Introduction
The epidermal growth factor receptor (EGFR) family
of growth receptors, consisting of EGFR, human epidermal growth
factor receptor (HER)2, HER3 and HER4 (also known as ErbB1-4,
respectively), is known to be involved in a number of cancer types.
Therefore, these receptors, particularly EGFR and HER2, are
important targets for tumour diagnostics and therapy.
HER2-overexpression is most common in breast and ovarian cancers,
and is also associated with poor prognosis (1).
Upon ligand binding, EGFR forms a dimer, which is
internalised via endosomes and eventually degraded in lysosomes
(2,3).
However, HER2, which has no natural ligand and therefore is
normally dependent on heterodimerisation with other members of the
HER family of receptors, is not internalised and degraded in this
way. Certain authors have reported that this receptor continuously
circulates between the cell membrane and early endosomes (4), whereas others regard HER2 as an
internalisation-resistant receptor that is actively excluded from
clathrin-coated pits (5). This may be
part of the reason why HER2 signalling is so potent (6,7).
However, HER2 may be forced into internalisation and
degradation using the ansamycin antibiotic geldanamycin (GA) or its
derivative 17-allylamino-17-demethoxygeldanamycin (17-AAG)
(8). By blocking its ATP-binding
site, these compounds inhibit heat shock protein 90 (Hsp90), a
chaperone required for stabilisation of HER2 at the cell membrane
(9). Without an active Hsp90
chaperone, HER2 is ubiquitinylated by the E3 ubiquitin ligase CHIP
(10,11) and internalised in a
proteasome-dependent process that ends in lysosomal degradation
(12). It is not clear whether the
increased internalisation results from the redirection of endosomes
from recirculation to degradation in lysosomes (13), or from faster endocytosis followed by
degradation (12).
Hsp90 acts as a chaperone for a number of other
proteins involved in receptor signalling, including Raf of the
Ras/mitogen-activated protein kinase pathway and p-Akt of the
phosphoinositide-3 kinase (PI3K)/Akt pathway (14). Since these pathways are often
upregulated in cancer cells, a feature believed to be associated
with cytotoxic drug resistance (15,16),
impaired signalling may potentiate the effect of cytotoxic agents.
17-AAG, which is less toxic than geldanamycin, has shown agonistic
tumour-cell killing effects in combination with, for instance,
paclitaxel or carboplatin (17,18) and γ
irradiation (19). 17-AAG (or
formulated as KOS-953/Tanespimycin) has been recently evaluated in
clinical trials (15).
The current study investigated the effects of 17-AAG
on a radiolabelled HER2-binding Affibody molecule, ABY-025.
Affibody molecules are three-helical proteins derived from the
staphylococcal IgG-binding protein A. By randomising 13 amino acids
in its binding site, it is possible to change the IgG-binding
properties of the Affibody molecule and generate Affibody molecules
specific for other proteins via phage display or other selection
systems (20). Affibody molecules
specific for EGFR and HER2 have shown promising results in
tumour-targeting studies (21,22).
The results indicate that 17-AAG treatment of SKOV-3
and SKBR-3 cells to some extent shifts the localisation of
111In-ABY-025 from the cell surface to intracellular
compartments in both cell lines. ABY-025 labelled with the
high-linear energy transfer (LET) α emitter 211At is
also internalised to a higher degree; due to its physiological
properties, however, this nuclide is excreted faster and thus does
not residualise intracellularly.
Materials and methods
Cell culture
SKOV-3 human ovarian carcinoma cells (ATCC,
Manassas, VA, USA) were grown in McCoy's medium (Biochrom; Merck
KGaA, Darmstadt, Germany) supplemented with 10% foetal bovine serum
(Sigma-Aldrich, St. Louis, MO, USA), 2 mM L-glutamine (Biochrom;
Merck KGaA), 100 U/ml penicillin and 100 µg/ml streptomycin
(Biochrom; Merck KGaA). SKBR-3 cells (ATCC) were cultured in the
same medium but with 20% foetal bovine serum.
Affibody molecules
The HER2-specific Affibody Cys-Z2891 was
used. This protein has a C-terminally located cysteine, which was
utilised for maleimide coupling of Alexa Fluor® 488 to
the protein (as described below). When this cysteine is instead
maleimide-coupled to DOTA, the Affibody is referred to as ABY-025
(23). Cys-Z2891 and
ABY-025 were kindly provided by Affibody AB (Solna, Sweden).
211At labelling of
ABY-025
The labelling was performed as previously described
by Lindegren et al (24). In
short, 250 µg (35.7 nmol) of ABY-025 in 0.1 M borate buffer (pH
8.5) was mixed with 71.4 nmol of m-MeATE in dimethyl sulfoxide
(DMSO) and incubated for 30 min with gentle agitation. After
elution with 0.2 M sodium acetate buffer (pH 5.5) on a NAP-5 column
(GE Healthcare Life Sciences, Uppsala, Sweden), ABY-025-MeATE was
added to 39.8 MBq of 211At (Rigshospitalet, Copenhagen,
Denmark) which had been activated with N-iodosuccinimide (NIS) and
incubated with agitation for 60 sec. More NIS was added, and the
mixture was incubated for a further 60 sec. Sodium ascorbate was
added in order to reduce unreacted astatine, and the
211At-labelled ABY-025 was purified on a NAP-5 column
using phosphate-buffered saline (PBS) as eluent.
