Introduction
In the USA, prostate cancer is the most prevalent
malignancy among men and is the second most common cause of
cancer-associated mortality (1).
Although prostate cancer mortality rates declined steadily over
recent years, it was estimated that there will be 180,890 new cases
and 26,120 mortalities reported in 2016, accounting for ~21% of
newly diagnosed male cancers in 2016 (2).
Histone acetylation/deacetylation is important in
the post-translational modification of histones and is a vital
mechanism in gene expression (3).
Histone acetyltransferase and histone deacetylase (HDAC) are the
key enzymes responsible for these reversible, post-translational
modifications (4). It has been
previously observed that several HDACs are aberrantly expressed or
mutated in human disease, particularly in cancer; thus, these have
been targeted therapeutically for the treatment of various forms of
human cancer (5). HDAC inhibitors
(HDACIs) are currently being assessed as anticancer drugs (6).
Valproic acid (VPA) is an established drug for the
long-term treatment of seizure disorders, which has recently been
classified among the HDACIs, resulting in an increasing interest in
its application in cancer therapy (7). Previous studies have investigated the
anti-tumor effect of VPA on prostate cancer, with results
demonstrating that VPA inhibits the growth and angiogenesis of
prostate cancer (8,9), whilst a further study elucidated the
induction of autophagy by VPA in prostate cancer (10). Autophagy has recently been discussed
as a potential target in cancer, and Akt is a vital kinase
implicated in the negative regulation of autophagy via mammalian
target of rapamycin (mTOR) (11).
In the present study, the inhibitory effect of VPA
on the Akt/mTOR signaling pathway and its relevance to the
induction of autophagy in prostate cancer was examined, providing a
novel mechanism and therapeutic target for the treatment of
prostate cancer, therefore, the current study aimed investigate
these mechanisms further.
Materials and methods
Cell culture
Three human prostate cancer cell lines, PC3, DU145
and LNCaP, were obtained from the Chinese Academy of Sciences
(Beijing, China) and Kunming Cell Bank of Chinese Academy of
Sciences (Kunming, China). Cells were cultured at 37°C, with an
atmosphere of 95% air and 5% CO2, in RPMI-1640 medium
with L-glutamine (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% heat-inactivated fetal bovine serum (Thermo
Fisher Scientific, Inc.) and 1% penicillin/streptomycin. Cells were
cultured until they achieved 70–80% confluence and were harvested
with 0.05% trypsin/0.25 mmol/l ethylenediaminetetraacetic acid
(Thermo Fisher Scientific, Inc.). VPA (1 mol/l; VPA sodium salt;
Sigma-Aldrich, St. Louis, MO, USA) stock was prepared in RPMI-1640,
and was filtered and sterilized through a 0.22-mm filter.
Flow cytometry for cell cycle
assay
PC3 and DU145 cell lines, which had been treated
with 0.6, 1.2 and 2.5 mmol/l VPA, were collected by trypsinization,
washed with PBS 3 times, for 10 minutes each wash, and resuspended
in cold PBS at 1×106 cells/ml. The cell suspension was
added dropwise to an equal volume of cold 70% ethanol with
continuous agitation. Resuspended cells were treated with PBS
containing 0.1% Triton X-100 and 10 µg/ml RNase (pancreatic
ribonuclease; Sigma-Aldrich) for 30 min at room temperature in the
dark. Propidium iodide (PI; Sigma-Aldrich) was subsequently added
to a final concentration of 50 µg/ml. The samples were analyzed
using an Accuri C6 Flow Cytometer (BD Biosciences, San Jose, CA,
USA). The fluorescent emissions were collected through a 575 nm
band-pass filter for PI. At least 10,000 cells/sec were analyzed
using FlowJo 7.6 software (FlowJo LLC, Ashland, OR, USA). .
Transmission electron microscopy
The effect of VPA treatment on ultrastructures in
PC3, DU145 and LNCaP cells was detected using transmission electron
microscopy. Briefly, each cell type (2×105) was plated
in a 100-mm Petri dish, incubated overnight at 37°C and treated
with VPA (2.5 mmol/l) for 48 h at 37°C. Samples without VPA served
as a control group. Cells were harvested, pelleted, fixed in 3%
glutaraldehyde (Sigma-Aldrich) for 2 h at 4°C and finally postfixed
with 1% osmium tetroxide (Sigma-Aldrich) at 37°C for 1 h. The
samples were then rinsed with water, dehydrated in a graded series
of alcohol (50, 70 and 90% alcohol) and kept in 90% alcohol/90%
acetone (dilution, 1:1), followed by dehydration in a graded series
of acetone (90–100%). Subsequent to rinsing with acetone/Poly Bed
(Polysciences, Warrington, PA, USA) at proportions of 3:1, 1:1 and
1:3, the samples were embedded in Poly Bed and cured at 37°C for 12
h, 45°C for 12 h and 60°C for 24 h. Ultrathin sections (50–70 nm)
were cut using a Leica RM2235 ultramicrotome (Leica Microsystems
GmbH, Wetzlar, Germany) and observed with a transmission electron
microscope (Hitachi H-800; Hitachi, Tokyo, Japan).
