Introduction
The epidermal growth factor receptor (EGFR) is an
attractive target for anticancer therapy, as EGFR signaling is a
pathway that has a significant role in the growth, proliferation
and survival of numerous solid tumors, including non-small cell
lung cancer (NSCLC) (1). Gefitinib
(Iressa®, also known as ZD1839; AstraZeneca, London,
UK), a synthetic anilinoquin- azoline and adenosine triphosphate
(ATP) mimetic, is the first commercially available EGFR tyrosine
kinase inhibitor (EGFR-TKI). Administered orally, gefitinib
competes with ATP for the tyrosine kinase binding site on the EGFR
and the resulting inhibition of autophosphorylation blocks
downstream signaling (2). Gefitinib
has minimal adverse effects, but tumor responses are observed in
only 10–19 % of patients with chemotherapy-refractory advanced
NSCLC (3). However, a subgroup of
patients with NSCLC possessing specific mutations in the tyrosine
kinase domain of the EGFR gene, which correlates with favorable
clinical responsiveness to gefitinib therapy, has been noted
(3). All mutations appear to be
limited to exons 18, 19, 20 and 21 of the EGFR gene (4). Missense mutations in exon 21 (L858R) and
in-frame deletions within exon 19 (delE746-A750) have been shown to
be the most frequent EGFR-TKI sensitive mutations (80%) in NSCLC
(5,6).
EGFR activation has been shown to be associated with
the stimulation of tumor angiogenesis, and angiogenesis is
essential to growth, proliferation and metastasis of cancer cells
(7–14). The EGFR ligands, EGF and transforming
growth factor (TGF)-α, demonstrated angiogenic properties.
Expression of EGFR has been reported to be associated with the
expression of angiogenic factors, such as TGF-α and VEGF in human
cancers (7,15). Activation of EGFR by EGF and TGF-α
also upregulated VEGF expression in human cancer cell lines
(8,9).
Gefitinib, which blocked the EGFR signaling pathway via inhibition
of phosphorylated AKT, was reported to exert anti-angiogenic
effects by blocking EGF induced upregulation of VEGF and
interleukin (IL)-8 in human cancer cell lines (10). Treatment of several
EGFR/TGF-α-coexpressing tumor cell lines with gefitinib also
resulted in growth inhibition that was accompanied by a decreased
production of VEGF, basic EGF and TGF-α (11). The above data suggest that the EGFR
signaling pathway modulates angiogenesis by way of upregulation of
VEGF or other key angiogenic factors. VEGF is a key stimulator of
angiogenesis, which induces proliferation, differentiation and
migration of endothelial cells (12).
VEGF also increases the vascular permeability and induces the
production of proteases involved in the modification of the
extracellular matrix (12). In NSCLC
patients, high serum VEGF level is associated with increasing
intratumoral angiogenesis and poor prognosis (13). As the mutations in EGFR may lead to
increased growth factor signaling, the present study hypothesized
that NSCLC with EGFR mutations may have more potential in induction
of angiogenesis. However, the association of EGFR mutations and the
activities of angiogenic factors in lung cancer have not been
previously studied to the best of our knowledge.
In the present study, the association of VEGF
expression with EGFR mutation was investigated in lung cancer cells
and NSCLC tissues. Lung cancer cell lines stably transfected with
wild-type and mutant EGFR genes were also established. VEGF
expression and inhibitory effects of gefitinib to VEGF expression
were also evaluated in these cells.
Materials and methods
Cell culture
The NSCLC cell lines A549 (ATCC CCL-185), H460 (ATCC
HTB-177), H1650 (ATCC CRL-5883) and H1975 (ATCC CRL-5908) were
purchased from American Type Culture Collection (Manassas, VA,
USA). H1650 and H1975 cell lines have EGFR mutations (delE746-A750
for H1650; L858R and T790 M for H1975) (14). Human umbilical vein endothelial cells
(HUVECs; H-UV001) were purchased from Bioresource Collection and
Research Center (Hsinchu City, Taiwan). Cells were grown in
complete growth medium [Dulbecco's modified Eagle's medium (Lonza,
Basel, Switzerland) for A549 and H460 cells; RPMI-1640 media
(Lonza) for H1650 and H1975 cells] supplemented with 10% fetal calf
serum (Thermo Fisher Scientific, Inc., Waltham, MA, USA), 30 ng/ml
EGF (Invitrogen; Thermo Fisher Scientific, Inc.), 10 U/ml
penicillin and 10 µg/ml streptomycin at 37°C and 5% CO2.
Cells were incubated for 72 h and the supernatant of growth medium
was collected for detection of VEGF levels. HUVECs were grown in
90% Medium 199 (Lonza) with 25 U/ml heparin (Lonza) and 30 µg/ml
endothelial cell growth supplement (Lonza), adjusted to contain 1.5
g/l sodium bicarbonate, 10 U/ml penicillin and 10 µg/ml
streptomycin, as well as 10% fetal calf serum. Gefitinib and G418
were purchased from Sigma Aldrich (EMD Millipore, Billerica, MA,
USA).