Specificity of
211At-ABY-025 uptake
Cells (25,000 of SKOV-3 and 100,000 of SKBR-3 per
well) were seeded into 6-well plates and allowed to grow in
complete medium for 5 days. Following 2 h of incubation with 2.3 nM
211At-ABY-025 with or without 230 nM unlabelled ABY-025,
the cells were washed, trypsinised and measured in a gamma counter
(Wizard 1480; Wallac Oy, Turku, Finland).
Uptake and internalisation of
211At-ABY-025 (acid wash assay)
SKOV-3 and SKBR-3 cells were seeded into 6-well
plates as described. The medium was replaced with 3 ml of 2.3 nM
(=30 × KD) ABY-025 in complete medium with either 100 nM
17-AAG (A.G. Scientific, Inc., San Diego, CA, USA) dissolved in
DMSO, or the corresponding volume of DMSO (control). After 2, 4 and
6 h, samples were taken using the acid wash internalisation assay
(25): After two washes in serum-free
medium, the cells were incubated with 0.5 ml ice-cold acid (0.2 M
glycine, 0.15 M NaCl, 4 M urea, pH 2) on ice for 5 min. The acid
(with the cell surface fraction of 211At) was collected
and cells were washed with additional 0.5 ml acidic solution. The
cells were treated with 1 M NaOH and removed from the petri dish
using a cell scraper. This cell suspension was retained as the
internalised fraction of 211At. For each time point,
triplicates were used for every treatment. Radioactivity was
measured in a gamma counter, with all samples in one reading.
111In labelling of
ABY-025
Labelling was performed as described previously
(23). In short, 50 µg of ABY-025 was
diluted in 50 µl 0.2 M ammonium acetate buffer (pH 5.3), mixed with
50 MBq 111InCl (Medtronic, Minneapolis, MN, USA) and
incubated at 60°C for 40 min. Labelling yield was determined on
chromatography strips (Biodex Medical Systems, Shirley, NY, USA) in
0.2 M citric acid and analysed in a Phosphor Imager (Cyclone
Storage Phosphor System; PerkinElmer, Inc., Waltham, MA, USA).
Uptake and internalisation of
111In-ABY-025 (acid wash assay)
Approximately 500,000 SKOV-3 or SKBR-3 cells were
seeded into 3.5-cm petri dishes and allowed to grow at least
overnight. Cells were incubated with 111In-ABY-025
±17-AAG, and surface-bound and internalised fractions were
separated using acid wash as described. SKOV-3 cells were treated
with 10 and 100 nM 17-AAG, while SKBR-3 cells were treated with 100
nM only. Samples were taken at 0, 1, 3, 5 and 7 h after the start
of incubation. Radioactivity was measured in a gamma counter, with
all samples in one reading.
Alexa Fluor® 488 labelling
of Cys-Z2891
Cys-Z2891 (700 µg) was diluted to 100 nM
and reduced with 20 mM dithiothreitol (DTT) for 45 min at 37°C. DTT
was removed in NAP-5 columns equilibrated in PBS, and 500 nmol (5X
molar excess) Alexa Fluor® 488 C5-maleimide (Molecular
Probes; Thermo Fisher Scientific, Inc., Waltham, MA, USA) dissolved
in DMSO was added. After incubation at 4°C overnight, unbound
Alexa488 was removed in a PD-10 column (GE Healthcare Life
Sciences) equilibrated with PBS. Degree of labelling and protein
concentration were determined using a NanoDrop ND-1000 (Thermo
Fisher Scientific, Inc., Wilmington, DE, USA).
Immunofluorescence microscopy
SKOV-3 (10,000 cells) and SKBR-3 (20,000 cells) were
seeded into 8-chamber slides (Nunc 154534) and allowed to grow for
3 days. The medium was exchanged for complete medium with 200 nM
Alexa488-Z2891 and 100 nM 17-AAG, or the corresponding volume DMSO
(control), and the cells were incubated for 3 h at 37°C. The slides
were gently washed twice with serum-free medium and the chambers
removed. Following a quick wash in PBS, the cells were fixed in 4%
formaldehyde for 15 min at room temperature. The slides were washed
in PBS, incubated for 10 min in 2 µM Hoechst 33342 and allowed to
dry. They were then mounted in Vactashield Antifade Mounting Medium
(Vector Laboratories, Inc., Burlingame, CA, USA). Images were taken
with a Zeiss LSM 510 Meta confocal microscope.
Pre-incubation with
111In-ABY-025
SKOV-3 cells were incubated with
111In-ABY-025 as described above, and 100 nM 17-AAG was
added either immediately or after 0.5 or 2 h. Samples were taken at
different time points using the acid wash method described
above.