Fluorescent microscopy
Prostate cancer cell lines, DU145 and LNCaP, were
grown in 6-well, flat-bottomed, microtiter plates (5×105
cells/well), incubated overnight and cultured in medium with VPA
(2.5 mmol/l for 48 h). Samples without VPA served as a control
group. Cells were washed with PBS and fixed for 15 min using 4%
paraformaldehyde (Merck & Co., Inc., Whitehouse Station, NJ,
USA). Subsequently, the cells were permeabilized with 0.1% Triton
X-100 (Sigma-Aldrich) for 25 min at room temperature, washed twice
with PBS for 10 min and blocked with PBS containing 0.5% (w/v)
bovine serum albumin (BSA; Biological Industries, Kibbutz
Beit-Haemek, Israel) for 30 min at room temperature. Cells were
treated with primary rabbit anti-human 1A/1B-light chain 3 (LC3)-II
(catalog no., sc-134226; dilution, 1:1,000 in BSA; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and Beclin-1 antibodies
(catalog no., sc-10086; dilution, 1:100 in BSA; Santa Cruz
Biotechnology, Inc.) at 4°C overnight, washed with PBS 3 times and
subsequently incubated with secondary goat anti-rabbit fluorescein
isothiocyanate-conjugated antibody DyLight 549-conjugated antibody
(catalog no., ab6721; dilution, 1:5,000 in BSA; Abcam, Cambridge,
UK) for 30 min at 37°C, avoiding light. The cells were then washed
with PBS 3 times, dyed in 4′,6-diamidino-2-phenylindole (Roche
Diagnostics, Basel, Switzerland) for 30 min and finally observed
under a fluorescent microscope (Olympus IX70; Olympus Corporation,
Tokyo, Japan).
Western blot analysis
PC3, DU145, and LNCaP cell lines, which had been
cultured to 70–80% confluence in 100-mm Petri dishes, were treated
with complete medium containing 0, 2.5 and 5.0 mmol/l VPA for 48 h.
Cells were harvested, washed in PBS 3 times for 10 min each and
lysed in 100 µl mammalian protein extraction reagent (Beyotime
Institute of Biotechnology, Haimen, China). A Bicinchoninic Acid
Protein assay kit (Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China) was used to determine total protein
concentration, and purified BSA was used to generate the standard
curve. Concentrated proteins were separated by gel electrophoresis
on a 15% Tris-hydrochloride (HCl) polyacrylamide (separating gel)
or 4–18% gradient gel (electrophoresis gel), and transferred to a
polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA,
