Overview of ubiquitin-proteasome system
(UPS) and ubiquitin-like proteins (UBL) pathway as a therapeutic
target
The UPS is important in regulating protein
homeostasis in human cells through programmed degradation. The
degradation of regulatory proteins can affect cell-cycle
regulation, cell proliferation, intracellular signaling, DNA repair
and apoptosis (1–4). As UPS degradation is a major clearance
system associated with proteolysis within the cells, its
deregulation can cause the pathogenesis of cancer and other
diseases through the inappropriate loss of regulatory proteins or
unintentional activation of certain specific signalling cascades.
For instance, cancerous cells develop when cell-cycle controls
break down, leading to unregulated cell proliferation. Cancer cells
can also evade apoptosis induced by a number of different cellular
stresses. Hence, an interruption to the normal regulation of the
UPS could lead to abnormal cell proliferation.
The 26S proteasome is a protease complex capable of
degrading polyubiquitinated proteins. The 26S complex is composed
of a barrel-shaped 20S proteasome core with a 19S regulatory
particle at either or both of its ends. The 20S proteasome contains
the enzymatic active sites, whilst the 19S regulatory particle
helps to control access of ubiquitin-like protein (UBL)-conjugated
substrates to the core. There are three proteasome active sites
within the 20S core, namely the caspase-like (β1), trypsin-like
(β2) and chymotrypsin-like (β5) domains. These sites use an
N-terminal threonine as the catalytic amino-acid residue (5,6).
Ubiquitin (Ub) and UBLs share certain common
structural elements, such as a three-dimensional structure called
the Ub or β-grasp fold (7). The UBLs
are a group of proteins encompassing neural precursor cell
expressed, developmentally down-regulated 8 (NEDD8), small
ubiquitin-like modifier 1/2/3, interferon-stimulated gene 15
(ISG15), HLA-F-adjacent transcript 10 (FAT10), autophagy-related
protein (ATG)8 and ATG12, which conjugate to their targets in a
manner similar to that of ubiquitination (8). Ub is a highly conserved protein of 76
amino acids that are able to attach to other proteins in a
reversible fashion. There are three vital structural domains within
Ub: i) the β-grasp fold, commonly found in all UBLs; ii) a
C-terminal tail; and iii) seven lysine residues that correspond to
polyubiquitin-linked chains (6).
NEDD8 has distinctive functions in cells due to its structural
differences, which mediate specialised interactions with target
proteins compared with Ub. The crystal structure of NEDD8 is
analogous to that of Ub, with the exception of two surface regions
(9,10). NEDD8 was initially discovered in fetal
mouse brain (11) and can be found
predominantly in adult tissues (11–13). NEDD8
and the neddylation pathway enzymes are overexpressed in human
cancers (13–15). FAT10 is a 165-amino acid protein that
comprises two Ub-like domains with 29% identity and 36% homology to
Ub at its N and C-termini, respectively (16). The protein is known to be involved in
apoptosis, immune responses and cancer (16–18).
Fig. 1A shows the domain structure of
Ub, NEDD8 and FAT10.
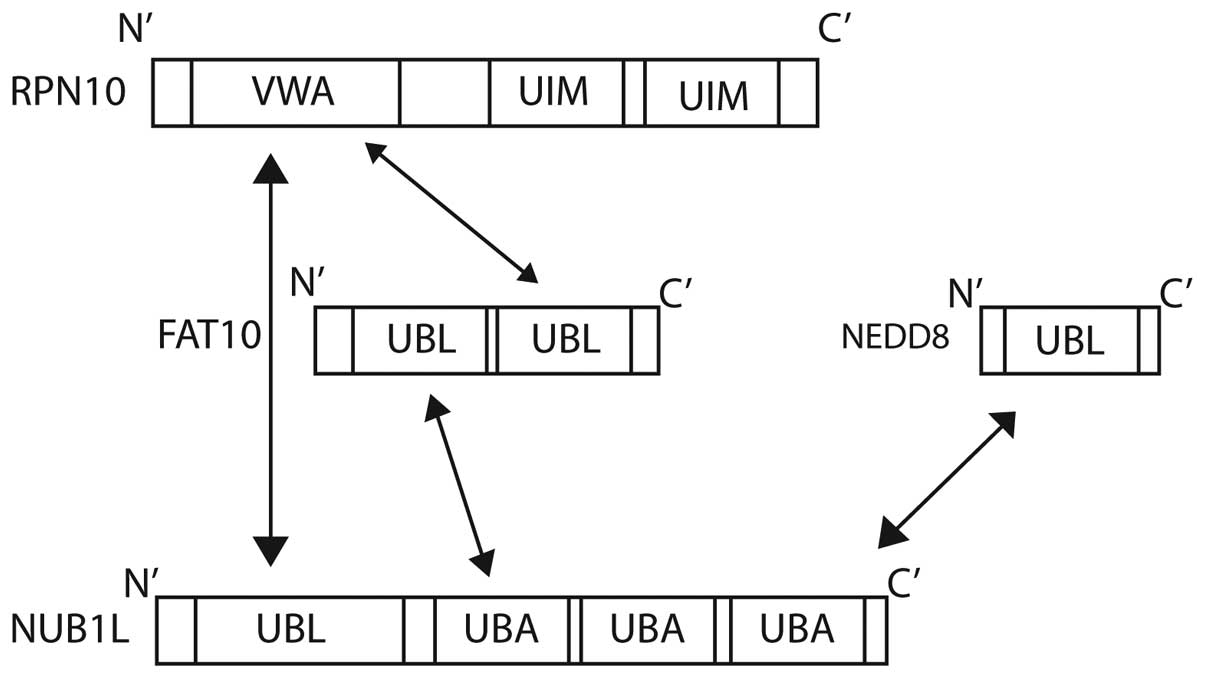
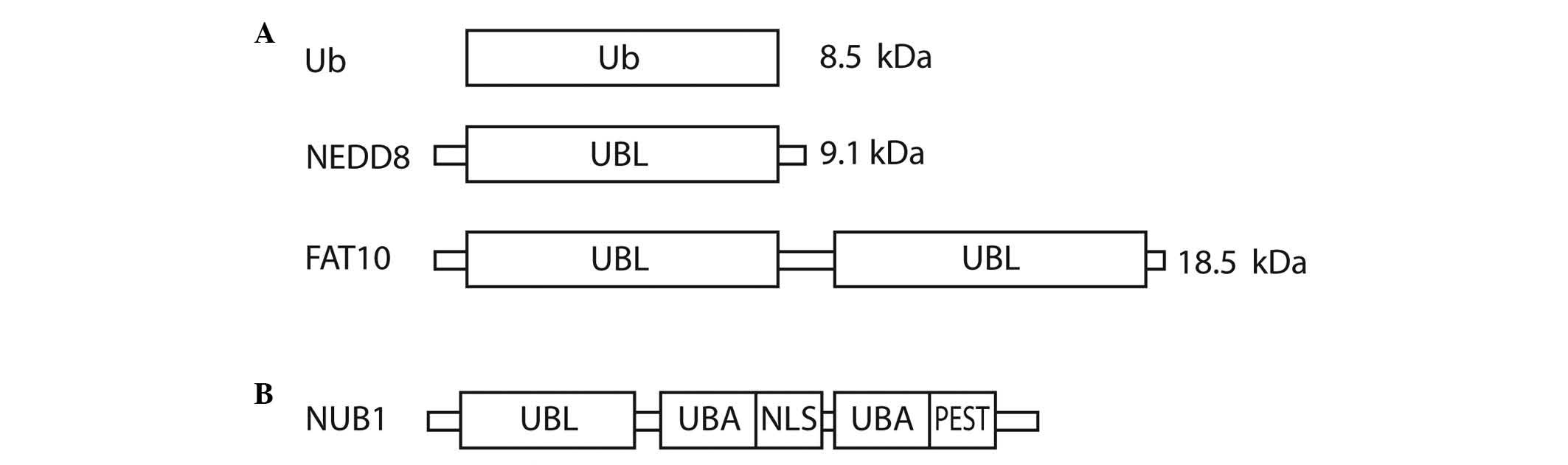 | Figure 1.Structural representation of (A) Ub,
NEDD8 and FAT10, and (B) NUB1. Ub, ubiquitin; NEDD8, neural
precursor cell expressed, developmentally down-regulated 8; FAT10,
HLA-F-adjacent transcript 10; NUB1, NEDD8 ultimate buster 1; UBL,
ubiquitin-like domain; UBA, ubiquitin-associated; NLS, nuclear
localisation signal; PEST, proline-enriched glutamic acid, serine
and threonine domain. |
The roles of UPS and UBL conjugation pathways in
normal cell function and in disease has prompted the search for
inhibitors that are able to selectively disrupt pathway function.