Results
211At-ABY-025
specificity
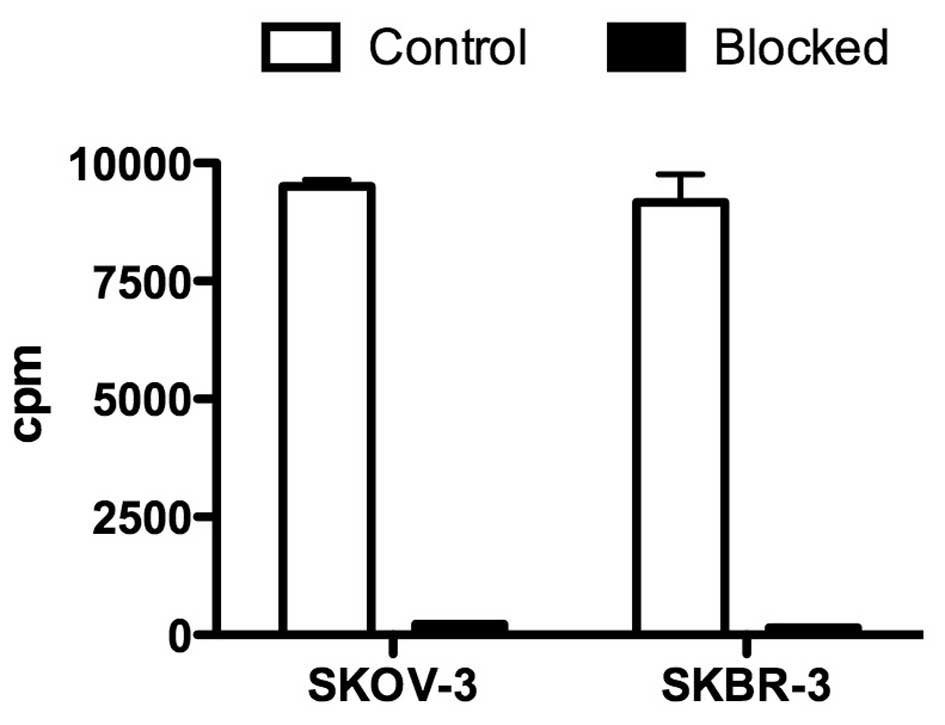
211At-ABY-025 was shown to bind to SKOV-3
and SKBR-3 cells, and this binding was completely blocked with 230
nM of ABY-025 (Fig. 1), confirming
certain specificity.
Uptake and internalisation of
211At-ABY-025
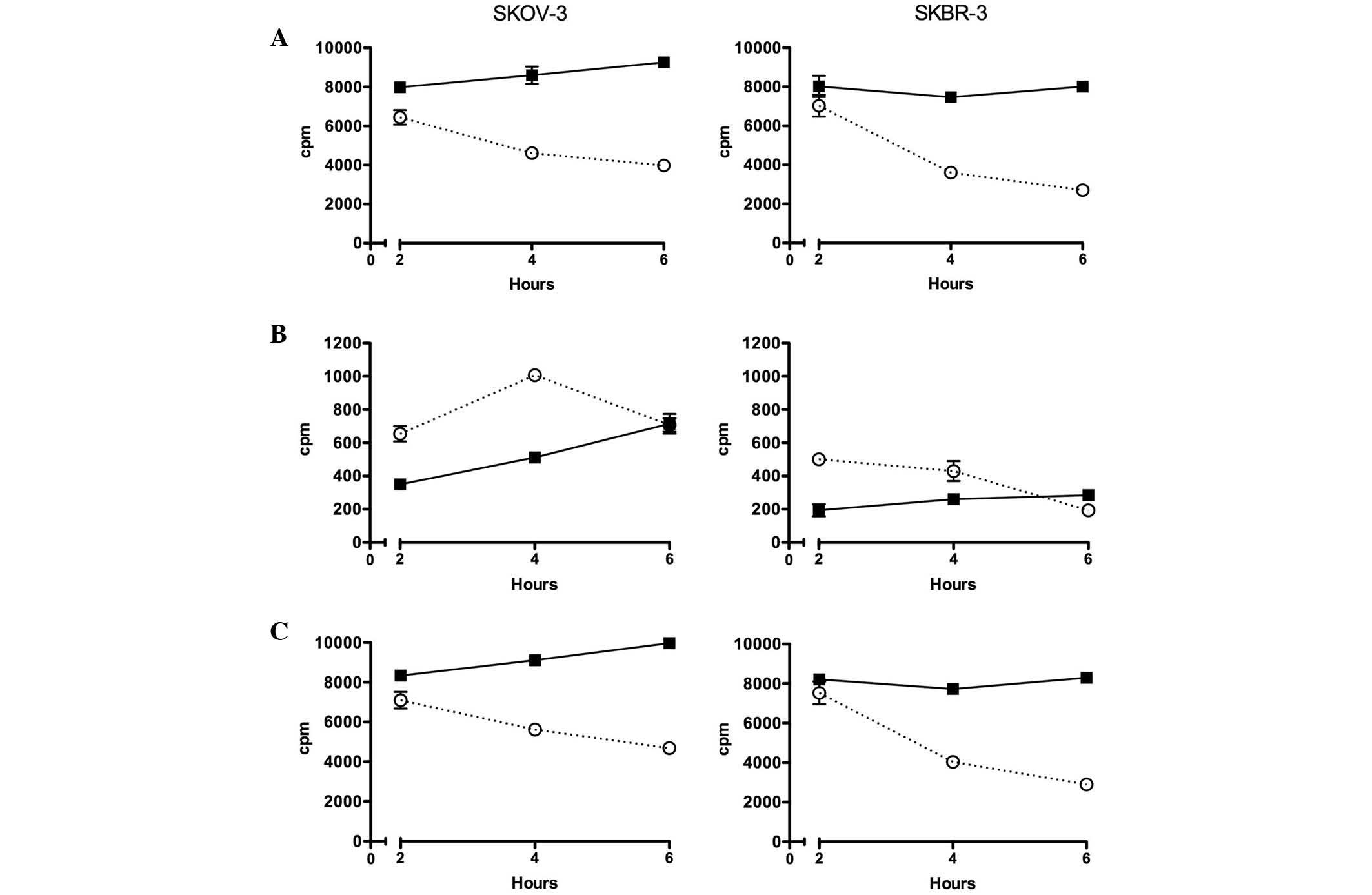
In untreated SKOV-3 cells, the amount of
surface-bound 211At-ABY-025 increased slightly between 2
and 6 h, whereas treatment with 17-AAG decreased cell surface
location during the same time (Fig.