USA). The membrane was blocked for 1 h in blocking buffer
(containing 100 mmol/l Tris-HCl, 150 mmol/l NaCl and 0.1% Tween 20)
with 5% nonfat dry milk, and incubated with rabbit anti-human
antibody (catalog no., ab6759; dilution, 1:100 in BSA; Abcam)
overnight, followed by incubation with anti-rabbit immunoglobulin G
horseradish peroxidase-conjugated antibody (Santa Cruz
Biotechnology, Inc.) for 1 h at room temperature. Immunoreactive
bands were detected using the enhanced chemiluminescence plus
western blotting detection system (EMD Millipore) according to the
manufacturer's protocol. Polyclonal anti-β-actin antibody (Santa
Cruz Biotechnology, Inc.) was used to detect β-actin in the same
blots, which served as a loading control. ImageJ 1.48v (National
Institutes of Health, Bethesda, MD, USA) was used to quantify the
results of western blot analysis.
Statistical analysis
Each experiment was performed in triplicate and data
are expressed as the mean ± standard deviation. One-way
analysis of variance with Scheffe post-hoc comparison test was used
for comparison of dose-dependent treatment effects. P<0.05 was
considered to indicate a statistically significant difference. All
statistical tests were performed using GraphPad Prism version 5.0
(GraphPad Software, Inc., La Jolla, CA, USA). The present study was
approved by the Ethics Committee of Shandong Provincial Hospital
Affiliated to Shandong University (Jinan, China).
Results
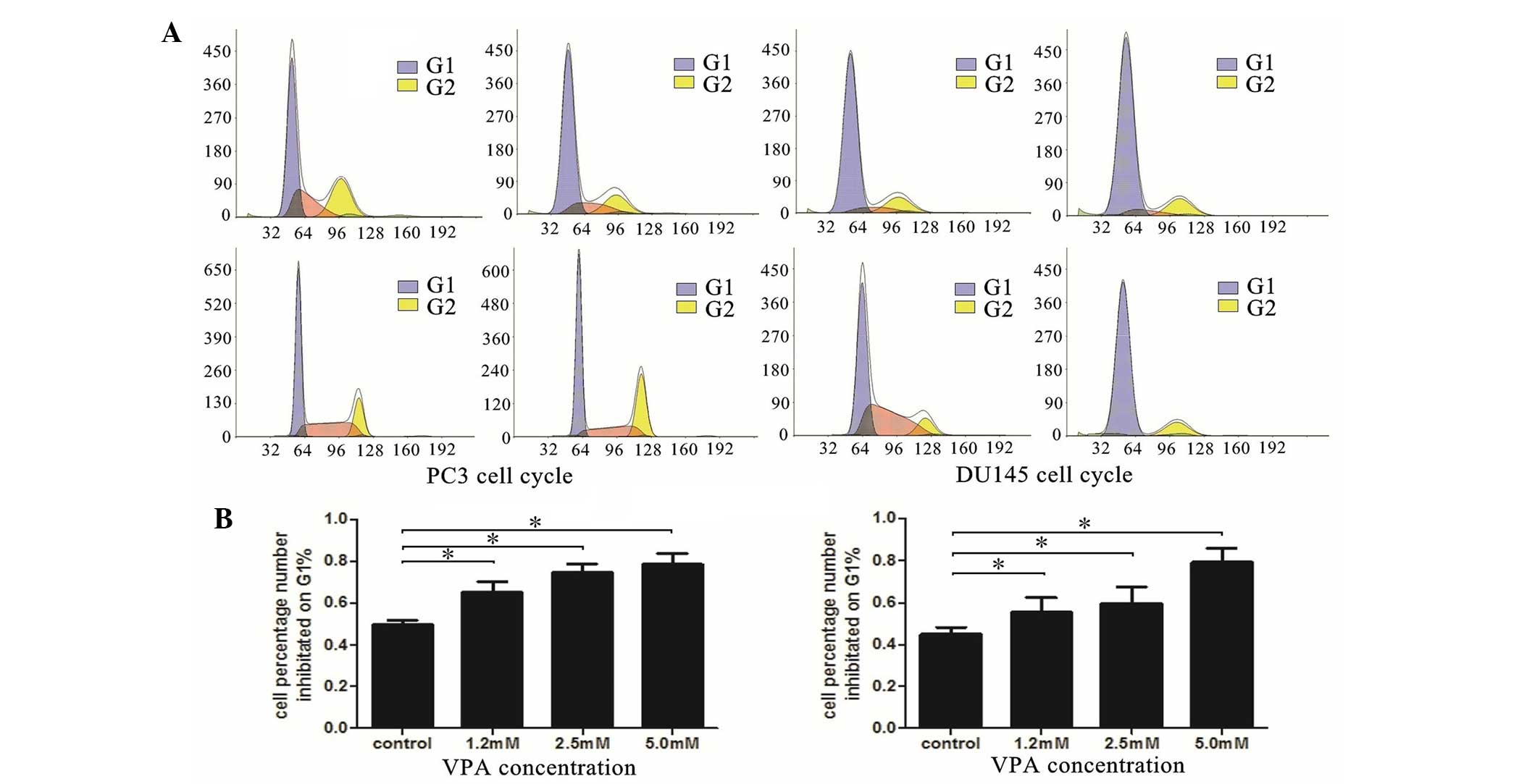
VPA induces cell cycle arrest
primarily at the G0-G1 phase
To further understand the mechanism underlying
VPA-induced cell growth inhibition, the impact of VPA on the
regulation of cell cycle distribution was investigated using flow
cytometry. As presented in Fig. 1,
the results demonstrated that VPA was able to induce cell cycle
arrest in the DU145 and LNCaP cell lines. The cell number
percentage in the G0-G1 phase significantly
increased in a dose-dependent manner compared with control groups
(PC3, P=0.035; DU145, P=0.037).
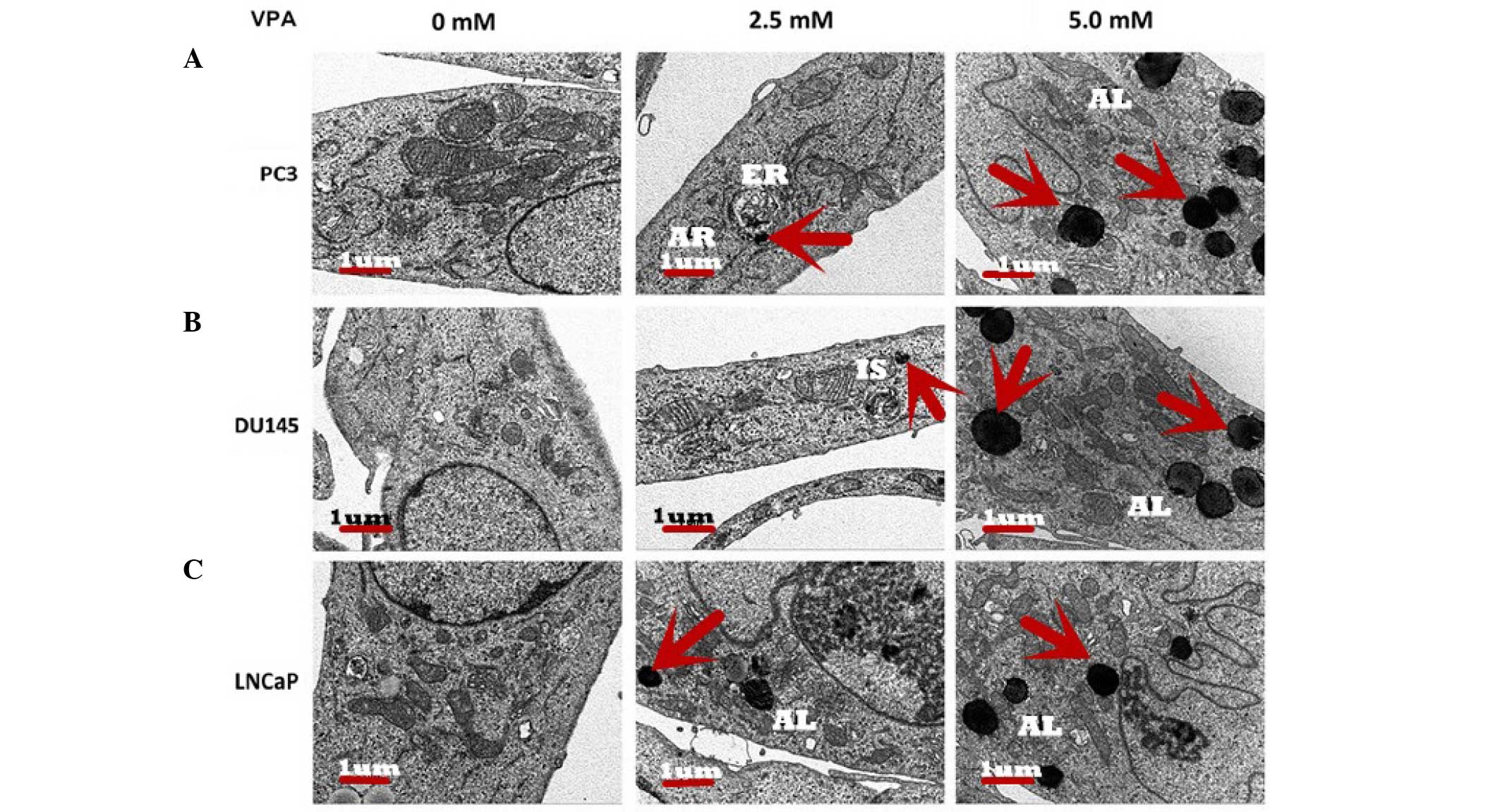
Ultrastructure of autophagy in
prostate cancer cells
Scanning electron micrographs illustrated that
following treatment with VPA, autophagosomes formed inside the
cytoplasm of the prostate cancer cells and appeared to contain
cellular organelles (Fig. 2).