Proteasome inhibitors have been synthesised to halt the function of
the proteasomal activities. As mentioned, the proteasome has three
active sites (β1, β2 and β5), which utilise N-terminal threonine as
the catalytic amino acid residue (7).
All UPS inhibitors were developed to covalently modify this
threonine residue in order to block the enzyme's kinetics. The
therapeutic value of UPS inhibition has been demonstrated with the
proteasome inhibitor bortezomib (Velcade®; Millennium
Pharmaceuticals, Inc., Cambridge, MA, USA). Bortezomib is used in
the treatment of patients suffering from multiple myeloma (19,20) and
mantle cell lymphoma (21).
As the first clinical proteasome inhibitor to target
the UPS, bortezomib was approved in 2003. Several second-generation
proteasome inhibitors are currently in development, such as
carfilzomib, oprozomib, ixazomib citrate, marizomib and delanzomib.
In comparison with UPS inhibition, blocking of UBL pathways may
provide a more specific effect by targeting the substrate proteins.
UPS inhibition is a common step in blocking the degradation of a
broader range of substrates. For instance, inhibition of
E1-activating enzymes may be achieved through covalent inactivation
(e.g. PYR-41) (22) and adduct
formation (e.g. MLN4924) (23).
Blocked E2 interactions offer a more selective inhibition (e.g.
synthetic peptide UBC12N26). Recent research has frequently
focussed on targeting deubiquitinating enzymes (DUBs), as this
class of proteins are capable of reversing the action of the Ub
conjugation cascade. Table I
summarises the current clinical development of second-generation
proteasome inhibitors, and Table II
lists the E1/2/3 inhibitors. Furthermore, a NEDD8-activating enzyme
(NAE) inhibitor, MLN4924, which targets the NEDD8 pathway, appears
to be a potentially important anticancer strategy (24). The development of DUB inhibitors is
more recent compared with that of the proteasome and E1/2/3
inhibitors. To the best of our knowledge, no DUB inhibitors have
entered clinical trials.
 | Table I.First- and second-generation
proteasome inhibitors. |
Table I.
First- and second-generation
proteasome inhibitors.
| Drug | Company | Status |
|---|
| First
generation |
|
|
|
Bortezomib | Millennium
Pharmaceuticals, Inc. (Cambridge, MA, USA) | FDA-approved for
multiple myeloma and relapsed mantle cell lymphoma |
| Second
generation |
|
|
|
Carfilzomib (Kyprolis) | Onyx
Pharmaceuticals, Inc. (San Francisco, CA, USA) | FDA-approved for
multiple myeloma |
|
Oprozomib (ONX0912) | Onyx
Pharmaceuticals, Inc. (San Francisco, CA, USA) | Phase I |
|
Ixazomib citrate
(MLN9708) | Millennium
Pharmaceuticals, Inc. (Cambridge, MA, USA) | Phase I/II |
|
Marizomib (NPI-0052) | Nereus
Pharmaceuticals (San Diego, CA, USA) | Phase I |
|
Delanzomib (CEP-18770) | Cephalon, Inc.
(Frazer, PA, USA) | Phase I |
|
Calpeptin (IPSI-001) | Lanospharma
Laboratories Co., Ltd. (Chongqing, China) | Phase I |
|
ONX0914 | Onyx
Pharmaceuticals (San Francisco, CA, USA) | Phase I |
| Table II.A summary of small molecule
inhibitors targeting E1s, E2s and E3s. |
Table II.
A summary of small molecule
inhibitors targeting E1s, E2s and E3s.
| Drug | Target | Company | Status |
|---|
| PYR-41 | E1 | Millennium
Pharmaceuticals, Inc. (Cambridge, MA, USA) | N/A |
| MLN4924 | E1 | Millennium
Pharmaceuticals, Inc. (Cambridge, MA, USA) | Phase II |
| CC0651 | E2 | N/A | N/A |
| NSC697923 | E2 | N/A | N/A |
| Nutlin | E3 | Roche Products
Limited (Pharmaceuticals) (Welwyn Garden City, UK) | Phase I |
| MI-773 | E3 | Sanofi S.A.
(Gentilly, France) | Phase I |
| CGM097 | E3 | Novartis
International AG (Basel, Switzerland) | Phase I |
NEDD8 ultimate buster 1 (NUB1), a
NEDD8- and FAT10-degrading enzyme, and approaches to anticancer
therapy
NUB1 is an interferon (IFN)-inducible protein of 69
kDa, composed of 601 amino acids. It also has a splice variant,
NUB1L, which possesses an extra 14 amino acids that encode an
additional Ub-associated (UBA) domain (Fig. 1B). NUB1 proteins can recruit FAT10-
and NEDD8-conjugated proteins to the proteasome for degradation and
negatively regulate the NEDD8-conjugation system (25–29). The
NUB1 proteins have been observed in various types of cancer cells,
including cervical adenocarcinoma, rectal adenocarcinoma,
neuroblastoma, malignant lymphoma and renal cell carcinoma (RCC)
(26). Upregulated NUB1 expression
has been linked to IFNα-induced antimitogenic actions.
Additionally, NUB1 has demonstrated anticancer properties in RCC
cell lines, where it was involved in apoptosis and S-phase
transition through its action on p27 and cyclin E (30,31).
Upregulation of NUB1 effectively inhibits the proliferation of
IFNα-resistant RCC cells (31).
NUB1 protein has been reported to play a role in
Huntington's disease (32) and
congenital amaurosis (33). In
cancers, NUB1 is an attractive candidate for inhibition of
p27KIP1 and p21CIP1 via the regulation of the
Skp, Cullin, F-box-containing (SCF)SKP2 ligase activity
(31). The upregulated
p21CIP1 in NUB1-knockdown cancer cells is thought to be
promising in directing the cells to senescence. NUB1 protein was
reported to be a tumour suppressor as it exerts growth inhibition
during its overexpression; upon IFNα treatment, overexpressed NUB1
induced apoptosis in IFNα-resistant A498 cells (31). However, its general lack of enzymatic
activities makes NUB1 less suitable for small molecule inhibition
(32). Thus, the low-molecular-weight
proteins FAT10 and NEDD8 could be key to developing novel
strategies in anticancer therapy, as they interact with NUB1
(34). The current review focuses on
the relevance of NUB1 protein in NEDD8 and FAT10 conjugation in
cancers, and the potential for targeting it as a novel therapeutic
approach.
