S, Nicaise C, Bleickardt E,
et al: Dasatinib in imatinib-resistant Philadelphia
chromosome-positive leukemias. N Engl J Med. 354:2531–2541. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Steinberg M: Dasatinib: A tyrosine kinase
inhibitor for the treatment of chronic myelogenous leukemia and
philadelphia chromosome-positive acute lymphoblastic leukemia. Clin
Ther. 29:2289–2308. 2007. View Article : Google Scholar : PubMed/NCBI
|