2). The intracellular concentration of 211At was
lower for treated and non-treated cells; however, the relative
increase with time was higher in both cases. In SKBR-3 cells, the
pattern was similar, but the surface uptake of untreated cells did
not increase over time, and 17-AAG induced a faster reduction of
surface-bound 211At-ABY-025 than in the SKOV-3 cells.
Total cellular uptake was simply calculated as the sum of counts of
the two fractions. Due to the relatively small intracellular
fraction, the total value was fairly similar to that of the surface
fraction.
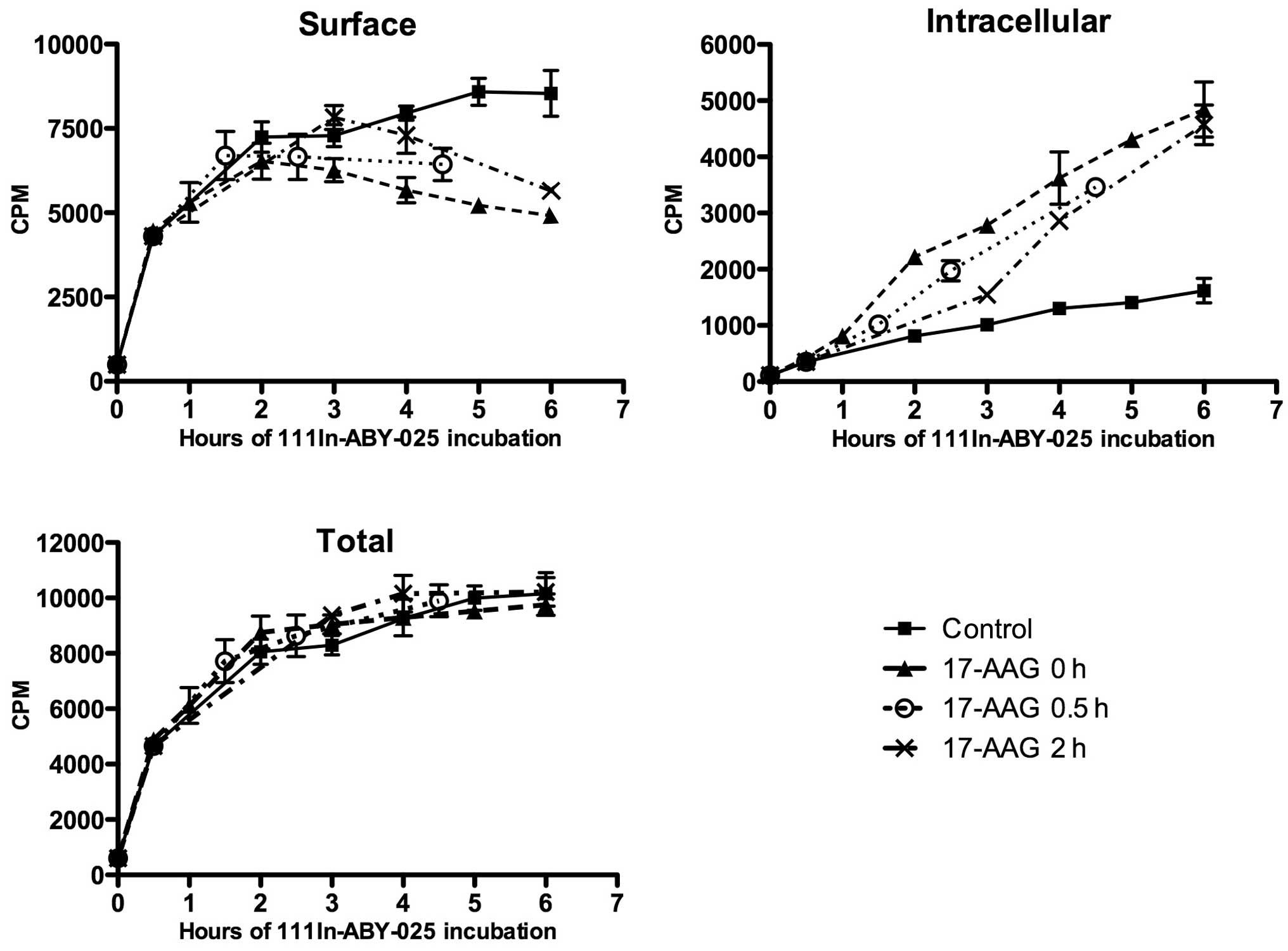
Uptake and internalisation of
111In-ABY-025
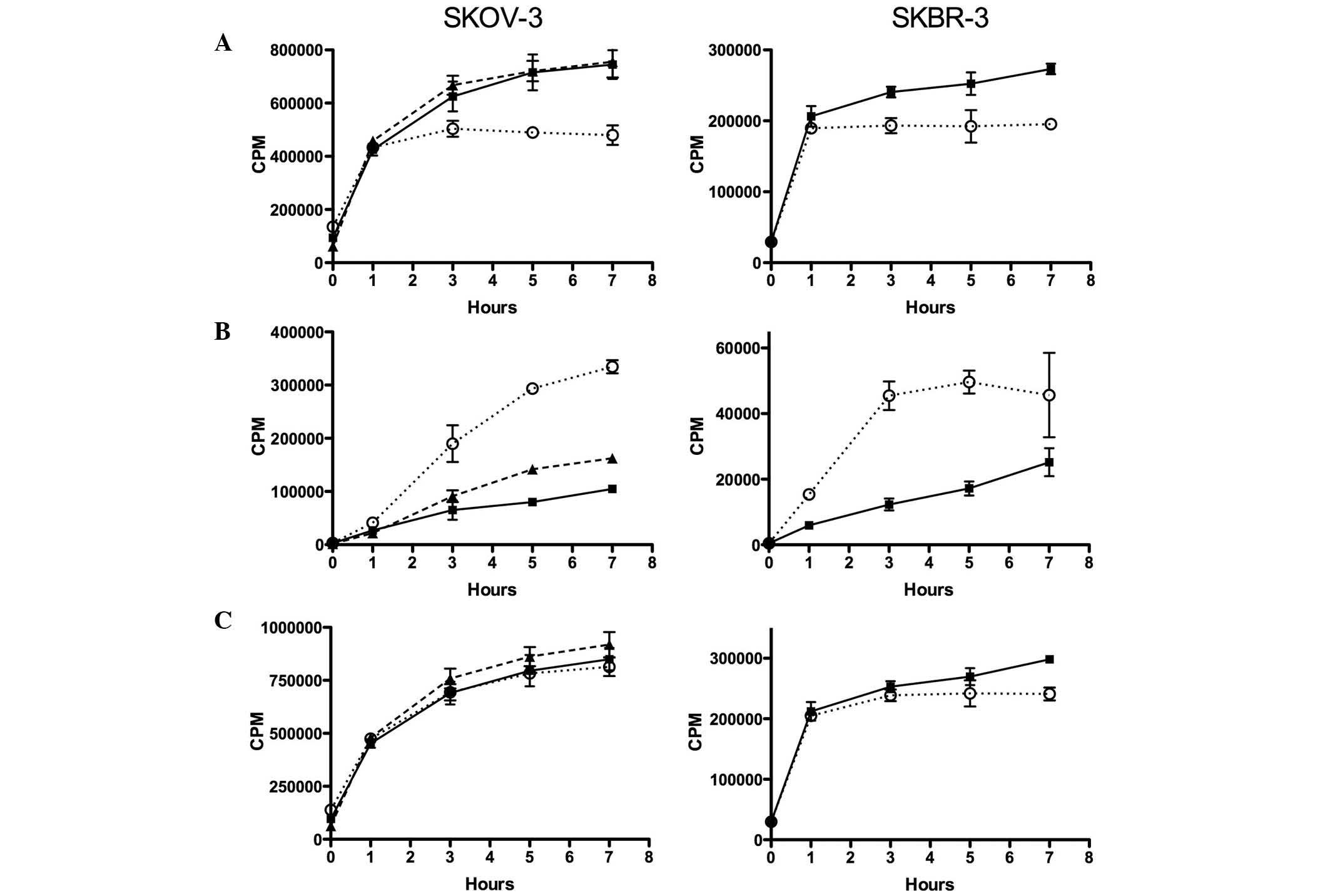
In untreated SKOV-3 cells, surface bound
111In-ABY-025 increased with time and reached a steady
level after 5–7 h (Fig. 3). With 100
nM 17-AAG present, the level of surface-bound
111In-ABY-025 stabilised at a lower level after ~3 h,
whereas 10 nM of 17-AAG had no detectable effect. The decreased
surface levels of 111In-ABY-025 observed with 100 nM
17-AAG corresponded to a continuous increase in intracellular
111In. A similar but weaker increase was observed with
10 nM 17-AAG. In SKBR-3 cells, the pattern was similar. In cells
treated with 100 nM 17-AAG, the surface levels of
111In-ABY-025 were stable from ~1 h, whereas this
fraction continued to increase in untreated cells. The increased
intracellular portion observed with 100 nM 17-AAG was faster than
in SKOV-3 cells. The total uptake was similar for 17-AAG-treated
and untreated SKOV-3 and SKBR-3 cells.
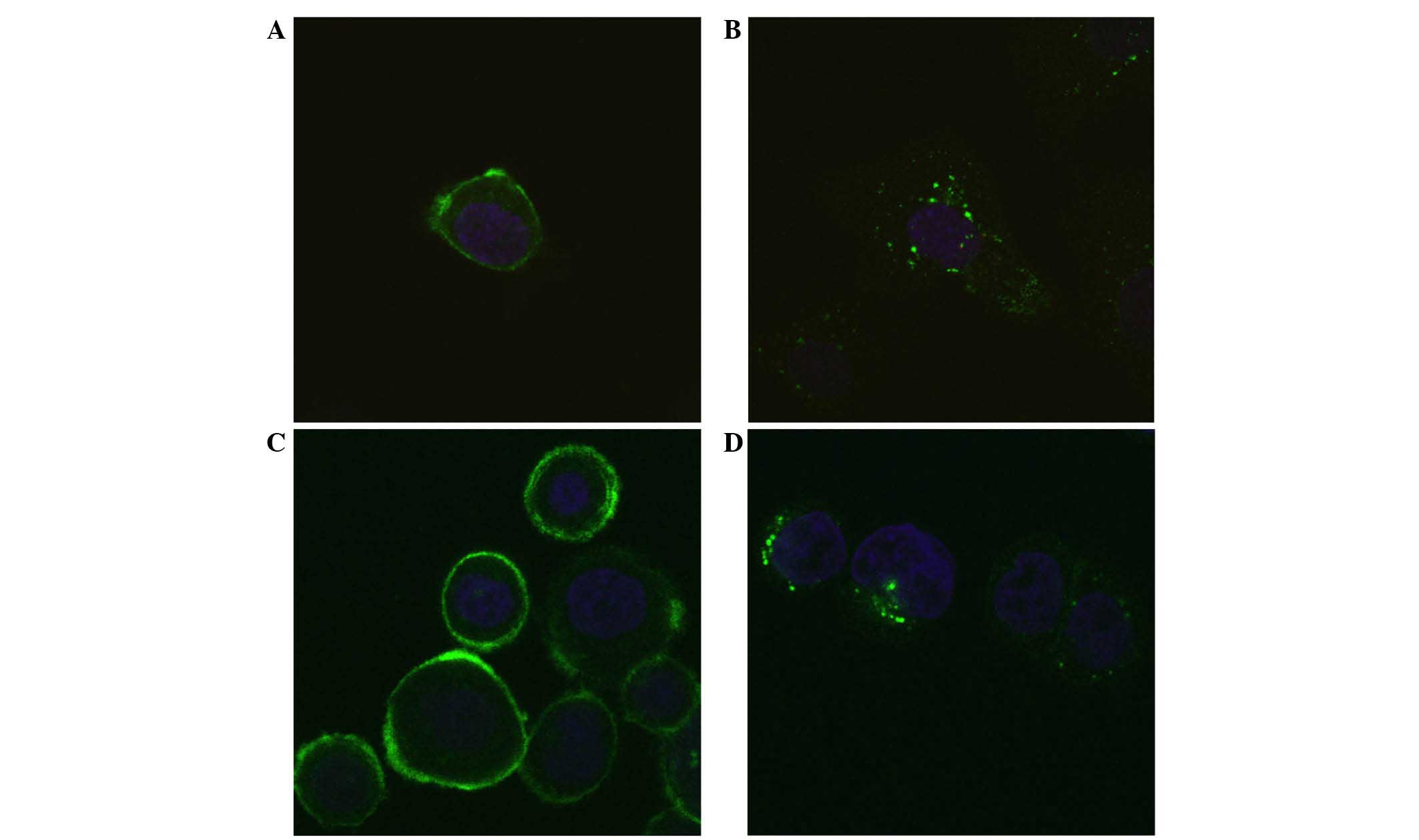
Immunofluorescence staining with
directly labelled Affibody molecules
Alexa488-labelled anti-HER2 Affibody molecules
(Z2891) were incubated with SKOV-3 or SKBR-3 cells for 3 h in the