Tissues
Thirty-two NSCLC tissue samples were obtained from
NSCLC patients undergoing surgical resection of the primary tumor
between July 2006 and May 2009, after approval from the
Institutional Review Board at Chang Gung Memorial Hospital (IRB
nos. 100-1405B and 103-6693C1) and patients' signed consent were
obtained. All tissue samples were requested from the tissue bank of
Chang Gung Memorial Hospital. Formalin-fixed, paraffin-embedded
tissue samples were converted into tissue microarray (TMA) blocks
using an AutoTiss 1000 arrayer (EverBio Technology, Inc., New
Taipei City, Taiwan). The quality of the TMA slides was confirmed
by the pathologist using hematoxylin- and eosin-stained slides.
DNA extraction and EGFR mutation
analysis
DNA was extracted from formalin-fixed paraffin
embedded tumors using the QIAamp DNA FFPE Tissue kit (Qiagen GmbH,
Hilden, Germany). A high sensitivity OncoFOCUS™ Panel version 1.0
developed by Sequenom (San Diego, CA, USA) was used for EGFR
mutation analysis with the mass-spectroscopy based MassArray device
(16).
RNA extraction, complementary (c)DNA
synthesis and reverse transcription-polymerase chain reaction
(RT-PCR)
RNA was extracted using the RNeasy Mini kit (Qiagen
GmbH) from cell pellets according to the manufacturers' protocol.
Total RNA was then reverse transcribed to cDNA using the iScript™
cDNA Synthesis kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
A total of 2 µl of reverse-transcribed cDNA was subjected to RT-PCR
using iQ™ SYBR® Green supermix at a total volume of 20
µl and the Bio-Rad CFX96™ quantitative PCR system (Bio-Rad
Laboratories, Munich, Germany). The following primers were used for
PCR: VEGF, 5′-TTCTGCTGTCTTGGGTGCATTGG-3′ (sense) and
5′-ATCTCTCCTATGTGCTGGCCTT-3′ (antisense) (17); and β-actin,
5′-CCTGGACTTCGAGCAAGAGATG-3′ (sense) and
5′-AGGAAGGAAGGCTGGAAGAGTG-3′ (antisense). A typical protocol
included a 95°C denaturation step for 3 min followed by 35 cycles
with 95°C denaturation for 20 sec, 60.3°C annealing and extension
for 30 sec. Detection of the fluorescent product was performed at
the extension step using a PCR machine. Melting curve detection and
analysis were performed by an additional 80 cycles with a 55°C
denaturation, with a 0.5°C increase following each cycle. Finally,
the RT-PCR products were kept at 4°C. Relative VEGF expression was
analyzed by the 2−ΔΔCq method, using β-actin as the
internal control (18).
Establishment of lung cancer stable
cell lines expressing wild-type and mutant EGFR genes
Mutant EGFR genes (L858R and Del E746-A750) were
generated by site-directed mutagenesis with specific primers from
an expression vector harboring EGFR cDNA (pUSEAmpEGFRWt) (Upstate
Biotechnology, Charlottesville, VA, USA). The following primers
were used for site-directed mutagenesis: L858R,
5′-GATCACAGATTTTGGGCGGGCCAAACTGCTGGG-3′ (sense) and
5′-CCCAGCAGTTTGGCCCGCCCAAAATCTGTGATC-3′ (antisense); and Del
E746-A750, 5′-CCCGTCGCTATCAAAACATCTCCGAAAGCC-3′ (sense) and
5′-GGCTTTCGGAGATGTTTTGATAGCGACGGG-3′ (antisense). A QuikChange™
Site-Directed Mutagenesis kit (Stratagene California, San Diego,
CA, USA) was used for site-directed mutagenesis according to the
manufacturer's protocol. Cells were transfected with expression
vectors using Lipofectamine® 2000 reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. A total of 48 h post transfection, cells were selected in
G418 for 4 weeks, and cell colonies were selected and amplified for
further studies.
Enzyme-linked immunosorbent assay
(ELISA) of VEGF
The levels of VEGF in cell culture media were
measured using a commercially available VEGF ELISA kit (Human VEGF
ELISA kit; Biosource International, Inc., Camarillo, CA, USA)
according to the manufacturer's protocol. The limits of sensitivity
were 5 pg/ml for VEGF.
Immunohistochemistry (IHC)
Formalin-fixed, paraffin-embedded tissues were cut
into 4-µm sections, mounted on slides, deparaffinized with xylene
and dehydrated using a gradient ethanol series. Stable lung cancer
cells were cultured in a Nunc Lab-Tek™ II-Chamber Slide system
(Thermo Fisher Scientific, Inc.), and subsequently fixed with
formalin for further IHC study. Antigen retrieval was performed
with citric acid (pH 6.0) at 97°C for 30 min, followed by treatment
with 3% hydrogen peroxide. The slides were incubated overnight at
4°C with antibodies against VEGF. The rabbit polyclonal VEGF
antibody (catalog no., sc-152; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) was used as primary antibody at a 1:50 dilution in
TBS with 1% BSA. Following TBST washes, endogenous peroxidase
activity was then quenched with 0.3% hydrogen peroxide in TBS.
Mouse and rabbit specific HRP/DAB (ABC) detection IHC kit (catalog
no., ab64264; Abcam, Cambridge, MA, USA) was then used according to
the manufacturer's protocol. Detection was achieved using a
biotinylated goat anti-rabbit secondary antibody (dilution, 1:1000;
catalog no., ab6720; Abcam) and DAB chromogen. The sections were
counterstained with hematoxylin before being mounted with organic
media and glass slides. The IHC data for the specimens were
assessed using the semi-quantitative immunoreactive score (IRS).