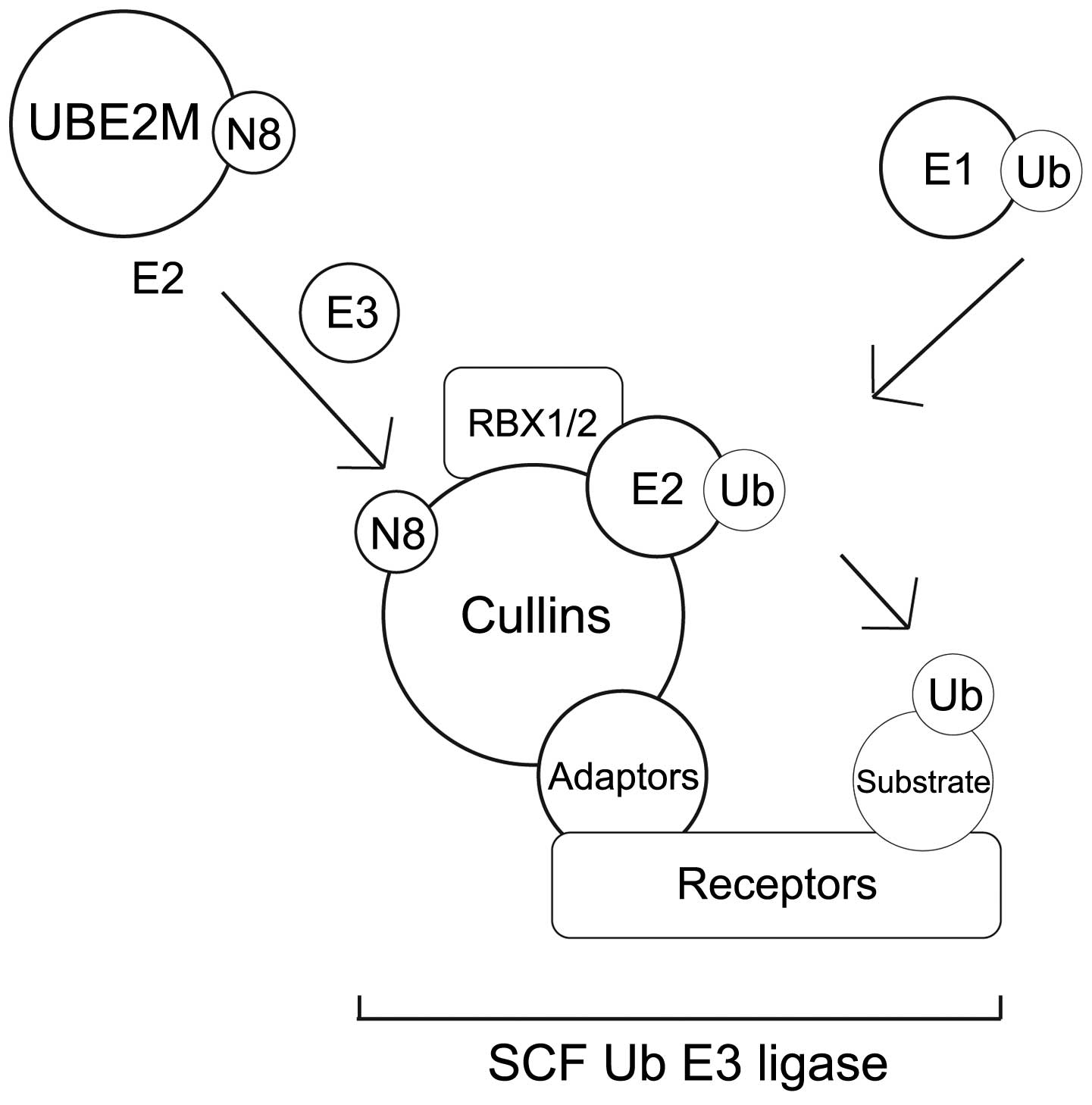
The NEDD8-conjugation (neddylation) pathway
and the effect of neddylation on transcription factors
The UBL enzymatic cascade scheme that results in UBL
conjugation and protein degradation involves several distinct
steps. Each step requires different classes of enzyme, as shown in
Table III.
| Table III.Overview of the enzymatic cascade
involved in UBL conjugation. |
Table III.
Overview of the enzymatic cascade
involved in UBL conjugation.
| UBL | Ub | NEDD8 | FAT10 and Ub |
|---|
| E1-activating
enzymes | UAE | APPBP1-UBA3
heterodimer | UBA6 |
| E2-conjugating
enzymes | UBCs | UBE2M and
UBE2F | USE1 |
| E3 ligases | Ub E3 ligases | NEDD8 E3
Ligases | N/A |
| Substrates | 1,000s | 200s | N/A |
Neddylation is a post-translational modification
process that conjugates NEDD8 to its target proteins, in a process
that is analogous to that observed for ubiquitination. However, the
neddylation process uses a distinct E1 and E2 enzyme reaction
scheme (5,8,11,35,36)
(Table III). Fig. 2A summarises the NEDD8 conjugation and
deconjugation steps (34,37). The C-terminal glycine of NEDD8 is
adenylated by an E1 NEDD8-activating enzyme (NAE), a heterodimer
composed of amyloid-β precursor protein-binding protein 1 (APPBP1)
and Ub-like modifier-activating enzyme (UBA) 3. NEDD8 is covalently
conjugated to the NAE via a thiolester linkage (38). The activated NEDD8 is consecutively
transferred to the E2 NEDD8-conjugation enzyme and then to the
specific substrates (i.e. cullin proteins) via an isopeptide bond
(39). The RING-box protein (RBX)
1/ROC1, mouse double minute 2 homolog (MDM2), F-box protein 11 and
c-Cbl proteins are neddylated in the same way (39).
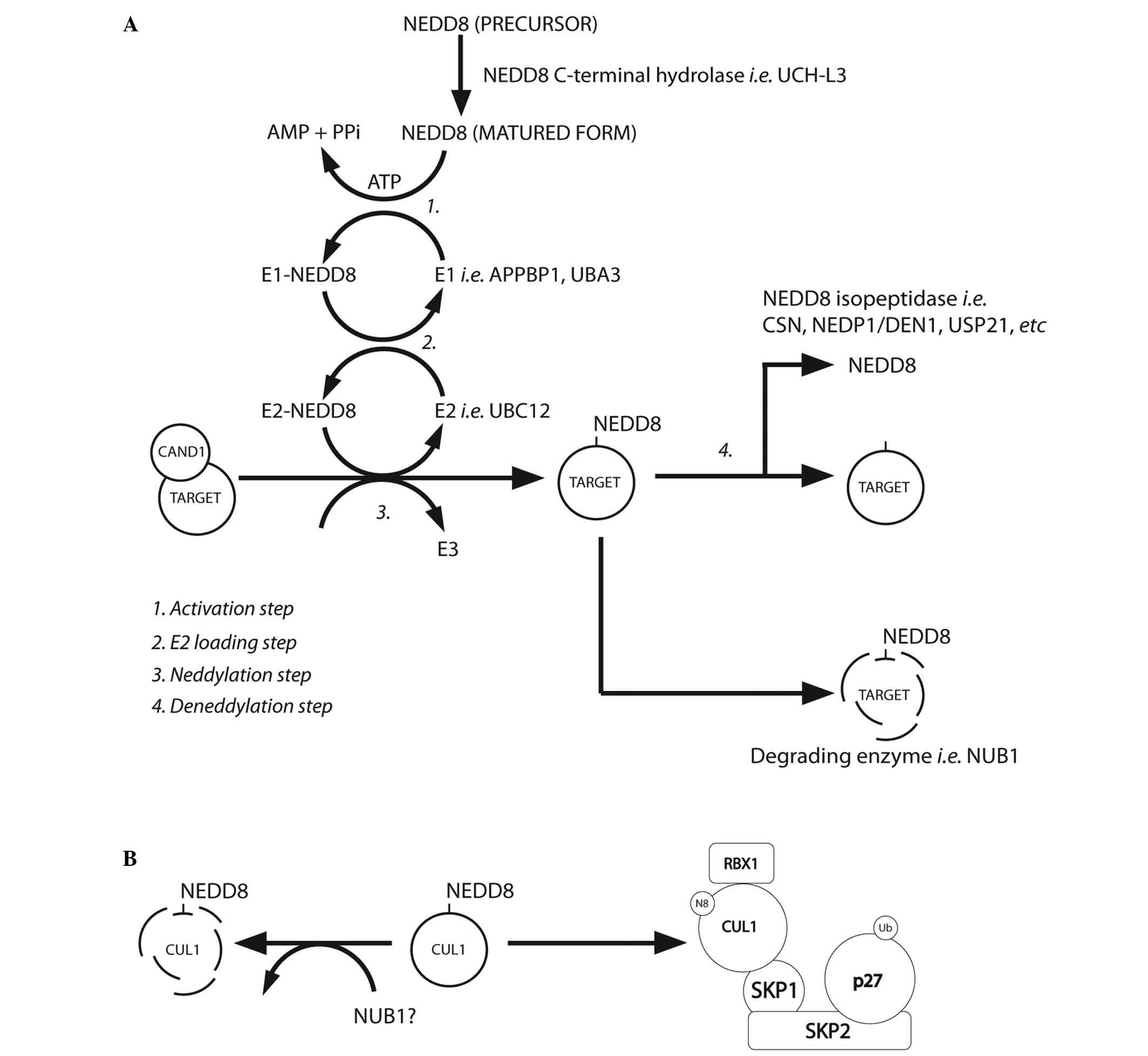 | Figure 2.(A) NEDD8 conjugation pathway.