presence or absence of 100 nM 17-AAG. It was clearly observed that
17-AAG increased intracellular localisation; the fluorescence was
could be detected mainly in vesicles around the nucleus (Fig. 4).
Pre-incubation with
111In-ABY-025
Pre-incubation with 111In-ABY-025 and the
later addition of 17-AAG delayed internalisation somewhat; however,
once 17-AAG was added, the internal uptake seemed to follow the
same pattern as when 17-AAG was added simultaneously (Fig. 5). After 4–6 h, the intracellular
fraction appeared to be similar in all 17-AAG-treated cells,
independently of pre-incubation time (0, 0.5 or 2 h). The total
amount of cell-bound 111In was approximately the same in
all cases, as 17-AAG treatment led to reduced amounts of
111In-ABY-025 at the cell membrane.
Discussion
The HER2 receptor is known to be resistant to
internalisation followed by degradation, and is believed to also
protect EGFR and HER3 from internalisation when forming
heterodimers with these receptors (6,7). Hence,
17-AAG may be used to reduce the number of receptors on the surface
of HER2-overexpressing tumour cells. Furthermore, a number of
downstream signalling proteins are dependent on the Hsp90
chaperone, and application of 17-AAG also leads to degradation of
these proteins (14). A number of
clinical studies on 17-AAG (or tanespimycin) have been published
(26–28) (www.clinicaltrials.gov).
As demonstrated in the present study, 17-AAG can
also be used to increase the internalisation of a HER2-targeted
molecule, such as the Affibody ABY-025. 211At-ABY-025
and 111In-ABY-025 were internalised to a higher extent
with 17-AAG present, and were most likely directed to the lysosomes
for degradation (12). However, the
retention of the nuclides differed. As 211At is a