The IRS was calculated by multiplying the staining intensity (0=no
staining, 1=weak staining, 2=moderate staining and 3=strong
staining) by the percentage of positively stained cells (0=0% of
cells stained, 1=<10% of cells stained, 2=11–50% of cells
stained, 3=51–80% of cells stained and 4=>81% of cells
stained).
Protein extraction and western blot
analysis
Whole cell proteins were extracted from lung cancer
cells using M-PER Mammalian Protein Extraction Reagent (Pierce,
Rockford, IL, USA) with Phosphatase Inhibitor Cocktail Set II
(Calbiochem, San Diego, CA, USA) and Complete Protease Inhibitor
Cocktail (Roche, Basel, Switzerland), according to the
manufacturer's protocols. In total, 40 µg protein were separated on
8% sodium dodecyl sulfate-polyacrylamide gels and transferred to
Immobilon-P membranes (EMD Millipore). Membranes were incubated
with primary antibodies against EGFR (catalog no., 2232; dilution,
1:500) phospho-EGFR (Tyr1068) (catalog no., 2220; dilution, 1:500)
(both from Cell Signaling Technology, Inc., Danvers, MA, USA),
β-actin (catalog no., A5441; dilution, 1:1,000) (Sigma-Aldrich; EMD
Millipore), Akt (catalog no., sc-8312; dilution, 1:500),
phospho-Akt (Ser473) (catalog no., sc-33437; dilution, 1:500), and
γ-tubulin (catalog no., sc-12881; dilution, 1:1,000) (all from
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) at 4°C overnight.
Subsequently, primary antibody and antigen complexes were bound to
specific HRP-conjugated secondary antibodies [anti-rabbit (catalog
no., sc-2340; dilution, 1:5,000), anti-goat (catalog no., sc-2953;
1:10,000), and anti-mouse (catalog no., sc-2371; dilution, 1:2,000)
(all from Santa Cruz Biotechnology, Inc.)] at room temperature for
1 h. An enhanced chemiluminescence blotting analysis system (GE
Healthcare Life Sciences, Piscataway, NJ, USA) was used for
antigen-antibody detection. The density of western blot bands was
semi-quantified by ImageJ software (version 1.46; National
Institutes of Health, Bethesda, MD, USA).
Transwell co-culture assay
HUVECs (3×104) were cultured in 35-mm 6
well dual-layered culture dishes at 37°C. After 24 h, wild-type and
mutant EGFR transfected cells (5×104) were seeded onto
the cell culture insert with 0.4-µm micropores on the bottom (BD