Schematic summary of the main steps of the neddylation pathway
[modified from Rabut and Peter, 2008 (37); Tanaka et al, 2012 (34)]. (B) Neddylated CUL1 locks the SCF
complex with phosphorylated p27 and cyclin E [as suggested by
Bornstein et al, 2006 (30)
and Tanaka et al, 2012 (34)].
CAND1, cullin-associated and neddylation-dissociated 1; AMP,
adenosine monophosphate; PPi, anion P2O74-; ATP,
adenosine triphosphate; UCH-L3, ubiquitin C-terminal hydrolase
isozyme L3; APPBP1, amyloid-β precursor protein binding protein 1;
DEN1, deneddylase 1, CSN, COP9 signalosome; NEDP1, NEDD8-specific
protease 1; NUB1, NEDD8 ultimate buster 1; USP21, ubiquitin
specific peptidase 21, CUL1, cullin 1; SKP1, S-phase
kinase-associated protein 1; SKP2, S-phase kinase-associated
protein 2; RBX1, ring-box 1, E3 ubiquitin protein ligase; SCF, Skp,
cullin, F-box-containing complex. |
There are two NEDD8-specific E2-conjugating enzymes,
namely ubiquitin conjugating enzyme E2 (UBE2)M (also known as
UBC12) and UBE2F. These E2 enzymes act to transfer NEDD8 to its
target protein through E3 enzymes. It has been reported that all
NEDD8 E3 enzymes can function as Ub E3 enzymes. The predominant
NEDD8 E3 ligases are the RING subunits RBX1 and RBX2 (38,40–43).
Meanwhile, the non-RBX family NEDD8 E3 ligases include c-CBL, ring
finger protein 111, MDM2 and inhibitor of apoptosis 1 (44).
Of the numerous NEDD8 substrates, neddylation has
been best described in the cullin family (45). In this mechanism, NEDD8 from the E2
cysteine active site is transferred onto a lysine residue in the
N-terminus of the target proteins (Fig.
2A) (46). Cullin neddylation is
further mediated by defective in cullin neddylation protein 1-like
proteins (44). It was reported that
RING E3 ligases could neddylate the same substrate on multiple
lysine residues (44). However, the
interaction between non-RBX RING E3 ligase and E2 enzymes remains
to be elucidated.
NEDD8-conjugated substrates are deneddylated by
various proteins that include COP9 signalosome (CSN),
NEDD8-specific protease 1 (NEDP1/DEN1) and ubiquitin specific
peptidase 21 (37,47–51).
Neddylation may be inhibited by cullin-associated and
neddylation-dissociated 1 through its direct binding to cullins
(52,53). NEDD8 and neddylated substrates are
recruited by NUB1 for proteasomal degradation (25,26)
(Fig. 2A). In the G1-S-phase
transition, Bornstein et al (30) demonstrated how neddylated cullin 1
cooperatively activates the SCFSKP2 Ub ligase complex,
which results in p27 degradation (Fig.
2B). Furthermore, previous studies found that cullin
neddylation increased the Ub E3 ligase activity of the SCF complex
(Fig. 3) (46,54).
NEDD8 is negatively regulated by NUB1, which links
the UBLs to the 26S proteasome for further UPS degradation. Reports
have described that NUB1 is able to recruit NEDD8 and
NEDD8-conjugated proteins to the proteasome for degradation, and
this may modulate the cell-cycle profile in response to stresses
(34). The capability of NEDD8 to
activate the Ub E3 ligase-SCF complex (by covalent binding to
cullins) adds further complexity to the ubiquitination machinery
(11,55–59).
Therefore, validation of NEDD8 targets would allow identification
of genuine NEDD8 substrates.
Challenges in identifying physiological
neddylation targets
Hjerpe et al (45) demonstrated that NEDD8 and Ub cascades
are independent of one another during normal cellular homeostasis.
NEDD8 conjugation onto Ub substrates through the Ub cascade has a
spurious role in normal physiological conditions. The single amino
acid change in the C-terminus of NEDD8 compared to Ub, from Arg72
to Ala72, confers the specificity between these two UBLs (44). This ensures that the correct UBL is
passed to the appropriate E2 enzyme, E3 enzyme and the substrate
respectively (Table III). However,
when NEDD8 is in excess, the NEDD8 E1 enzyme UBA1 can activate
NEDD8, which is then transthiolated to Ub E2 enzymes. This
phenomenon results in the neddylation of Ub-specific substrates
(10,45). NEDD8 can form NEDD8 chains or mixed
Ub-NEDD8 chains (39,60). An increase of NEDD8 over Ub, as a
result of cellular stresses, cellular diversity or pathological
conditions, could exert different effects on neddylated substrates
(44). This raises concerns, since
the majority of research performed to date to identify neddylated
substrates in cells relies on the overexpression of NEDD8; as this
would cause an imbalance between cellular NEDD8 and Ub levels, it
could result in the aberrant neddylation of proteins via the Ub
pathway (45).
Enchev et al (44) therefore revised and proposed a set of
criteria to define the search for physiological neddylation
targets: A neddylation substrate must demonstrate the covalent
attachment of NEDD8 through the carboxyl-terminal glycine to the
lysine residue of the substrates; and the neddylation must be
detected under homeostatic conditions under endogenous NEDD8 levels
and substrate expression. The NAE inhibitor MLN4924 should be
incorporated into the study, as it blocks cullin neddylation but
not ubiquitination (44). It remains
optional to examine the possible NEDD8 E2 and E3 enzymes (44,45). It is
also advisable to look at the regulation and biological
consequences of neddylation (44). In
endogenous protein experiments, immunoprecipitation with specific
antibodies is a recommended approach (44). Genome editing techniques, such as a
CRISPR/cas9 approach, may be used to introduce affinity-tagged
versions of a particular gene product (44). The NEDD8 substrate should also be
confirmed using mass spectrometry, using LysC protease as the
cleavage enzyme, as it can discriminate between Ub, NEDD8 and ISG15
conjugates (61). Mass spectrometry
can also be used to determine the site of the neddylated Lys
residue, and the type of NEDD8 chains that are formed. The
neddylated Lys residue needs further study if it is also targeted
by Ub. The relative abundance of Ub, NEDD8 and FAT10 must be
examined for its physiological relevance (44). A mutant form of the substrate that can
no longer be neddylated must also be included to serve as a
negative experimental control (44).
Overexpression of NEDD8 and the aberrant activation
of the neddylation pathway and cullin-RING Ub ligase (CRL) activity
can drive the progression of cancers (4,13),
inflammatory and autoimmune diseases (7). Mainstream research focuses on the
effects of CRL inhibition, neddylation and deneddylation. The
small-molecule NAE inhibitor MLN4924 is undergoing clinical trials.
MLN4924 is an analog of adenosine monophosphate that competitively
binds to the enzymatic pocket of NAE. This small molecule therefore
inhibits neddylation and CRL activity. MLN4924 treatment causes DNA
replication by stabilising chromatin licensing and DNA replication
factor 1, a DNA replication licensing factor and CRL substrate.
MLN4924-treated cells accumulate DNA damage due to DNA repair
failure, leading to apoptosis (62)
or senescence (19). Neddylation is
able to inhibit the transcriptional activity of the tumour
suppressors p53 and p73, and to stabilise Hu-antigen R (63), cell division cycle 6 and
hypoxia-inducible factors (64). One
of the important outcomes of MLN4924 treatment is that it causes
the cancer cells to undergo apoptosis and senescence (44).
Transcriptional regulation via the
neddylation of transcription factors
Several studies have suggested that neddylation of
transcription factors can lead to the suppression of their
transcriptional activity (44,65).