Micrographs demonstrated that the control group exhibited normal
organelle morphology. Autophagic vesicles and autolysosomes were
observed in organelles, including the mitochondria, endoplasmic
reticulum and cytoplasmic materials, in the cells treated with VPA.
At the initiation of cell autophagy in all three cell lines,
numerous smooth endoplasmic reticulum-like isolation membranes were
observed, which formed portions of the cytosol. These
double-membrane structures allow the fusion of autophagosomes with
lysosomes, resulting in the formation of autophagic lysosomes
(12). Finally, the majority of
mitochondria and other organelles had disappeared from the cells,
and remnants of organelles were identified in the autophagic
lysosomes of VPA-treated cells whose nuclear chromatin had
condensed into small, irregular masses.
Expression of specific autophagic
proteins, LC3-II and Beclin-1, in DU145 and LNCaP cells
Accumulation of LC3-II is associated with the extent
of autophagosome formation (13).
Following DyLight-549 staining, the cells in the VPA treatment
group exhibited red fluorescence in the cytoplasm, but not in the
nucleolus, and the fluorescence was stronger than that of the
non-treated control group, thus indicating the formation of
autophagolysosomes (Fig. 3A). These
autophagic changes were further confirmed by western blotting as
presented in Fig. 3C and D (LNCaP,
P=0.019; DU145, P=0.043). Furthermore, Beclin-1 and LC3-II
exhibited strong immunofluorescence in the cytoplasm of the DU145
and LNCaP cells compared with the non-treated control group
(Fig. 3A and B), which was also
detected by western blot analysis (LNCaP, P=0.033; DU145, P=0.027)
(Fig. 3C and 3D).
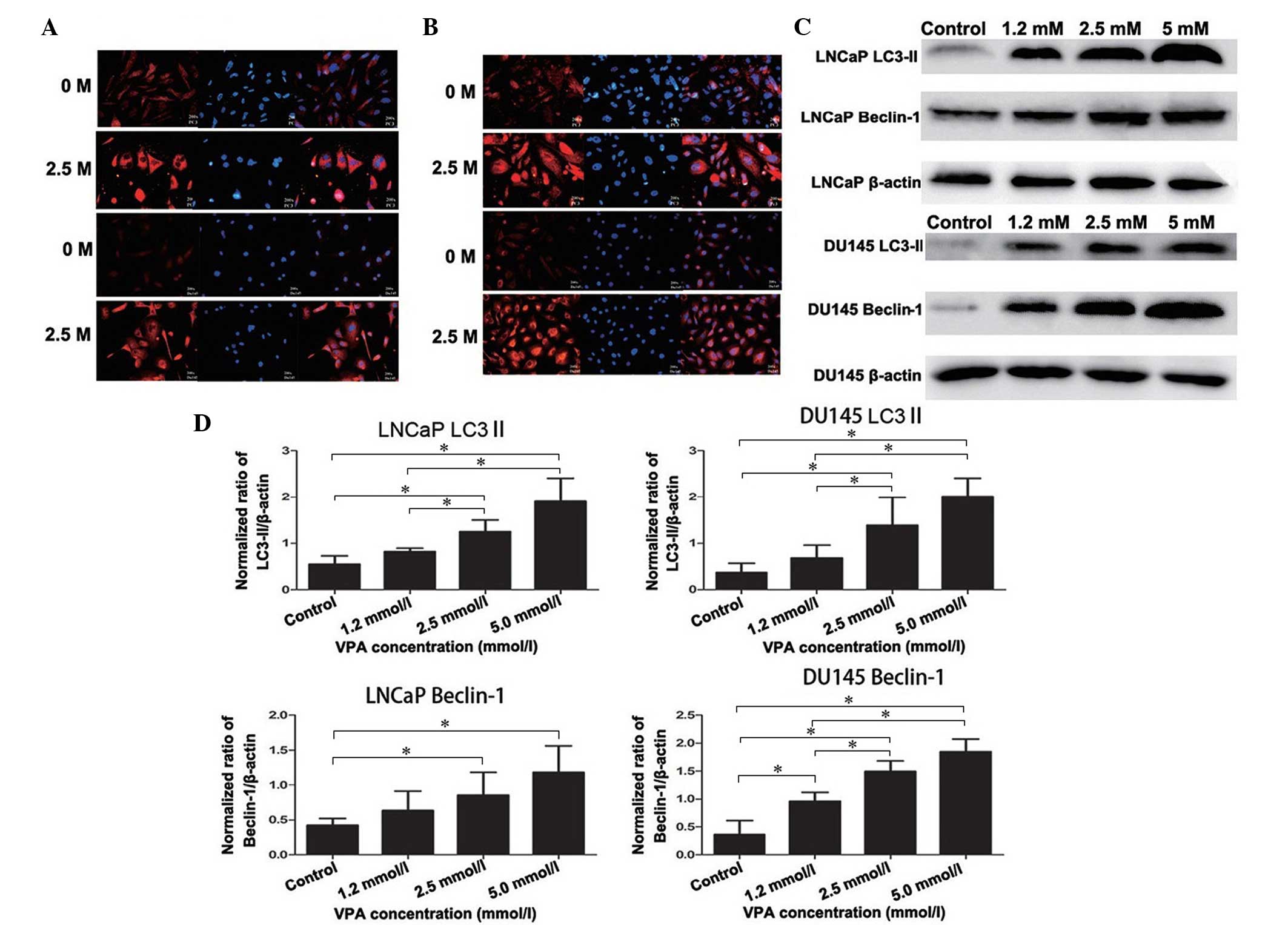 | Figure 3.Expression of specific autophagic
proteins, LC3-II and Beclin-1, in prostate cancer cells. There was
an overlap in the distribution and expression of (A) LC3 and (B)
Beclin-1 protein, which merged with DAPI nuclear staining (blue).