Biosciences, Franklin Lakes, NJ, USA) and placed in the wells
growing HUVECs. HUVECs were collected on day 5 following
co-culturing, and viable cells were then counted with a
hemocytometer.
Statistical analysis
The Student's t-test was used to compare VEGF
expression in various groups of samples. Statistical analysis was
performed using SPSS (version 10.0; SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a significant difference, with
two-sided analysis. All experiments were performed in
triplicate.
Results
EGFR mutations and VEGF expression in
lung cancer cells and NSCLC tissues
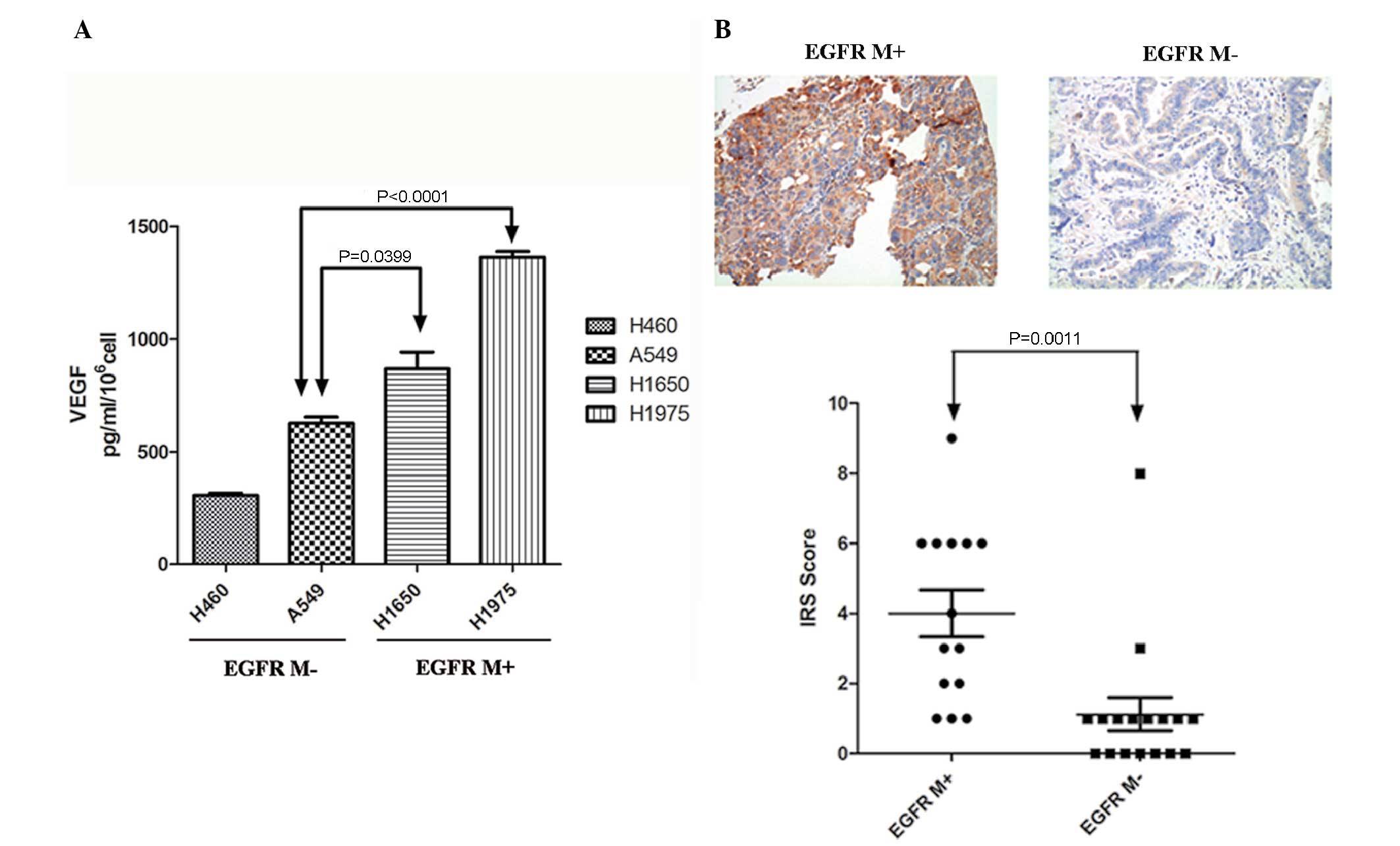
The association between EGFR mutations and VEGF
expression was detected in lung cancer cell lines. VEGF levels in
the culture media of lung cancer cell lines with mutant (H1975 and
H1650) and wild-type EGFR (A549 and H460) were measured by ELISA
(Fig. 1A). Significantly increased
VEGF levels were noted in H1975 and H1650 lung cancer cell lines
compared to the A549 cell line (Fig.
1A).
EGFR mutation statuses were analyzed in 32 NSCLC
tissue samples. EGFR mutations were detected in 14 (41.6%) samples,
including 5 exon 21 L858R, 5 exon 19 deletions, 2 exon 21 L861Q and
1 exon 20 insertion mutation. Expression of VEGF was subsequently
detected using IHC staining. Significantly increased expression of
VEGF was noted in lung cancer tissues with EGFR mutations (Fig. 1B).
Establishment of lung cancer stable
cells expressing wild-type and mutant EGFR genes
To further validate the association of mutant EGFR
and expression of VEGF in an isogenic background, stable lung
cancer cell lines expressing wild-type and mutant EGFR genes were
subsequently established. A549 lung cancer cells were transfected
with vectors containing wild-type and mutant EGFR genes. Following
selection with G418, resistant cell colonies were selected and
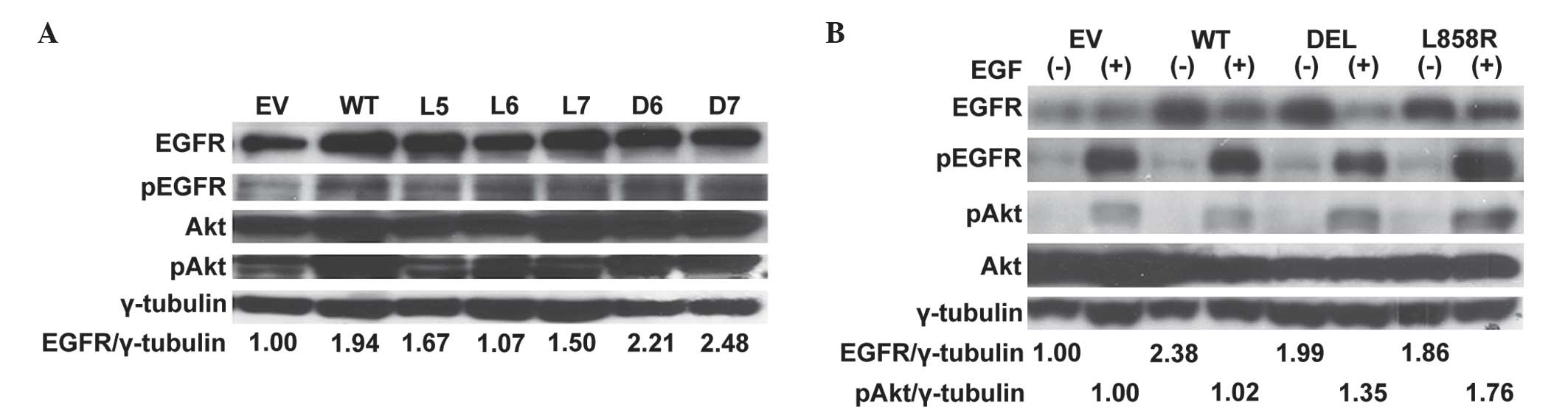
amplified. EGFR expression in cell colonies was detected by western
blot analysis (Fig. 2A). Colonies
with overexpression of EGFR were subsequently pooled and amplified
for further analysis. Compared to wild-type EGFR gene transfected
cells, increased expression of EGFR and phosphor-Akt proteins
following EGF stimulation were detected in mutant EGFR gene
transfected A549 lung cancer stable cells (Fig. 2B).