halogen, it is rapidly excreted by the cells following lysosomal
degradation of the Affibody. The radiometal 111In, on
the other hand, is known to be trapped in the cell. This was
observed when 17-AAG increased the amount of intracellular
111In without decreasing the total amount of
cell-associated 111In (29). The difference in cellular retention
between radiometal- and halogen-labelled proteins has been
demonstrated previously, and is probably a consequence of the
lipophilicity, size and charge of the nuclide, linker and amino
acid to which it is bound (29,30).
The 17-AAG-induced internalisation of ABY-025 could
be useful in several different applications. As long as the
receptor binder is surface-bound, it is in equilibrium with the
extracellular concentration of the binder. When extracellular
concentration decreases, there is a risk of detachment of the
binder, particularly if the internalisation rate is slow, as for
HER2 (5). A triggered internalisation
could increase the cellular retention of radionuclides provided
that the nuclide is retained within the cell. It is well known, and
was demonstrated in this study, that radiohalogens are excreted
rapidly and are therefore not suitable for this approach (30). By contrast, radiometals are
well-suited for this purpose due to their long intracellular
retention. Furthermore, in a situation where metal radionuclides
are used for therapeutic applications, efficacy could be improved
by internalisation. The chance of a alpha or beta particle track
from a radionuclide decay to ionise the DNA strand increases the
closer to DNA the decay occurs due to pure geometry. A dose to the
nucleus of a single cell will be higher if the radionuclide is
positioned in the cytoplasm, compared with a dose from a
radionuclide that is cell surface-bound (31).