Cells untreated or treated with VPA (2.5 mmol/l for 48 h) were
fixed and probed with polyclonal rabbit anti-LC3-II and
anti-Beclin-1 antibodies, in addition to DyLight 549-conjugated
(red) secondary antibodies. (C) Western blot analysis detected the
induction of LC3-II and Beclin-1 protein expression in cells
treated with VPA (1.2, 2.5 and 5.0 mmol/l) compared with the
non-treated control. LC3-II and Beclin-1 expression increased in a
dose-dependent manner in all cell lines after 48 h incubation with
VPA. (D) Histograms representing the level of LC3-II and Beclin-1
expression in LNCaP and DU145 cells (normalized to β-actin). In
LNCaP cells, LC3-II and Beclin-1 protein levels in the 2.5 mmol/l
and 5.0 mmol/l VPA treatment groups were significantly increased
compared with the control groups. In addition, 1.2 mmol/l VPA
treatment did not significantly increase LC3-II and Beclin-1
protein levels. In DU145 cells, LC3-II protein levels in 2.5 mmol/l
and 5.0 mmol/l VPA treatment groups were significantly increased
compared with the control groups. Beclin-1 protein levels in all
three VPA treatment groups were significantly increased compared
with the control groups. *P<0.05. LC3, 1A/1B-light chain 3; VPA,
valproic acid. |
VPA inhibits activation of the
Akt/mTOR pathway in prostate cancer cell lines
The Akt/mTOR signaling pathway was investigated in
PC3, DU145 and LNCaP cell lines treated with VPA. The relative fold
of phosphorylated Akt and mTOR increased as determined by scanning
densitometry of the western blot normalized to β-actin. VPA-treated
cells were observed to induce a dose-dependent decrease in the
phosphorylation of Akt (PC3, P=0.048; DU145, P=0.045; LNCaP,
P=0.039) and mTOR (PC3, P=0.012; DU145, P=0.41; LNCaP, P=0.35)
protein compared to the control group (Fig. 4) in all cell lines. However, the total
protein levels of Akt (PC3, P=0.082; DU145, P=0.065; LNCaP,
P=0.059) and mTOR (PC3, P=0.12; DU145, P=0.095; LNCaP, P=0.089) did
not change following VPA treatment (P>0.05) (Fig. 4D), which indicates that VPA inflicts a
potent inhibitory effect on Akt/mTOR signaling.
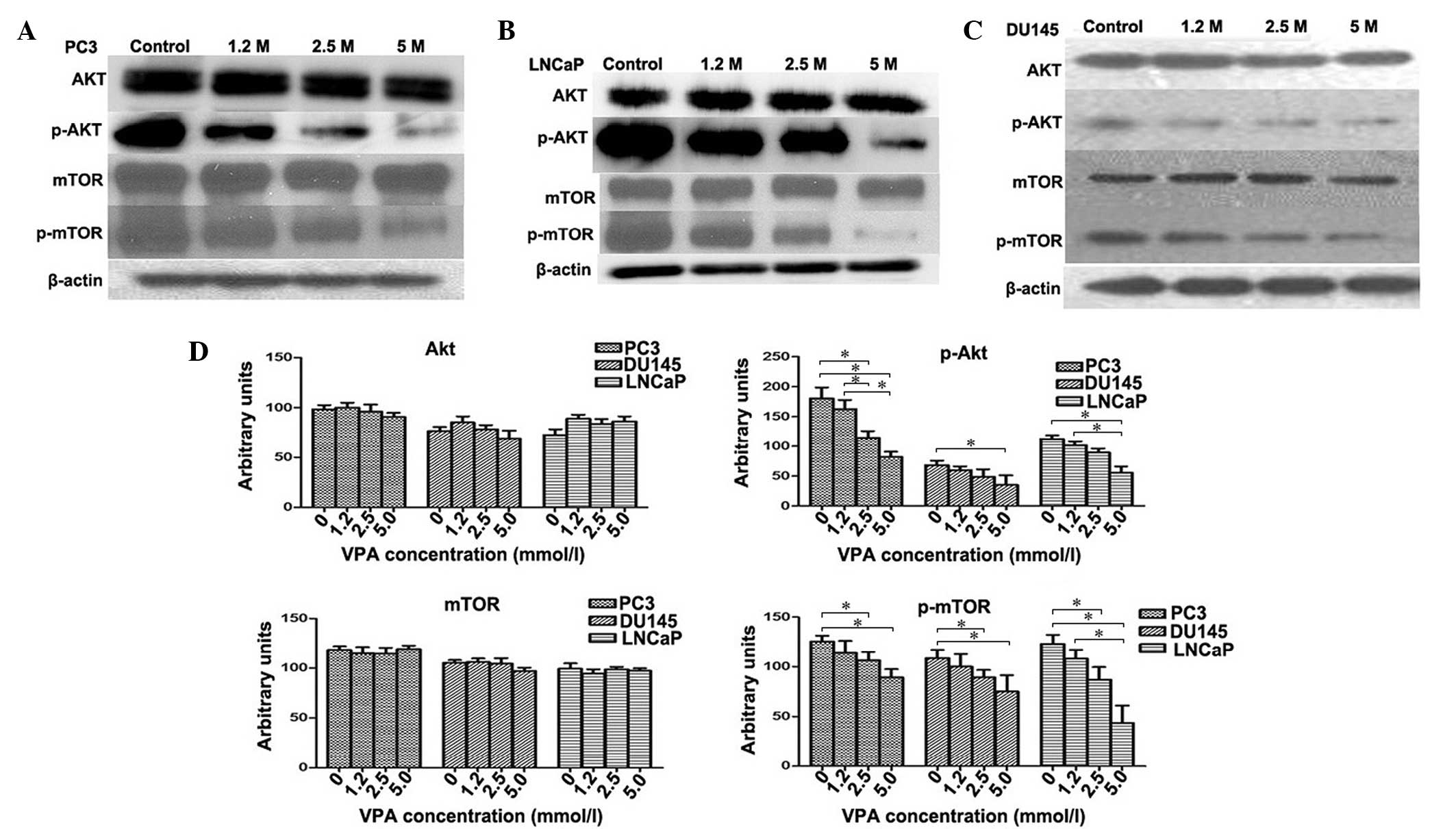 | Figure 4.Effect of VPA on Akt, mTOR and their
phosphorylated forms. Western blotting measured the levels of Akt,
mTOR, and their phosphorylated forms in (A) PC3, (B) LNCaP and (C)
DU145 cells subsequent to VPA treatment at various concentrations.