EGFR mutations and expression of VEGF
in lung cancer stable cells
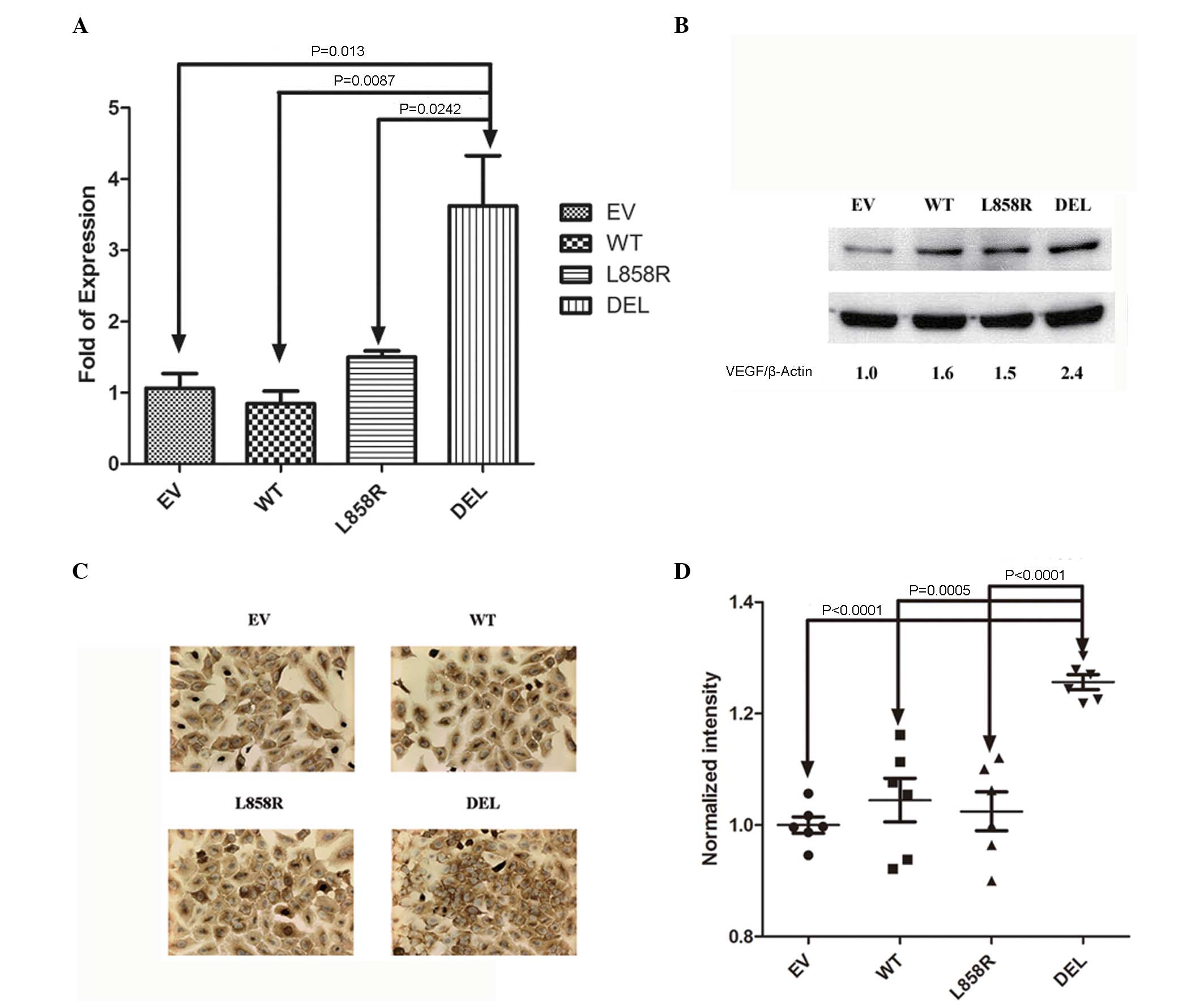
The expression of VEGF mRNA and protein were
detected in mutant and wild type EGFR gene transfected stable lung
cancer cells. Significantly increased expression of VEGF mRNA was
noted in stable cells transfected with exon 19 deletion (E746-A750)
mutant EGFR gene compared to those transfected with empty vector,
wild-type and exon 21 missense (L858R) EGFR genes (Fig. 3A). Expression of VEGF protein was also
observed in mutant and wild-type EGFR gene transfected stable lung
cancer cells. Increased expression of VEGF protein was noted in
stable cells transfected with exon 19 deletion (E746-A750) mutant
EGFR gene by western blot (Fig. 3B)
and IHC (Fig. 3C and D) analysis.