E2F transcription factor 1 (E2F1)
Neddylation of E2Fs reduces their transcriptional
activity (66,67). E2F1 was shown to be neddylated in the
DNA-binding domain and its protein levels reduced following
neddylation (67). DEN1 deneddylates
E2Fs and consequently activates E2F-mediated transcription. DNA
damage promotes the expression of DEN1, which subsequently
deneddylates E2F and causes its stabilisation (68). Neddylation specifically regulates a
subset of E2F target genes; for example, E2F1 deneddylation upon
DNA damage triggers the transcription of proapoptotic factors
(66).
p53 and p73
p53 acts by inhibiting cell cycle progression or
triggering senescence or apoptosis (33). It is inhibited by the RING-domain E3
ligase MDM2, which targets ubiquitinated p53 for degradation. MDM2
is able to neddylate p53 and inhibit its transcriptional activity
(33). The neddylated p53 is further
recruited by NUB1, leading to its inactivation (69). p73 that is neddylated by MDM2
undergoes cytoplasmic relocalisation and downregulation of
transcriptional activity (70). In
addition, the Ub E3 ligase SKP1-CUL1-F-box protein 11
(SCFFbox11) may neddylate p73 and downregulate its
transcriptional activities (71).
Nuclear factor κB (NF-κB)
When extracellular signaling is absent, NF-κB is
distributed in the cytoplasm and inhibited by inhibitor of NF-κB
(IκB) family members. Upon stimulation by proinflammatory cytokines
[such as tumour necrosis factor (TNF)], IκB kinases (IKKs) -α, -β
and -γ phosphorylate IκB, which is then ubiquitylated and targeted
for degradation by SCFβTrCP. Under the same conditions,
IKKγ was reported to be neddylated and degraded by the proteasome,
which reduces NF-κB activation and inhibits NF-κB activity in
gastrointestinal neoplasia (72).
Therefore, neddylated IKKγ may exert a tumour suppressor
function.
Amyloid precursor protein intracellular
domain (AICD)
Amyloid precursor protein is cleaved by secretase to
become amyloid-β peptide and AICD. AICD is a component of a
transcription factor complex with amyloid-β (A4) precursor
protein-binding family B member 1 (FE65) and TAT-interactive
protein 60 (73). Neddylated AICD
blocks its interaction with FE65 and prevents the formation of the
transcription factor complex, thereby reducing its transcriptional
activity (2,74). Thus, neddylated AICD inhibits the
transcription of downstream targets.
The FAT10-conjugation (FAT10ylation) pathway
and its function
FAT10 was discovered by Sherman Weissman in 1996
(5). Due to the poor solubility of
the protein at high concentrations, the structure of FAT10 protein
was only recently defined (75).
FAT10 consists of two β-grasp fold domains connected by a short
linker (75). FAT10 protein was found
to be expressed predominantly in immune tissue, including the
thymus, lymph nodes and spleen (76–78). Its
expression is stimulated by proinflammatory cytokines, namely IFNγ
and TNFα (79). FAT10 protein is
found in mature dendritic cells and it demonstrates oncogenic
characteristics; ectopic expression of FAT10 causes malignant
transformation and promotes tumour growth (80), and it is known to be upregulated in
several tumour types, including liver and colon tumours (18,81).
FAT10 shares the same E1 and E2 enzymes with the Ub
conjugation pathway. The FAT10 E1 enzyme UBA6 is able to activate
Ub and FAT10 (29,82–84). The
adenylation and transthiolation reactions of FAT10 are kinetically
slower than those for Ub. UBA6 protein is thought to be the only
FAT10 E1 enzyme in cells, since UBA6 knockdown can effectively
abolish the formation of FAT10-conjugates in vitro (84,85).
Similarly, UBA6-specific E2 enzyme (USE1) is the only UBA6-specific
E2 enzyme discovered to be involved in FAT10 conjugation, although
it also functions in a similar fashion to the conjugation of Ub
(83). USE1 may only bind to
activated Ub from UBA6, not UBE1 (83).
Little is currently known about FAT10, and research
to identify possible FAT10 E3 ligases and deconjugating enzymes is
ongoing. One study demonstrated that ectopically expressed FAT10
was not degraded over time, suggesting the possibility that a group
of FAT10-deconjugating enzymes may not exist (86). It is believed that FAT10 is capable of
promoting its own proteasomal-dependent degradation without the aid
of deconjugating enzymes (86).
FAT10-conjugated proteins were found to have a reduced half-life,
similar to that observed for Ub-conjugated proteins (21). Conversely, it was demonstrated that
FAT10-conjugated p62 accumulated under proteasome inhibition
(79).
The interferon-inducible protein NUB1 interacts with
FAT10 non-covalently (25), and
significantly accelerates the degradation of FAT10 by the
proteasome (25). NUB1 binds to the
proteasome subunit S5a (28), and
also to FAT10 via its three C-terminal UBA domains (Fig. 4) (87).
NUB1 is also able to interact with the von Willebrand A (VWA)
domain of RPN10 (S5a), one of the subunits of the 26S proteasome
(25,28). The degradation of FAT10 is accelerated
further by NUB1 splicing variant, NUB1L, which is able to bind to
regulatory particle non-ATPase (RPN)10 in addition to the 19S
regulator subunit, RPN1 (S2) (87).
The 26S proteasome subunit Rpn10 (S5a) is the
docking site for FAT10, NUB1L and polyubiquitin. Ub interaction
motifs 1 and 2 of Rpn10 are bound by lysine 48-linked polyubiquitin
chains. FAT10 is able to target substrate proteins to the
proteasome independently of poly-FAT10ylation. FAT10 interacts
directly with the VWA domain of RPN10, and no ubiquitination is
required (28). The co-expression of
NUB1L has been shown to accelerate the degradation of FAT10,
suggesting a preference for proteasomal degradation (28).
Substrates of FAT10 conjugation. The biological
function of FAT10 remains poorly understood. FAT10 overexpression
has been demonstrated to induce apoptosis in mouse fibroblasts
(67), HeLa cells (68) and renal tubular epithelial cells
(70). FAT10 is synergistically
induced by IFNγ and TNF-α, which leads to the induction of
apoptosis (25). Several
FAT10-interacting proteins have been identified, and are summarised
in the following paragraphs.
The inflammatory mediator leucine-rich repeat
Fli-I-interacting protein 2 (LRRFIP2) is covalently modified by
FAT10 (6). LRRFIP2 positively
regulates the activity of NF-κB in the inflammatory response
mediated by toll-like receptor (TLR)4 (6). FAT10ylation of LRRFIP2 hinders its
recruitment to the plasma membrane, which results in the inhibition
of lipopolysaccharides (LPS)/TLR4-mediated NF-κB activation
(6). This consequently leads to the
reduced expression of NF-κB-responsive genes, including apoptosis
inhibitors (6). Overexpression of
FAT10 can induce apoptosis, causing FAT10NULL mice to be
hypersensitive to LPS challenge due to NF-κB inhibition (33). However, FAT10 was observed to protect
leukocytes in the spleen, thymus and bone marrow from apoptosis in
a mouse model (33). In another
study, the colon cancer cell line HCT116 was protected from
TNF-α-induced apoptosis in the presence of FAT10 (88). The induction of apoptosis by FAT10 is
therefore cell type-specific; however, the mechanisms involved
remain unknown.
Mitotic arrest-deficient 2 (MAD2), a spindle
assembly checkpoint protein, binds to FAT10 protein non-covalently
(33). In prometaphase, overexpressed
FAT10 in HCT116 cells was found to reduce the localisation of MAD2
at the kinetochore (88,89). Ren et al (88) reported that TNF-α-induced upregulation
of FAT10 also delocalised MAD2 from kinetochores in a similar way
and accelerated cell mitosis. The mis-segregation of chromosomes
was shown to be abolished when FAT10 levels were reduced by siRNA
(89). Hence, FAT10 is considered to
cause mis-segregation of chromosomes during cell division (88,89).
FAT10 has been found to be highly expressed in
colorectal, ovarian, hepatocellular and uterine carcinomas,
suggesting that FAT10 expression may promote oncogenesis (89). A study found that 72% of
hepatocellular carcinoma and 53% of colon carcinoma tissues
overexpressing FAT10 also expressed the IFNγ/TNF-α-dependent
immunoproteasome subunit low molecular mass protein 2 (90), suggesting that the pro-inflammatory
cytokine response may be responsible for FAT10 overexpression in
carcinoma tissues.