(D) In contrast to the control group, VPA-treated cells were found
to demonstrate a dose-dependent decrease in the phosphorylation of
Akt and mTOR proteins in DU145, PC3 and LNCaP cells. There was no
significant change in the protein levels of total Akt and mTOR. In
addition, 5.0 mmol/l VPA treatment induced statistically
significant decreases in p-mTOR protein levels in all three cell
lines; 2.5 mmol/l and 5.0 mmol/l VPA treatment induced
statistically significant decreases in p-mTOR protein levels in all
three cell lines. *P<0.05. p-, phosphorylated; mTOR, mammalian
target of rapamycin; VPA, valproic acid. |
Discussion
The two forms of programmed cell death, autophagic
cell death and apoptosis, are not mutually exclusive and may
contribute to oncogenesis and mortality (14). Traditionally, autophagic cell death is
characterized by the formation of autophagic vacuoles, and
regulation of the caspase family performs diversified roles in
pathophysiology and physiology in various cell types (11,15). In
the tumor microenvironment, metabolic stress, including hypoxia and
ischemia, may induce autophagy, which provokes cancer cells to
autodigest and recycle damaged organelles by balancing its
intracellular homeostasis to survive (16). By contrast, autophagy may also be
involved in the degeneration of cell structures, inducing cell
death subsequent to the activation of death signals. In addition,
it was previously determined that there may be an association
between tumorigenesis and the absence of autophagy genes, including
Beclin-1, autophagy related (ATG) 5 and ATG7 (17).
HDACIs have previously undergone testing against
oncogenesis due to their potential ability to reverse epigenetic
changes associated with cancer cells (18). It has been reported that HDACIs are
able to induce tumor cell death with similar physiological and
morphological characteristics to those of apoptosis, particularly
as they are selective and exert minimal toxic effects on the host
(19). In a previous study, it was
demonstrated that VPA significantly decreased the proliferation of
prostate cancer cells in vitro and reduced tumor volume
(20). It was considered that this
antitumor activity may be associated with temporary cell arrest and
downregulated expression of androgen receptors induced by chronic
VPA treatment (11). The autophagy
phenomenon has also been observed following a short period (<4
days) of VPA administration in PC3 cells (10). In the present study, VPA was
identified to inhibit prostate cancer cell growth in a
dose-dependent manner, affirming the antineoplastic effect of VPA.
Flow cytometry analysis demonstrated that the cell number
percentage in the G1 phase significantly increased in
the higher dose drug group, but the cell number in the
G2% peak did not show an increase or decrease with
increasing drug doses. The induction of cell cycle arrest by VPA
highlights its antineoplastic role. This phenomenon may be
explained by the knowledge that HDACIs activate cell death,
resulting in cell cycle arrest in G1-G2. The
G1 arrest induced by VPA in the present study was in
accordance with the reported effect of this drug in endometrial
carcinoma cell lines (15).
Chronic VPA treatment is expected to produce a more
profound effect on cancer cell proliferation compared with standard
VPA treatment, which may have limited activity. The
cyclin-dependent kinase inhibitor, p21WAF/CIP1, is consistently
induced by VPA and is key for the inhibition of cell growth
(21). Cell death is typically
associated with apoptosis; however, it may also occur through
alternative mechanisms, including non-lysosomal vesiculate cell
death and autophagy (22). The
phenomenon of autophagy in response to antitumor therapies may be
monitored by immunohistochemical analysis employing
anti-lysosome-associated membrane protein 1 and anti-LC3B
antibodies (23). Previously, it was
reported that VPA was able to initiate a moderate apoptotic
response through preferential activation of the mitochondrial
pathway in prostate cancer cell lines (24). The results of the present study
demonstrated that VPA may also induce prostate cancer cell death
through the autophagy pathway. The presence of autophagic vacuoles
in cancer cells following VPA treatment indicated that they were
undergoing autophagy-related cell death (Fig. 2). Electron microscopy identified a
number of large vacuoles in the cytoplasm in VPA treated groups,
which were seldom observed in the control group (Fig. 2). These vacuoles exhibited typical
morphological features of autophagy with a double ‘C’ formation at
the membrane origin of autophagosomes. It is generally considered
that the mitochondria, plasma membrane or Golgi bodies may function
as the primary membrane source for autophagosomes and other related
structures (25). The present study
observed that the initial autophagic ultrastructures emerged around
these organelles in the VPA treatment group (Fig. 2), and the bulk of the cytoplasm and
certain organelles were observed to be wrapped into the vacuole,
and the autophagosome had merged with the lysosome.
Autophagosomes appear in the cytoplasm at the first
stage of autophagy-associated cell death, and
microtubule-associated LC3, particularly LC3-II, serves as an
autophagosome-specific protein (26).