EGFR mutations and VEGF expression in
lung cancer stable cells
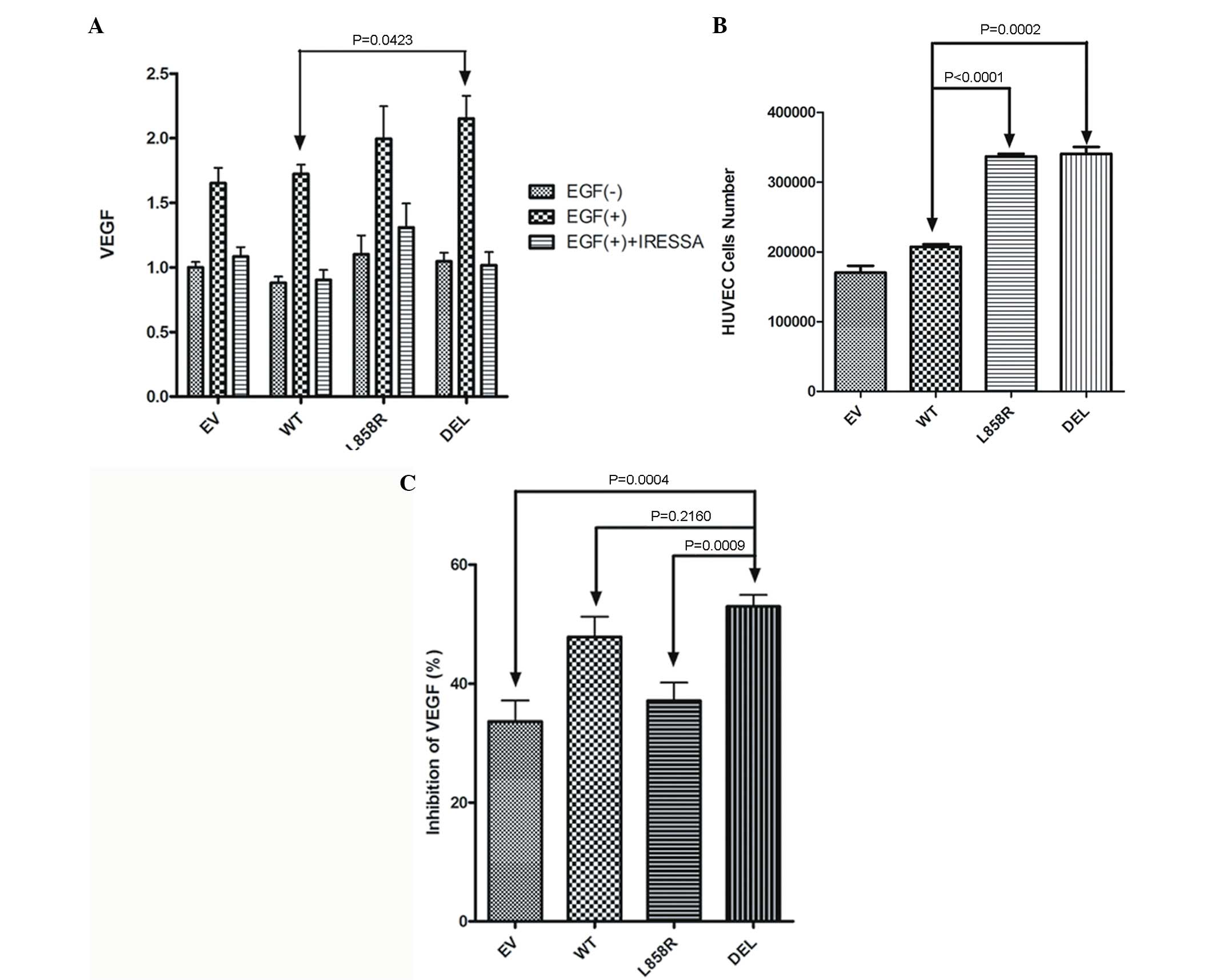
Expression of VEGF in culture medium of EGFR
transfected A549 stable lung cancer cells with/without EGF
stimulation was measured. Significantly increased VEGF levels were
noted in stable cells transfected with exon 19 deletion (Del
E746-A750) mutant EGFR gene compared to those transfected with
wild-type EGFR gene (Fig. 4A). A
Transwell co-culture system was subsequently used to evaluate the
ability of mutant EGFR genes to promote HUVEC growth. HUVECs were
co-cultured with EGFR transfected A549 stable lung cancer cells in
a Transwell system for 5 days. Significantly increased numbers of
HUVECs were also noted in cells co-cultured with exon 21 missense
(L858R) and exon 19 deletion (Del E746-A750) mutant EGFR genes
(Fig. 4B).
EGFR mutations and angiogenesis in
lung cancer stable cell lines
Inhibitory effects of gefitinib to VEGF in EGFR
transfected A549 stable lung cancer cells were also evaluated.
Expression of VEGF in culture medium of EGFR transfected A549
stable lung cancer cells with EGF stimulation and gefitinib
treatment was measured (Fig. 4C).
Expression of VEGF following EGF stimulation was partially
inhibited by gefitinib treatment. However, a significantly
increased inhibition of VEGF was observed in stable cells
transfected with exon 19 deletion (Del E746-A750) mutant EGFR gene
(56%) compared to those transfected with empty vector (33.6%),
wild-type EGFR (47.8%) and exon 21 missense (L858R) mutation
(37.1%) genes (Fig. 4C).