Autophagy adaptor p62 or sequestosome-1 protein can
regulate aggresome formation, which protects cells from
aggregation-prone protein-induced toxicity (79). FAT10ylated p62 tends to be
proteasomally degraded (79). A
previous study revealed that FAT10 expression induced by
pro-inflammatory cytokines leads to a decrease in endogenous p62
(79). FAT10 was found to be
transported by histone deacetlyase 6 along microtubules into
aggresomes, causing p62 degradation (79). Under pathological conditions, p62 is
localised in aggresomes along with the aggregated proteins found in
neuronal diseases, including Alzheimers (91). The impact of FAT10 on P62-induced
pathogenesis remains unresolved. However, there is no evidence that
FAT10ylated p62 has a role in autophagic pathways (91).
NEDD8 and FAT10 pathway perspectives
The SCF Ub E3 ligases have been shown to be
deregulated in various cancers; this results in unlimited cell
proliferation and carcinogenesis via accumulation of their
substrate proteins (34).
Consequently, the E3 ligases are the subject of research into
potential strategies for anticancer therapy (34). It is believed that the NEDD8-Ub-SCF
complexes and the NEDD8-FAT10-degrading enzyme NUB1 are potential
candidates for therapy (Fig. 2B)
(34).
The search for neddylation targets requires further
experimental validation. The conventional ectopic overexpression of
UBLs is thought to lead to false positive conjugation of substrates
(45). However, genome editing
techniques, such as CRISPR-Cas9 technology, could overcome this, as
it permits the neddylated substrates to be examined endogenously
(44). In addition to NEDD8 and Ub
chain formation, proteomic studies have reported phosphorylation,
acetylation and succinylation sites on NEDD8. The functional
significance of this observation remains unknown. There is a
general lack of information on non-cullin protein neddylation under
homeostatic conditions. Furthermore, the physiological relevance of
several reported NEDD8 substrates, including p62/sequestosome,
remains unknown. Whether neddylation is functionally distinct from
ubiquitination is a question that remains unresolved. For example,
polyneddylation and polyubiquitination at DNA damage sites or in
response to other stress conditions are functionally redundant, and
NEDD8 and Ub may be recognised by the same interaction motifs.
However, in certain circumstances, such as the neddylation or
ubiquitination of TGFβRII, these two modifications can elicit
distinct biological responses. Efforts are clearly needed to
identify and characterise NEDD8-interacting domains and
proteins.
NUB1 proteins cause the degradation of FAT10- and
NEDD8-conjugated targets. Their expression regulates NEDD8- and
FAT10-based signalling in response to cellular stresses (34,45).
However, the structural mechanisms of NUB1 protein and its clinical
relevance in UBL pathways remain to be explored. Hosono et
al (31) found that
overexpression of NUB1 inhibits cell growth, and the same study
demonstrated lower NUB1 mRNA expression in IFNα-sensitive 4THUR
cells. The same study highlighted that NUB1 is not induced in
IFNα-resistant cells, although transiently expressed NUB1
sensitised the same cells and induced apoptosis (31). Therefore, killing IFNα-resistant cells
by increasing NUB1 activity is a potential strategy (31).
FAT10 research is still in an early stage and the
biological consequences of FAT10ylation are poorly described.
De-FAT10ylating enzymes are under active investigation as drug
targets in the pharmaceutical industry at present, based on the
fact that a number of putative FAT10 targets are oncogenes or
inhibitors of apoptosis. Future works should focus on the
FAT10-modulated proteasome system and mechanisms of
cytokine-induced reactions.
Conclusion
Experimental studies have demonstrated that negative
regulation of the Ub, NEDD8 and FAT10-conjugation pathways have
great potential in the context of cancer suppression. SCF complexes
are often deregulated in cancer and could be modulated through
manipulation of cullin neddylation. Neddylation and FAT10ylation
inhibitors have recently been developed as a novel class of
anticancer agent. These compounds are expected to exhibit better
specificity for cancer cells and have reduced toxicity. Degrading
enzymes, such as CSN and NUB1/NUB1L, are attractive candidates for
the inhibition of Ub, NEDD8 and FAT10-ligase activities (34). These are expected to provide new
strategies in anticancer therapy.
Acknowledgements
Mr. Ka-Liong Tan is financially supported by the
Academic Staff Training Scheme (SLAI scheme), Ministry of
Education, Malaysia.
References
|
1
|
Ciechanover A: The ubiquitin-proteasome
pathway: On protein death and cell life. EMBO J. 17:7151–7160.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ciechanover A and Schwartz AL: The
ubiquitin-proteasome pathway: The complexity and myriad functions
of proteins death. Proc Natl Acad Sci USA. 95:2727–2730. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hershko A: The ubiquitin system for
protein degradation and some of its roles in the control of the
cell division cycle. Cell Death Differ. 12:1191–1197. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hershko A and Ciechanover A: The ubiquitin
system. Annu Rev Biochem. 67:425–479. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu YC, Pan J, Zhang C, Fan W, Collinge M,
Bender JR and Weissman SM: A MHC-encoded ubiquitin-like protein
(FAT10) binds noncovalently to the spindle assembly checkpoint
protein MAD2. Proc Natl Acad Sci USA. 96:4313–4318. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gunawardena HP, Huang Y, Kenjale R, Wang
H, Xie L and Chen X: Unambiguous characterization of site-specific
phosphorylation of leucine-rich repeat Fli-I-interacting protein 2
(LRRFIP2) in Toll-like receptor 4 (TLR4)-mediated signaling. J Biol
Chem. 286:10897–10910. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hochstrasser M: Origin and function of
ubiquitin-like proteins. Nature. 458:422–429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bawa-Khalfe T and Yeh ET: SUMO losing
balance: SUMO proteases disrupt SUMO homeostasis to facilitate
cancer development and progression. Genes Cancer. 1:748–752. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rao-Naik C, delaCruz W, Laplaza JM, Tan S,
Callis J and Fisher AJ: The rub family of ubiquitin-like proteins.
Crystal structure of Arabidopsis rub1 and expression of multiple
rubs in Arabidopsis. J Biol Chem. 273:34976–34982. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Whitby FG, Xia G, Pickart CM and Hill CP:
Crystal structure of the human ubiquitin-like protein NEDD8 and
interactions with ubiquitin pathway enzymes. J Biol Chem.
273:34983–34991. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kamitani T, Kito K, Nguyen HP and Yeh ET:
Characterization of NEDD8, a developmentally down-regulated
ubiquitin-like protein. J Biol Chem. 272:28557–28562. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim DY, Kwon E, Hartley PD, Crosby DC,
Mann S, Krogan NJ and Gross JD: CBFβ stabilizes HIV Vif to
counteract APOBEC3 at the expense of RUNX1 target gene expression.
Mol Cell. 49:632–644. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hori T, Osaka F, Chiba T, Miyamoto C,
Okabayashi K, Shimbara N, Kato S and Tanaka K: Covalent
modification of all members of human cullin family proteins by
NEDD8. Oncogene. 18:6829–6834. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Salon C, Brambilla E, Brambilla C,
Lantuejoul S, Gazzeri S and Eymin B: Altered pattern of Cul-1
protein expression and neddylation in human lung tumours:
Relationships with CAND1 and cyclin E protein levels. J Pathol.
213:303–310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chairatvit K and Ngamkitidechakul C:
Control of cell proliferation via elevated NEDD8 conjugation in
oral squamous cell carcinoma. Mol Cell Biochem. 306:163–169. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Raasi S, Schmidtke G and Groettrup M: The
ubiquitin-like protein FAT10 forms covalent conjugates and induces
apoptosis. J Biol Chem. 276:35334–35343. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fan W, Cai W, Parimoo S, Schwarz DC,
Lennon GG and Weissman SM: Identification of seven new human MHC
class I region genes around the HLA-F locus. Immunogenetics.
44:97–103. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee CG, Ren J, Cheong IS, Ban KH, Ooi LL,
Tan S Yong, Kan A, Nuchprayoon I, Jin R, Lee KH, et al: Expression
of the FAT10 gene is highly upregulated in hepatocellular carcinoma
and other gastrointestinal and gynecological cancers. Oncogene.