LC3 is one of the most credible markers of autophagosomes in
mammalian cells (27). LC3-I is
cytoplasmic, whilst LC3-II is a tight membrane-bound protein that
attaches to autophagosomes, which subsequently fuse with lysosomes
(28). Relative amounts of
membrane-bound LC3-II reflects the abundance of autophagosomes with
the process that transforms LC3-I into LC3-II; thus, the induction
and inhibition of autophagy is able to be monitored by immunoassay
through the measurement of LC3-II levels (29). It has been reported that autophagy is
suppressed in various types of cancer cells, and that cellular
autophagic activity is inversely correlated with malignancy
(30). Beclin-1 may also function as
a marker of autophagy, which has been expressed in a monoallelic
manner in human prostate, ovarian and breast cancer, which
suggested that the process of autophagy may possess
tumor-suppressor properties (31,32). In
the current study, western blot analysis demonstrated that LC3-II
and Beclin-1 expression increased with VPA in a dose-dependent
manner in prostate cancer cells, which was also observed by
fluorescence microscopy (Fig. 3).
Various signaling pathways, such as
autophagy-related (Atg) proteins, ULK and the Bcl-2 family, were
involved in this process, were involved in this process, which
consist of the core autophagic delivery to cell death (33). In yeast and mammalian cells, the Ras
and mTOR pathways are two well-known signaling cascades that are
sensitive to nutrient status, cell growth and differentiation, and
are negatively regulated during programmed cell death (34). The phosphoinositide 3-kinase/Akt/mTOR
pathway exists in several types of cancer and may be activated by
the loss of tumor suppressor phosphatase and tensin homolog (PTEN)
function (35). The formation of an
autophagosome membrane may be affected by regulating the
recruitment of the transmembrane protein ATG9, which facilitates
lipid assembly to expand autophagosomes (36). This step is regulated by mTOR kinase,
but the intracellular mechanism is remains unclear. The activation
of Akt and its phosphorylation induces the expression of p21WAF and
p27Kip1, which are associated with cell cycle growth through the
acetylation of relevant genes (37).
The present study demonstrated that treatment with VPA inhibited
the activity of Akt and mTOR, resulting in a depletion of
phosphorylated (p)-AKT and p-mTOR, which is considered to occur due
to VPA concomitantly inducing p27 and p21. This may subsequently
result in cell cycle arrest, growth inhibition and
PTEN-loss-induced activation of the Akt pathway in prostate cancer
cells. It is suspected that VPA-induced autophagic cell death may
be involved in this process.
In conclusion, the present study provided evidence
that VPA may function as an HDACI to suppress the growth of
prostate cancer cells through the modulation of autophagic
pathways, including inhibition of the Akt/mTOR pathway. Further
experiments are required to determine the significance of each
pathway involved in VPA-induced growth inhibition.
Acknowledgements
The present study was sponsored by the Department of
Science and Technology of Shandong Province (grant no.
BS2010YY047).
References
|
1
|
Antonarakis ES, Armstrong AJ, Dehm SM and
Luo J: Androgen receptor variant-driven prostate cancer: Clinical
implications and therapeutic targeting. Prostate Cancer Prostatic
Dis. May 17–2016.(Epub ahead of print). View Article : Google Scholar
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang W, Zheng Y, Huang Z, Wang M, Zhang
Y, Wang Z, Jin X and Xia Q: Role of SMAD4 in the mechanism of
valproic acid's inhibitory effect on prostate cancer cell
invasiveness. Int Urol Nephrol. 46:941–946. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hegde M, Mantelingu K, Pandey M,
Pavankumar CS, Rangappa KS and Raghavan SC: Combinatorial study of
a novel poly (ADP-ribose) polymerase inhibitor and an HDAC
inhibitor, SAHA, in leukemic cell lines. Target Oncol. May
17–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barneda-Zahonero B and Parra M: Histone
deacetylases and cancer. Mol Oncol. 6:579–589. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Giannini G, Cabri W, Fattorusso C and
Rodriquez M: Histone deacetylase inhibitors in the treatment of
cancer: Overview and perspectives. Future Med Chem. 4:1439–1460.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chateauvieux S, Morceau F, Dicato M and
Diederich M: Molecular and therapeutic potential and toxicity of
valproic acid. J Biomed Biotechnol. 2010:4793642010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xia Q, Sung J, Chowdhury W, Chen CL, Höti
N, Shabbeer S, Carducci M and Rodriguez R: Chronic administration
of valproic acid inhibits prostate cancer cell growth in vitro and
in vivo. Cancer Res. 66:7237–7244. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gao D, Xia Q, Lv J and Zhang H: Chronic
administration of valproic acid inhibits PC3 cell growth by
suppressing tumor angiogenesis in vivo. Int J Urol. 14:838–845.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang ZX, Jin XB, Wang MW, Zhang YN, Zheng
Y and Xia QH: Effect of valproic acid on autophagy in prostate
cancer PC3 cells. Shandong Da Xue Xue Bao. 49:44–47. 2011.(In
Chinese).
|
|
11
|
Nho RS and Hergert P: IPF fibroblasts are
desensitized to type I collagen matrix-induced cell death by
suppressing low autophagy via aberrant Akt/mTOR kinases. PLoS One.
9:e946162014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lauritzen I, Pardossi-Piquard R, Bourgeois
A, Pagnotta S, Biferi MG, Barkats M, Lacor P, Klein W, Bauer C and
Checler F: Intraneuronal aggregation of the β-CTF fragment of APP
(C99) induces Aβ-independent lysosomal-autophagic pathology. Acta
Neuropathol. Apr 30–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Goode A, Butler K, Long J, Cavey J, Scott
D, Shaw B, Sollenberger J, Gell C, Johansen T, Oldham NJ, et al:
Defective recognition of LC3B by mutant SQSTM1/p62 implicates
impairment of autophagy as a pathogenic mechanism in ALS-FTLD.