The quality of the radiation, as well as the range
of the particle emitted, will also be important factors determining
the damage to the DNA of the cell (30). High LET from short-range particle
tracks, such as α-emitting nuclides, will produce more
lethal double-strand breaks than from low LET β-emitting
nuclides; however, only a few of the α-emitting radionuclides
fulfil the criteria for nuclear medicine applications, the most
studied being 211At, 213Bi, 225Ac
and 223Ra (32). With the
exception of 223Ra, which is injected as the radium
salt, all other proposed α-emitting radionuclides require a
tumour-specific carrier molecule. In the present study,
211At was used for labelling of ABY-025. However, due to
lack of residualising properties of At-211, a better choice in this
case would likely be the α-emitting metal radionuclide
213Bi (32).
Other approaches, such as targeted cytotoxic agents,
siRNAs or RNase, may also benefit from internalisation when aiming
for HER2-expressing cells. Intracellular distribution of these
agents is necessary to effectuate their therapeutic capacity.
Although a method by which to release the agents from the lysosome
to the cytoplasm, or even for further transport into the nucleus,
must be determined, a common risk for all of the aforementioned
approaches is the risk of triggering internalisation of HER2 before
the receptor binder reaches the cells. As shown in the current
study, when 111In-ABY-025 was allowed to bind HER2 prior
to 17-AAG treatment, the internalisation appeared to follow the
same pattern as for simultaneous treatment. It therefore seems
possible to avoid the risk of 17-AAG inducing HER2 internalisation
and degradation before ABY-025 has bound to the receptor.
In conclusion, 17-AAG may be used to facilitate
cell-specific intracellular localisation of a suitable cytotoxic or
radioactive agent coupled to ABY-025 in HER2-overexpressing
cells.17-AAG treatment of SKOV-3 and SKBR-3 cells to some extent
shifts the localisation of 111In-ABY-025 from the cell surface to
intracellular compartments in both cell lines. ABY-025 labelled
with the high-LET alpha emitter 211At is also internalised to a
higher degree, but due to its physiological properties this nuclide
is excreted faster
Acknowledgements
The authors wish to thank Affibody AB for providing
ABY-025; Associate Professor Kecke Elmroth and Helena Kahu of
Gothenburg University (Gothenburg, Sweden) for assisstance with
experimental setups; and Professor Vladimir Tolmachev of Uppsala
University for valuable discussions. This work was supported by the
Swedish Cancer Society.
References
|
1
|
Slamon DJ, Godolphin W, Jones LA, Holt JA,
Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J and Ullrich A:
Studies of the HER-2/neu proto-oncogene in human breast and ovarian
cancer. Science. 244:707–712. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carpenter G and Cohen S: 125I-labeled
human epidermal growth factor. Binding, internalization and
degradation in human fibroblasts. J Cell Biol. 71:159–171. 1976.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stoscheck CM and Carpenter G: Down
regulation of epidermal growth factor receptors: Direct
demonstration of receptor degradation in human fibroblasts. J Cell
Biol. 98:1048–1053. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hendriks BS, Opresko LK, Wiley HS and
Lauffenburger D: Coregulation of epidermal growth factor
receptor/human epidermal growth factor receptor 2 (HER2) levels and
locations: Quantitative analysis of HER2 overexpression effects.
Cancer Res. 63:1130–1137. 2003.PubMed/NCBI
|
|
5
|
Hommelgaard AM, Lerdrup M and van Deurs B:
Association with membrane protrusions makes ErbB2 an
internalization-resistant receptor. Mol Biol Cell. 15:1557–1567.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Worthylake R, Opresko LK and Wiley HS:
ErbB-2 amplification inhibits down-regulation and induces
constitutive activation of both ErbB-2 and epidermal growth factor
receptors. J Biol Chem. 274:8865–8874. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Citri A, Skaria KB and Yarden Y: The deaf
and the dumb: The biology of ErbB-2 and ErbB-3. Exp Cell Res.
284:54–65. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tikhomirov O and Carpenter G: Geldanamycin
induces ErbB-2 degradation by proteolytic fragmentation. J Biol
Chem. 275:26625–26631. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu W, Mimnaugh E, Rosser MF, Nicchitta C,
Marcu M, Yarden Y and Neckers L: Sensitivity of mature Erbb2 to
geldanamycin is conferred by its kinase domain and is mediated by
the chaperone protein Hsp90. J Biol Chem. 276:3702–3708. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou P, Fernandes N, Dodge IL, Reddi AL,
Rao N, Safran H, DiPetrillo TA, Wazer DE, Band V and Band H: ErbB2
degradation mediated by the co-chaperone protein CHIP. J Biol Chem.
278:13829–13837. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Raja SM, Clubb RJ, Bhattacharyya M, Dimri
M, Cheng H, Pan W, Ortega-Cava C, Lakku-Reddi A, Naramura M, Band V
and Band H: A combination of Trastuzumab and 17-AAG induces
enhanced ubiquitinylation and lysosomal pathway-dependent ErbB2
degradation and cytotoxicity in ErbB2-overexpressing breast cancer
cells. Cancer Biol Ther. 7:1630–1640. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lerdrup M, Hommelgaard AM, Grandal M and
van Deurs B: Geldanamycin stimulates internalization of ErbB2 in a
proteasome-dependent way. J Cell Sci. 119:85–95. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Austin CD, De Mazière AM, Pisacane PI, van
Dijk SM, Eigenbrot C, Sliwkowski MX, Klumperman J and Scheller RH:
Endocytosis and sorting of ErbB2 and the site of action of cancer
therapeutics trastuzumab and geldanamycin. Mol Biol Cell.