22:2592–2603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Soucy TA, Dick LR, Smith PG, Milhollen MA
and Brownell JE: The NEDD8 conjugation pathway and its relevance in
cancer biology and therapy. Genes Cancer. 1:708–716. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Richardson PG, Sonneveld P, Schuster MW,
Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D,
Lonial S, Goldschmidt H, et al: Bortezomib or high-dose
dexamethasone for relapsed multiple myeloma. N Engl J Med.
352:2487–2498. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
San Miguel JF, Schlag R, Khuageva NK,
Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT,
Palumbo A, Samoilova OS, et al: Bortezomib plus melphalan and
prednisone for initial treatment of multiple myeloma. N Engl J Med.
359:906–917. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kane RC, Dagher R, Farrell A, Ko CW,
Sridhara R, Justice R and Pazdur R: Bortezomib for the treatment of
mantle cell lymphoma. Clin Cancer Res. 13:5291–5294. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang Y, Kitagaki J, Dai RM, Tsai YC,
Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P,
et al: Inhibitors of ubiquitin-activating enzyme (E1), a new class
of potential cancer therapeutics. Cancer Res. 67:9472–9481. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao Y, Xiong X, Jia L and Sun Y:
Targeting Cullin-RING ligases by MLN4924 induces autophagy via
modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis.
3:e3862012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kamitani T, Kito K, Fukuda-Kamitani T and
Yeh ET: Targeting of NEDD8 and its conjugates for proteasomal
degradation by NUB1. J Biol Chem. 276:46655–46660. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kito K, Yeh ET and Kamitani T: NUB1, a
NEDD8-interacting protein, is induced by interferon and
down-regulates the NEDD8 expression. J Biol Chem. 276:20603–20609.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tanaka T, Kawashima H, Yeh ET and Kamitani
T: Regulation of the NEDD8 conjugation system by a splicing
variant, NUB1L. J Biol Chem. 278:32905–32913. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tanji K, Tanaka T and Kamitani T:
Interaction of NUB1 with the proteasome subunit S5a. Biochem
Biophys Res Commun. 337:116–120. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Groettrup M, Pelzer C, Schmidtke G and
Hofmann K: Activating the ubiquitin family: UBA6 challenges the
field. Trends Biochem Sci. 33:230–237. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bornstein G, Ganoth D and Hershko A:
Regulation of neddylation and deneddylation of cullin1 in SCFSkp2
ubiquitin ligase by F-box protein and substrate. Proc Natl Acad Sci
USA. 103:11515–11520. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hosono T, Tanaka T, Tanji K, Nakatani T
and Kamitani T: NUB1, an interferon-inducible protein, mediates
anti-proliferative actions and apoptosis in renal cell carcinoma
cells through cell-cycle regulation. Br J Cancer. 102:873–882.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lu B, Al-Ramahi I, Valencia A, Wang Q,
Berenshteyn F, Yang H, Gallego-Flores T, Ichcho S, Lacoste A, Hild
M, et al: Identification of NUB1 as a suppressor of mutant
Huntington toxicity via enhanced protein clearance. Nat Neurosci.
16:562–570. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kanaya K, Sohocki MM and Kamitani T:
Abolished interaction of NUB1 with mutant AIPL1 involved in Leber
congenital amaurosis. Biochem Biophys Res Commun. 317:768–773.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tanaka T, Nakatani T and Kamitani T:
Inhibition of NEDD8-conjugation pathway by novel molecules:
Potential approaches to anticancer therapy. Mol Oncol. 6:267–275.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Haas AL, Ahrens P, Bright PM and Ankel H:
Interferon induces a 15-kilodalton protein exhibiting marked
homology to ubiquitin. J Biol Chem. 262:11315–11323.
1987.PubMed/NCBI
|
|
36
|
Ichimura Y, Kirisako T, Takao T, Satomi Y,
Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi
M, et al: A ubiquitin-like system mediates protein lipidation.
Nature. 408:488–492. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rabut G and Peter M: Function and
regulation of protein neddylation. ‘Protein modifications: Beyond
the usual suspects’ review series. EMBO Rep. 9:969–976. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Walden H, Podgorski MS, Huang DT, Miller
DW, Howard RJ, Minor DL Jr, Holton JM and Schulman BA: The
structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis
for selective ubiquitin-like protein activation by an E1. Mol Cell.
12:1427–1437. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Leidecker O, Matic I, Mahata B, Pion E and
Xirodimas DP: The ubiquitin E1 enzyme Ube1 mediates NEDD8
activation under diverse stress conditions. Cell cycle.
11:1142–1150. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Olsen SK, Capili AD, Lu X, Tan DS and Lima
CD: Active site remodelling accompanies thioester bond formation in
the SUMO E1. Nature. 463:906–912. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang DT, Hunt HW, Zhuang M, Ohi MD,
Holton JM and Schulman BA: Basis for a ubiquitin-like protein
thioester switch toggling E1-E2 affinity. Nature. 445:394–398.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang DT, Ayrault O, Hunt HW, Taherbhoy
AM, Duda DM, Scott DC, Borg LA, Neale G, Murray PJ, Roussel MF and
Schulman BA: E2-RING expansion of the NEDD8 cascade confers
specificity to cullin modification. Mol Cell. 33:483–495. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kamura T, Conrad MN, Yan Q, Conaway RC and
Conaway JW: The Rbx1 subunit of SCF and VHL E3 ubiquitin ligase
activates Rub1 modification of cullins Cdc53 and Cul2. Genes Dev.
13:2928–2933. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Enchev RI, Schulman BA and Peter M:
Protein neddylation: Beyond cullin-RING ligases. Nat Rev Mol Cell
Biol. 16:30–44. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hjerpe R, Thomas Y, Chen J, Zemla A,
Curran S, Shpiro N, Dick LR and Kurz T: Changes in the ratio of
free NEDD8 to ubiquitin triggers NEDDylation by ubiquitin enzymes.
Biochem J. 441:927–936. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Saha A and Deshaies RJ: Multimodal
activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol
Cell. 32:21–31. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chan Y, Yoon J, Wu JT, Kim HJ, Pan KT, Yim
J and Chien CT: DEN1 deneddylates non-cullin proteins in vivo. J
Cell Sci. 121:3218–3223. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gong N, Li XY, Xiao Q and Wang YX:
Identification of a novel spinal dorsal horn astroglial D-amino
acid oxidase-hydrogen peroxide pathway involved in morphine
antinociceptive tolerance. Anesthesiology. 120:962–975. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lyapina S, Cope G, Shevchenko A, Serino G,
Tsuge T, Zhou C, Wolf DA, Wei N, Shevchenko A and Deshaies RJ:
Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome.
Science. 292:1382–1385. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mendoza HM, Shen LN, Botting C, Lewis A,
Chen J, Ink B and Hay RT: NEDP1, a highly conserved cysteine
protease that deNEDDylates Cullins. J Biol Chem. 278:25637–25643.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Schwechheimer C, Serino G, Callis J,
Crosby WL, Lyapina S, Deshaies RJ, Gray WM, Estelle M and Deng XW:
Interactions of the COP9 signalosome with the E3 ubiquitin ligase
SCFTIRI in mediating auxin response. Science. 292:1379–1382. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Goldenberg SJ, Cascio TC, Shumway SD,
Garbutt KC, Liu J, Xiong Y and Zheng N: Structure of the
Cand1-Cul1-Roc1 complex reveals regulatory mechanisms for the
assembly of the multisubunit cullin-dependent ubiquitin ligases.
Cell. 119:517–528. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu J, Furukawa M, Matsumoto T and Xiong
Y: NEDD8 modification of CUL1 dissociates p120 (CAND1), an
inhibitor of CUL1-SKP1 binding and SCF ligases. Mol Cell.
10:1511–1518. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Duda DM, Borg LA, Scott DC, Hunt HW,
Hammel M and Schulman BA: Structural insights into NEDD8 activation
of cullin-RING ligases: Conformational control of conjugation.