Discussion
Association of EGFR mutations and VEGF expression
has been reported in lung cancer tissues (19). In the present study, it was
additionally observed that increased expression of VEGF is
associated with EGFR mutations in lung cancer cells and NSCLC
tissues. In order to elucidate these findings, stable lung cancer
cell lines expressing exon 19 deletion (E746-A750), exon 21
missense (L858R) mutations and wild-type EGFR genes were
established. Increased phosphorylation of EGFR and Akt were
observed in stable lung cancer cell lines expressing exon 19
deletion (E746-A750) and exon 21 missense (L858R) mutations. It was
observed that increased expression of VEGF is associated with
overexpression of exon 19 deletion (E746-A750) EGFR gene and the
expression of VEGF is inhibited by gefitinib, an EGFR-TKI. As a
result, EGFR mutation may enhance VEGF expression through increased
phosphorylation of EGFR and Akt in lung cancer cells. Increased
proliferation rate was also observed in HUVECs co-cultured with
exon 19 deletion and (E746-A750) exon 21 missense (L858R) mutation
stable lung cancer cells. Furthermore, it was observed that the
expression of VEGF in stable lung cancer cells is inhibited by
gefitinib, which demonstrated increased inhibitory effects to
expression of VEGF in stable lung cancer cell lines expressing exon
19 deletion (E746-A750) mutation. EGFR-TKIs have been proposed to
inhibit lung cancer cells through oncogenic shock by inhibiting
phosphorylation of EGFR tyrokinase domain and downstream Akt
signaling pathways (20). The results
of the present study demonstrated that in addition to oncogenic
shock, EGFR-TKIs may exert their effects through inhibition of VEGF
expression and angiogenesis in lung cancer cells.
Both exon 19 deletion and exon 21 missense mutations
are common EGFR mutations, and have been proven to be associated
with a favorable response to gefitinib as well as other EGFR-TKIs,
including erlotinib and afatinib (21–23).
However, emerging evidence has suggested that exon 19 deletion
mutation is associated with an improved outcome following afatinib
therapy compared with exon 21 L858R mutation, which may suggest
that these mutations have distinct features (22). In the present study, it was observed
that exon 19 deletion mutation is associated with increased
expression of VEGF. The expression of VEGF was inhibited by
gefitinib in all lung cancer cells investigated. Notably, an
increased inhibitory effect of gefitinib to the secretion of VEGF
was observed in exon 19 deletion stable cells. As a result, the
present study supports the finding that exon 19 deletion and exon
21 L858R mutation have distinct features. The results of the
present study demonstrated that lung cancer cells harboring exon 19
deletion EGFR mutation may exert increased expression of VEGF and
angiogenesis ability. In addition, gefitinib, as well as other
EGFR-TKIs, may have increased inhibitory effects on these
cells.
In the present study, both exon 19 deletion and exon
21 L858R mutation cells were observed to promote proliferation of
HUVECs. As a result, other angiogenic factors in addition to VEGF
may be associated with EGFR mutations. For example, mutant EGFR was
reported to activate the gp130/JAK/STAT3 signaling pathway by means
of IL-6 upregulation in primary human lung adenocarcinomas
(24). In pancreatic cancer cells,
inhibition of EGFR activation resulted in decreased expression of
IL-8 (25). Taken together, mutant
EGFR may contribute to the production of VEGF, IL-8, IL-6 and other
angiogenic factors in lung cancer cells. The expression of VEGF was
only partially inhibited by gefitinib in the present study, which
implies that a combination of EGFR-TKI with anti-VEGF therapy may
provide an improved response compared with EGFR-TKI treatment alone
in lung cancer cells. This hypothesis is supported by a recent
meta-analysis study, which demonstrated that the combination of
erlotinib and bevacizumab, an anti-VEGF antibody, has an improved
response compared with erlotinib treatment alone in advanced NSCLC
(26). Further studies to investigate
this hypothesis are ongoing in our laboratory.
In summary, the present study demonstrated that
mutant EGFR is associated with increased expression of VEGF in
NSCLC cells and tissues. Exon 19 deletion mutation stable cells
were observed to have increased expression of VEGF and to be more
susceptible to EGFR-TKI inhibition of VEGF expression. The results
of the present study may provide an insight into the association
between mutant EGFR and VEGF expression in lung cancer, and lead to
further development of combined anti-EGFR and anti-VEGF therapy for
NSCLC in the future.
Acknowledgements
The present study was supported by grant nos.,
CMRPG650181 (to MSH and CTY), CMRPG6B0231 (to MSH), CMRPG6B0232 (to
MSH), CORPG6B0353 (to YHT and MSH) and CORPG6B0363 (to YCL and MSH)
in Chang Gung Memorial Hospital (Chiayi, Taiwan). In addition, the
authors would like to acknowledge the tissue microarray service
provided by the Expensive Advanced Instrument Core Laboratory,
Department of Medical Research and Development, Chang Gung Memorial
Hospital.
References
|
1
|
Arteaga CL: Epidermal growth factor
receptor dependence in human tumors: More than just expression?
Oncologist. 4:(Supp 7). S31–S39. 2002. View Article : Google Scholar
|
|
2
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Maemondo M, Inoue A, Kobayashi K, et al:
Gefitinib or chemotherapy for non-small-cell lung cancer with
mutated EGFR. N Engl J Med. 362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gazdar AF, Shigematsu H, Herz J and Minna
JD: Mutations and addiction to EGFR: The Achilles ‘heal’ of lung
cancers? Trends Mol Med. 10:481–486. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hsieh RK, Lim KH, Kuo HT, Tzen CY and
Huang MJ: Female sex and bronchioloalveolar pathologic subtype
predict EGFR mutations in non-small cell lung cancer. Chest.
128:317–321. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shih JY, Gow CH, Yu CJ, et al: Epidermal
growth factor receptor mutations in needle biopsy/aspiration
samples predict response to gefitinib therapy and survival of
patients with advanced nonsmall cell lung cancer. Int J Cancer.