Autophagy. May 9–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Castagna C, Merighi A and Lossi L: Cell
death and neurodegeneration in the postnatal development of
cerebellar vermis in normal and Reeler mice. Ann Anat. Feb
28–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao X, Yang W, Shi C, Ma W, Liu J, Wang Y
and Jiang G: The G1 phase arrest and apoptosis by intrinsic pathway
induced by valproic acid inhibit proliferation of BGC-823 gastric
carcinoma cells. Tumour Biol. 32:335–346. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kouidhi S, Noman MZ, Kieda C, Elgaaied AB
and Chouaib S: Intrinsic and tumor microenvironment-induced
metabolism adaptations of T cells and impact on their
differentiation and function. Front Immunol. 7:1142016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Y, Liu XR, Yin YQ, Lee CJ, Wang FT,
Liu HQ, Wu XT and Liu J: Unravelling the multifaceted roles of Atg
proteins to improve cancer therapy. Cell Prolif. 47:105–112. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Damaskos C, Karatzas T, Nikolidakis L,
Kostakis ID, Karamaroudis S, Boutsikos G, Damaskou Z, Kostakis A
and Kouraklis G: Histone deacetylase (HDAC) inhibitors: Current
evidence for therapeutic activities in pancreatic cancer.
Anticancer Res. 35:3129–3135. 2015.PubMed/NCBI
|
|
19
|
Carafa V, Miceli M, Altucci L and Nebbioso
A: Histone deacetylase inhibitors: A patent review (2009-2011).
Expert Opin Ther Pat. 23:1–17. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee JE and Kim JH: Valproic acid inhibits
the invasion of PC3 prostate cancer cells by upregulating the
metastasis suppressor protein NDRG1. Genet Mol Biol. 38:527–533.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nagai H, Fujioka-Kobayashi M, Ohe G, Hara
K, Takamaru N, Uchida D, Tamatani T, Fujisawa K and Miyamoto Y:
Antitumour effect of valproic acid against salivary gland cancer in
vitro and in vivo. Oncol Rep. 31:1453–1458. 2014.PubMed/NCBI
|
|
22
|
Cerella C, Teiten MH, Radogna F, Dicato M
and Diederich M: From nature to bedside: Pro-survival and cell
death mechanisms as therapeutic targets in cancer treatment.
Biotechnol Adv. 32:1111–1122. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Natsumeda M, Aoki H, Miyahara H, Yajima N,
Uzuka T, Toyoshima Y, Kakita A, Takahashi H and Fujii Y: Induction
of autophagy in temozolomide treated malignant gliomas.
Neuropathology. 31:486–493. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen X, Wong JY, Wong P and Radany EH:
Low-dose valproic acid enhances radiosensitivity of prostate cancer
through acetylated p53-dependent modulation of mitochondrial
membrane potential and apoptosis. Mol Cancer Res. 9:448–461. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mari M, Tooze SA and Reggiori F: The
puzzling origin of the autophagosomal membrane. F1000 Biol Rep.
3:252011. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Levine B and Kroemer G: Autophagy in
aging, disease and death: The true identity of a cell death
impostor. Cell Death Differ. 16:1–2. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Talaber G, Miklossy G, Oaks Z, Liu Y,
Tooze SA, Chudakov DM, Banki K and Perl A: HRES-1/Rab4 promotes the
formation of LC3(+) autophagosomes and the accumulation of
mitochondria during autophagy. PLoS One. 9:e843922014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lai SC and Devenish RJ: LC3-associated
phagocytosis (LAP): Connections with host autophagy. Cells.
1:396–408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Palmisano NJ and Meléndez A: Detection of
autophagy in Caenorhabditis elegans by western blotting
analysis of LGG-1. Cold Spring Harb Protoc.
2016:pdb.prot0865122016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lv ZQ, Han JJ, Liu YQ, Wang LL, Tang QL,
Sun Q and Li HG: Expression of beclin 1 in non-small cell lung
cancer: An immunohistochemical study. Clin Respir J. 9:359–365.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liang C, Feng P, Ku B, Dotan I, Canaani D,
Oh BH and Jung JU: Autophagic and tumour suppressor activity of a
novel Beclin1-binding protein UVRAG. Nat Cell Biol. 8:688–699.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li L, Chen X and Gu H: The signaling
involving in autophagy machinery in keratinocytes and therapeutic
approaches for skin diseases. Oncotarget. May 12–2016.(Epub ahead
of print).
|
|
34
|
Taneike M, Nishida K, Omiya S,
Zarrinpashneh E, Misaka T, Kitazume-Taneike R, Austin R, Takaoka M,
Yamaguchi O, Gambello MJ, et al: mTOR hyperactivation by ablation
of tuberous sclerosis complex 2 in the mouse heart induces cardiac
dysfunction with the increased number of small mitochondria
mediated through the down-regulation of autophagy. PLoS One.
11:e01526282016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Riquelme I, Tapia O, Espinoza JA, Leal P,
Buchegger K, Sandoval A, Bizama C, Araya JC, Peek RM and Roa JC:
The gene expression status of the PI3K/AKT/mTOR pathway in Gastric
Cancer Tissues and cell lines. Pathol Oncol Res. May 7–2016.(Epub
ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Feng Y, Backues SK, Baba M, Heo JM, Harper
JW and Klionsky DJ: Phosphorylation of Atg9 regulates movement to
the phagophore assembly site and the rate of autophagosome
formation. Autophagy. 12:648–658. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Malfitano AM, Laezza C, Galgani M,
Matarese G, D'Alessandro A, Gazzerro P and Bifulco M: The CB1
receptor antagonist rimonabant controls cell viability and ascitic
tumour growth in mice. Pharmacol Res. 65:365–371. 2012. View Article : Google Scholar : PubMed/NCBI
|