Cell. 134:995–1006. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Morimoto M, Nishida T, Nagayama Y and
Yasuda H: Nedd8-modification of Cul1 is promoted by Roc1 as a
Nedd8-E3 ligase and regulates its stability. Biochem Biophys Res
Commun. 301:392–398. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Osaka F, Kawasaki H, Aida N, Saeki M,
Chiba T, Kawashima S, Tanaka K and Kato S: A new NEDD8-ligating
system for cullin-4A. Genes Dev. 12:2263–2268. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Podust VN, Brownell JE, Gladysheva TB, Luo
RS, Wang C, Coggins MB, Pierce JW, Lightcap ES and Chau V: A Nedd8
conjugation pathway is essential for proteolytic targeting of
p27Kip1 by ubiquitination. Proc Natl Acad Sci USA. 97:4579–4584.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Read MA, Brownell JE, Gladysheva TB,
Hottelet M, Parent LA, Coggins MB, Pierce JW, Podust VN, Luo RS,
Chau V and Palombella VJ: Nedd8 modification of cul-1 activates SCF
(beta (TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell
Biol. 20:2326–2333. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wada H, Yeh ET and Kamitani T:
Identification of NEDD8-conjugation site in human cullin-2. Biochem
Biophys Res Commun. 257:100–105. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kim W, Bennett EJ, Huttlin EL, Guo A, Li
J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al: Systematic
and quantitative assessment of the ubiquitin-modified proteome. Mol
Cell. 44:325–340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jeram SM, Srikumar T, Zhang XD, Eisenhauer
H Anne, Rogers R, Pedrioli PG, Matunis M and Raught B: An improved
SUMmOn-based methodology for the identification of ubiquitin and
ubiquitin-like protein conjugation sites identifies novel
ubiquitin-like protein chain linkages. Proteomics. 10:254–265.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
van der Veen AG and Ploegh HL:
Ubiquitin-like proteins. Annu Rev Biochem. 81:323–357. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
McLarnon A: Cancer: Mdm2-regulated
stabilization of HuR by neddylation in HCC and colon cancer-a
possible target for therapy. Nat Rev Gastroenterol Hepatol.
9:42011. View Article : Google Scholar
|
|
64
|
Ryu JH, Li SH, Park HS, Park JW, Lee B and
Chun YS: Hypoxia-inducible factor alpha subunit stabilization by
NEDD8 conjugation is reactive oxygen species-dependent. J Biol
Chem. 286:6963–6970. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Freiberg RA, Hammond EM, Dorie MJ, Welford
SM and Giaccia AJ: DNA damage during reoxygenation elicits a
Chk2-dependent checkpoint response. Mol Cell Biol. 26:1598–1609.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ma T, Chen Y, Zhang F, Yang CY, Wang S and
Yu X: RNF111-dependent neddylation activates DNA damage-induced
ubiquitination. Mol Cell. 49:897–907. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Le Moan N, Houslay DM, Christian F,
Houslay MD and Akassoglou K: Oxygen-dependent cleavage of the p75
neurotrophin receptor triggers stabilization of HIF-1α. Mol Cell.
44:476–490. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Pawlus MR, Wang L and Hu CJ: STAT3 and
HIF1α cooperatively activate HIF1 target genes in MDA-MB-231 and
RCC4 cells. Oncogene. 33:1670–1679. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Choo YS, Vogler G, Wang D, Kalvakuri S,
Iliuk A, Tao WA, Bodmer R and Zhang Z: Regulation of parkin and
PINK1 by neddylation. Hum Mol Genet. 21:2514–2523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kandala S, Kim IM and Su H: Neddylation
and deneddylation in cardiac biology. Am J Cardiovasc Dis.
4:140–158. 2014.PubMed/NCBI
|
|
71
|
Abida WM, Nikolaev A, Zhao W, Zhang W and
Gu W: FBXO11 promotes the Neddylation of p53 and inhibits its
transcriptional activity. J Biol Chem. 282:1797–1804. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Noguchi K, Okumura F, Takahashi N, Kataoka
A, Kamiyama T, Todo S and Hatakeyama S: TRIM40 promotes neddylation
of IKKγ and is downregulated in gastrointestinal cancers.
Carcinogenesis. 32:995–1004. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Cao X and Sudhof TC: A transcriptionally
[correction of transcriptively] active complex of APP with Fe65 and
histone acetyltransferase Tip60. Science. 293:115–120. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lee MR, Lee D, Shin SK, Kim YH and Choi
CY: Inhibition of APP intracellular domain (AICD) transcriptional
activity via covalent conjugation with Nedd8. Biochem Biophys Res
Commun. 366:976–981. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Theng SS, Wang W, Mah WC, Chan C, Zhuo J,
Gao Y, Qin H, Lim L, Chong SS, Song J and Lee CG: Disruption of
FAT10-MAD2 binding inhibits tumor progression. Proc Natl Acad Sci
USA. 111:E5282–E5291. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Michel L, Diaz-Rodriguez E, Narayan G,
Hernando E, Murty VV and Benezra R: Complete loss of the tumor
suppressor MAD2 causes premature cyclin B degradation and mitotic
failure in human somatic cells. Proc Natl Acad Sci USA.
101:4459–4464. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Tapia C, Kutzner H, Mentzel T, Savic S,
Baumhoer D and Glatz K: Two mitosis-specific antibodies, MPM-2 and
phospho-histone H3 (Ser28), allow rapid and precise determination
of mitotic activity. Am J Surg Pathol. 30:83–89. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wagenaar-Miller RA, Gorden L and Matrisian
LM: Matrix metalloproteinases in colorectal cancer: Is it worth
talking about? Cancer Metastasis Rev. 23:119–135. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Aichem A, Kalveram B, Spinnenhirn V, Kluge
K, Catone N, Johansen T and Groettrup M: The proteomic analysis of
endogenous FAT10 substrates identifies p62/SQSTM1 as a substrate of
FAT10ylation. J Cell Sci. 125:4576–4585. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Gao Y, Theng SS, Zhuo J, Teo WB, Ren J and
Lee CG: FAT10, an ubiquitin-like protein, confers malignant
properties in non-tumorigenic and tumorigenic cells.
Carcinogenesis. 35:923–934. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lukasiak S, Schiller C, Oehlschlaeger P,
Schmidtke G, Krause P, Legler DF, Autschbach F, Schirmacher P,
Breuhahn K and Groettrup M: Proinflammatory cytokines cause FAT10
upregulation in cancers of liver and colon. Oncogene. 27:6068–6074.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Pelzer C, Kassner I, Matentzoglu K, Singh
RK, Wollscheid HP, Scheffner M, Schmidtke G and Groettrup M:
UBE1L2, a novel E1 enzyme specific for ubiquitin. J Biol Chem.
282:23010–23014. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Jin J, Li X, Gygi SP and Harper JW: Dual
E1 activation systems for ubiquitin differentially regulate E2
enzyme charging. Nature. 447:1135–1138. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Chiu YH, Sun Q and Chen ZJ: E1-L2
activates both ubiquitin and FAT10. Mol Cell. 27:1014–1023. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Aichem A, Pelzer C, Lukasiak S, Kalveram
B, Sheppard PW, Rani N, Schmidtke G and Groettrup M: USE1 is a
bispecific conjugating enzyme for ubiquitin and FAT10, which
FAT10ylates itself in cis. Nat Commun. 1:132010. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hipp MS, Kalveram B, Raasi S, Groettrup M
and Schmidtke G: FAT10, a ubiquitin-independent signal for
proteasomal degradation. Mol Cell Biol. 25:3483–3491. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hipp MS, Raasi S, Groettrup M and
Schmidtke G: NEDD8 ultimate buster-1L interacts with the
ubiquitin-like protein FAT10 and accelerates its degradation. J
Biol Chem. 279:16503–16510. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Ren J, Kan A, Leong SH, Ooi LL, Jeang KT,
Chong SS, Kon OL and Lee CG: FAT10 plays a role in the regulation
of chromosomal stability. J Biol Chem. 281:11413–11421. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Merbl Y, Refour P, Patel H, Springer M and
Kirschner MW: Profiling of ubiquitin-like modifications reveals
features of mitotic control. Cell. 152:1160–1172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Gong P, Canaan A, Wang B, Leventhal J,
Snyder A, Nair V, Cohen CD, Kretzler M, D'Agati V, Weissman S and
Ross MJ: The ubiquitin-like protein FAT10 mediates NF-kappaB
activation. J Am Soc Nephrol. 21:316–326. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Bjorkoy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|