118:963–969. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bancroft CC, Chen Z, Yeh J, Sunwoo JB, Yeh
NT, Jackson S, Jackson C and Van Waes C: Effects of pharmacologic
antagonists of epidermal growth factor receptor, PI3K and MEK
signal kinases on NF-kappaB and AP-1 activation and IL-8 and VEGF
expression in human head and neck squamous cell carcinoma lines.
Int J Cancer. 99:538–548. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goldman CK, Kim J, Wong WL, King V, Brock
T and Gillespie GY: Epidermal growth factor stimulates vascular
endothelial growth factor production by human malignant glioma
cells: A model of glioblastoma multiforme pathophysiology. Mol Biol
Cell. 4:121–133. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ravindranath N, Wion D, Brachet P and
Djakiew D: Epidermal growth factor modulates the expression of
vascular endothelial growth factor in the human prostate. J Androl.
22:432–443. 2001.PubMed/NCBI
|
|
10
|
Hirata A, Ogawa S, Kometani T, Kuwano T,
Naito S, Kuwano M and Ono M: ZD1839 (Iressa) induces antiangiogenic
effects through inhibition of epidermal growth factor receptor
tyrosine kinase. Cancer Res. 62:2554–2560. 2002.PubMed/NCBI
|
|
11
|
Ciardiello F, Caputo R, Bianco R, Damiano
V, Fontanini G, Cuccato S, De Placido S, Bianco AR and Tortora G:
Inhibition of growth factor production and angiogenesis in human
cancer cells by Z (Iressa), a selective epidermal growth factor
receptor tyrosine kinase inhibitor. Clin Cancer Res. 7:1459–1465.
2001.PubMed/NCBI
|
|
12
|
Ferrara N: Vascular endothelial growth
factor: Basic science and clinical progress. Endocr Rev.
25:581–611. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shimanuki Y, Takahashi K, Cui R, Hori S,
Takahashi F, Miyamoto H and Fukurchi Y: Role of serum vascular
endothelial growth factor in the prediction of angiogenesis and
prognosis for non-small cell lung cancer. Lung. 183:29–42. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Maeshima Y, Sudhakar A, Lively JC, Ueki K,
Kharbanda S, Kahn CR, Sonenberg N, Hynes RO and Kalluri R:
Tumstatin, an endothelial cell-specific inhibitor of protein
synthesis. Science. 295:140–143. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cote ML, Haddad R, Edwards DJ, Atikukke G,
Gadgeel S, Soubani AO, Lonardo F, Bepler G, Schwartz AG and Ethier
SP: Frequency and type of epidermal growth factor receptor
mutations in African Americans with non-small cell lung cancer. J
Thorac Oncol. 6:627–630. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jia SF, Guan H, Duan X and Kleinerman ES:
VEGF165 is necessary to the metastatic potential of Fas (−)
osteosarcoma cells but will not rescue the Fas (+) cells. J Exp
Ther Oncol. 7:89–97. 2008.PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reinmuth N, Jauch A, Xu EC, Muley T,
Granzow M, Hoffmann H, Dienemann H, Herpel E, Schnabel PA, Herth
FJ, et al: Correlation of EGFR mutations with chromosomal
alterations and expression of EGFR, ErbB3 and VEGF in tumor samples
of lung adenocarcinoma patients. Lung cancer. 62:193–201. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Scaltriti M and Baselga J: The epidermal
growth factor receptor pathway: A model for targeted therapy. Clin
Cancer Res. 12:5268–5272. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rosell R, Ichinose Y, Taron M, Sarries C,
Queralt C, Mendez P, Sanchez JM, Nishiyama K, Moran T, Cirauqui B,
et al: Mutations in the tyrosine kinase domain of the EGFR gene
associated with gefitinib response in non-small-cell lung cancer.
Lung Cancer. 50:25–33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang JC, Wu YL, Schuler M, Sebastian M,
Popat S, Yamamoto N, Zhou C, Hu CP, O'Byrne K, Feng J, et al:
Afatinib versus cisplatin-based chemotherapy for EGFR
mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6):
Analysis of overall survival data from two randomised, phase 3
trials. Lancet Oncol. 16:141–151. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao SP, Mark KG, Leslie K, Pao W, Motoi N,
Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B and Bromberg
JF: Mutations in the EGFR kinase domain mediate STAT3 activation
via IL-6 production in human lung adenocarcinomas. J Clin Invest.
117:3846–3856. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bruns CJ, Solorzano CC, Harbison MT, Ozawa
S, Tsan R, Fan D, Abbruzzese J, Traxler P, Buchdunger E, Radinsky R
and Fidler IJ: Blockade of the epidermal growth factor receptor
signaling by a novel tyrosine kinase inhibitor leads to apoptosis
of endothelial cells and therapy of human pancreatic carcinoma.
Cancer Res. 60:2926–2935. 2000.PubMed/NCBI
|
|
26
|
Sun L, Ma JT, Zhang SL, Zou HW and Han CB:
Efficacy and safety of chemotherapy or tyrosine kinase inhibitors
combined with bevacizumab versus chemotherapy or tyrosine kinase
inhibitors alone in the treatment of non-small cell lung cancer: A
systematic review and meta-analysis. Med Oncol. 32:4732015.
View Article : Google Scholar : PubMed/NCBI
|