15:5268–5282. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Powers MV and Workman P: Targeting of
multiple signalling pathways by heat shock protein 90 molecular
chaperone inhibitors. Endocr Relat Cancer. 13(Supply 1): S125–S135.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fukuyo Y, Hunt CR and Horikoshi N:
Geldanamycin and its anti-cancer activities. Cancer Lett.
290:24–35. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sain N, Krishnan B, Ormerod MG, De Rienzo
A, Liu WM, Kaye SB, Workman P and Jackman AL: Potentiation of
paclitaxel activity by the HSP90 inhibitor
17-allylamino-17-demethoxygeldanamycin in human ovarian carcinoma
cell lines with high levels of activated AKT. Mol Cancer Ther.
5:1197–1208. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Banerji U, Sain N, Sharp SY, Valenti M,
Asad Y, Ruddle R, Raynaud F, Walton M, Eccles SA, Judson I, et al:
An in vitro and in vivo study of the combination of the heat shock
protein inhibitor 17-allylamino-17-demethoxygeldanamycin and
carboplatin in human ovarian cancer models. Cancer Chemother
Pharmacol. 62:769–778. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu L, Hofmann J, Lu Y, Mills GB and Jaffe
RB: Inhibition of phosphatidylinositol 3′-kinase increases efficacy
of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer
Res. 62:1087–1092. 2002.PubMed/NCBI
|
|
19
|
Wu X, Wanders A, Wardega P, Tinge B, Gedda
L, Bergstrom S, Sooman L, Gullbo J, Bergqvist M, Hesselius P, et
al: Hsp90 is expressed and represents a therapeutic target in human
oesophageal cancer using the inhibitor
17-allylamino-17-demethoxygeldanamycin. Br J Cancer. 100:334–343.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nygren PA: Alternative binding proteins:
Affibody binding proteins developed from a small three-helix bundle
scaffold. FEBS J. 275:2668–2676. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Friedman M and Ståhl S: Engineered
affinity proteins for tumour-targeting applications. Biotechnol
Appl Biochem. 53:1–29. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Baum RP, Prasad V, Müller D, Schuchardt C,
Orlova A, Wennborg A, Tolmachev V and Feldwisch J: Molecular
imaging of HER2-expressing malignant tumors in breast cancer
patients using synthetic 111In- or 68Ga-labeled affibody molecules.
J Nucl Med. 51:892–897. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ahlgren S, Orlova A, Wallberg H, Hansson
M, Sandström M, Lewsley R, Wennborg A, Abrahmsén L, Tolmachev V and
Feldwisch J: Targeting of HER2-expressing tumors using
111In-ABY-025, a second-generation affibody molecule with a
fund-amentally reengineered scaffold. J Nucl Med. 51:1131–1138.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lindegren S, Frost S, Bäck T, Haglund E,
Elgqvist J and Jensen H: Direct procedure for the production of
211At-labeled antibodies with an epsilon-lysyl-3
(trimethylstannyl)benzamide immunoconjugate. J Nucl Med.
49:1537–1545. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wallberg H and Orlova A: Slow
internalization of anti-HER2 synthetic affibody monomer
111In-DOTA-ZHER2:342-pep2: Implications for development of labeled
tracers. Cancer Biother Radiopharm. 23:435–442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gartner EM, Silverman P, Simon M, Flaherty
L, Abrams J, Ivy P and Lorusso PM: A phase II study of
17-allylamino-17-demethoxygeldanamycin in metastatic or locally
advanced, unresectable breast cancer. Breast Cancer Res Treat.
131(3): 933–7. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pedersen KS, Kim GP, Foster NR,
Wang-Gillam A, Erlichman C and McWilliams RR: Phase II trial of
gemcitabine and tanespimycin (17AAG) in metastatic pancreatic
cancer: a Mayo Clinic Phase II Consortium study. Invest New Drugs.
33(4): 963–8. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Oki Y, Copeland A, Romaguera J, Fayad L,
Fanale M, Sde C Faria, Medeiros LJ, Ivy P and Younes A: Clinical
experience with the heat shock protein-90 inhibitor, tanespimycin,
in patients with relapsed lymphoma. Leuk Lymphoma. 53(5): 990–2.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shih LB, Thorpe SR, Griffiths GL, Diril H,
Ong GL, Hansen HJ, Goldenberg DM and Mattes MJ: The processing and
fate of antibodies and their radiolabels bound to the surface of
tumor cells in vitro: A comparison of nine radiolabels. J Nucl Med.
35:899–908. 1994.PubMed/NCBI
|
|
30
|
Tolmachev V: Chapter 8: Choice of
Radionuclides and Radiolabelling TechniquesTargeted Radionuclide
Tumor Therapy - Biological Aspects. Stigbrand T, Carlsson J and
Adams GP: Springer; pp. 145–174. 2008, View Article : Google Scholar
|
|
31
|
Hartman T, Lundqvist H, Westlin JE and
Carlsson J: Radiation doses to the cell nucleus in single cells and
cells in micro-metas-tases in targeted therapy with (131)I labeled
ligands or antibodies. Int J Radiat Oncol Biol Phys. 46:1025–1036.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wilbur S: Chemical and Radiochemical
Considerations in Radiolabeling with-Emitting Radionuclides.
Current Pharmaceuticals. 4:214–247. 2011. View Article : Google Scholar
|