Introduction
Ovarian cancer is one of the most common types of
gynecological malignancy and has a poor prognosis. Historically,
ovarian cancer was considered a silent cancer, as the majority of
patients present with late-stage disease (1). Despite advances in surgery and the
development of more effective chemotherapy, ovarian cancer remains
the leading cause of mortality from a gynecological cancer
(2). Drug resistance is the
predominant cause of mortality in late-stage patients. In total,
~30% of patients whose tumors are platinum-resistant will generally
either progress during primary therapy or shortly thereafter.
Additionally, there is no preferred standard second-line
chemotherapy to offer these patients (3,4). Thus,
elucidation of mechanisms and identification of new therapeutic
targets for ovarian cancer is critical to reduce fatality.
Histone deacetylase inhibitors (HDACis) show promise
as a novel class of anticancer agents in a wide spectrum of tumors,
including ovarian caner (5).
Previously, the present study investigated whether the HDACi
trichostatin A (TSA) induces apoptosis of ovarian cancer A2780
cells in a dose-dependent manner (6).
Thus far, numerous HDACis are being tested in over 100 clinical
trials and have exhibited encouraging therapeutic responses with
good safety profiles (7,8). The clinical potential of HDACis has been
well documented by the successful development of vorinostat
(suberoylanilide hydroxamic acid), which has been approved by the
U.S. Food and Drug Administration (9). Despite the rapid progress achieved,
clinical data has shown that there is limited efficacy for HDACi as
a single agent. The majority of current clinical trials are
combination studies looking at HDACi in combination with other
agents (10,11). These combination trials seek to
increase the antitumor activity of the treatments. Although these
combination strategies follow a rational molecular approach in
certain cases, in the majority of instances, they are relatively
empirical. Accordingly, synergism in antitumor efficacy may be
accompanied by adverse effects that are rarely observed with HDACis
alone, such as severe myelosuppression (5,12).
Therefore, revealing the molecular mechanisms underlying the low
potency of HDACi is pivotal in determining the optimal application
of this class of therapeutic agents in the treatment of ovarian
cancer.
In our previous study, it was reported that the
adhesion molecule cluster of differentiation 146 (CD146) is
significantly induced in HDACi-treated tumor cells (13). In the current study, it was found that
the induction of CD146 expression was significant in ovarian cancer
cells. CD146 is one of the adhesion molecules belonging to the
immunoglobulin superfamily (14). In
numerous types of cancer, including melanoma (15), prostate cancer (16) and ovarian cancer, elevated expression
of CD146 promotes tumor progression and is associated with poor
prognosis. Previously, targeting CD146 with antibody against the
molecule has been shown to inhibit tumor growth and angiogenesis in
several types of cancer. Based on these findings (15,17,18), the
present study chose to additionally explore whether the induced
expression of CD146 protected ovarian cancer cells from
HDACi-induced death. In addition, the current study tested whether
the antitumoral activity of HDACi may be significantly enhanced in
combination with the targeting of CD146 in ovarian cancer cells
in vitro and in vivo.
Materials and methods
Cells and reagents
The human ovarian cancer cell lines A2780, SKOV3 and
Caov3 were purchased from the American Type Culture Collection
(Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's
medium containing 10% fetal calf serum (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). All cells were cultured at
37°C in a humidified 5% CO2 atmosphere. The HDACi TSA
and vorinostat were purchased from Sigma-Aldrich (Merck Millipore,
Darmstadt, Germany), and dissolved in dimethyl sulfoxide (DMSO).
The mouse anti-human CD146 monoclonal antibody (mAb) AA98 and the
control mIgG were provided by Dr Xiyun Yan (Institute of
Biophysics, Chinese Academy of Sciences, Beijing, China) (19). The mouse anti-human CD146 mAb (1
mg/ml; ab24577) was purchased from Abcam (Cambridge, UK).
Fluorescein isothiocyanate-conjugated mouse anti-human CD146 mAb
(1:100; 11-1469-42) was purchased from eBioscience, Inc. San Diego,
CA, USA). Anti protein kinase B (Akt) rabbit anti-human polyclonal
antibody (1:1,000; #9272S), anti-phosphorylated Akt rabbit
anti-human mAb (1:1,000; #4058), anti-human phosphorylated glycogen
synthase kinase 3β (GSK3β) rabbit mAb, (1:1,000; #5558), anti-human
phosphorylated 4E-binding protein 1 (4E-BP1) rabbit mAb (1:1,000;
#2855) and anti-human phosphorylated ribosomal protein S6 kinase-1
(S6K1) mouse mAbs (1:1,000; #9206) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Triciribine was
purchased from Cayman Chemical Company (Ann Arbor, MI, USA).
Full-length Akt2 complementary DNA (cDNA) was cloned into pcDNA3.1
plasmid and termed the AAkt2 vector, which has been described
previously (13).
Cell viability assays
Cell viability was determined using a MTT assay. In
brief, 5×103 cells were plated into each well of 96-well
plates for 72 h following the indicated treatments. Subsequently, 5
mg/ml MTT was added and incubated at 37°C for 4 h. The medium was
then removed, and 1 ml DMSO was added to solubilize the
MTT-formazan product. The MTT absorbance was then determined at 570
nm on a Multiscan JX ver1.1 (Thermo Labsystems, Santa Rosa, CA,
USA). Results are expressed as a percentage of the viable cells in
the DMSO-treated group. Each data point is the mean ± standard
error of the mean of 6 replicates.
Apoptosis assays
Cells were stained with Annexin V and propidium
iodide and the percentage of apoptotic cells were determined by
flow cytometry, as described previously (20). CELL Quest software (BD Biosciences,
Franklin Lakes, NJ, USA) was used for data acquisition and
analysis.
Quantitative polymerase chain reaction
(qPCR)
Total RNA was isolated from A2780 or SKOV3 cells
after vorinostat treatment using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's
instructions. RNA quantitation was determined using a NanoDrop
micro-volume spectrophotometer (Thermo Fisher Scientific, Inc.),
and the messenger RNA (mRNA) integrity was verified by agarose gel
electrophoresis. Reverse transcription (RT)-qPCR was then performed
on 2 µg total RNA using a PrimeScript RT Reagent kit with gDNA
Eraser (Takara Bio, Inc., Otsu, Japan). qPCR was performed in ABI
Prism 7000 (Applied Biosystems; Thermo Fisher Scientific, Inc.)
with the SYBR Green PCR Master Mix (Sigma-Aldrich; Merck Millipore)
using the following thermocycler program for all genes: 5 min of
pre-incubation at 95°C, followed by 40 cycles of 15 sec at 95°C, 15
sec at 60°C, and 30 sec at 72°C. The primers for were as follows:
CD146 forward, 5′-CAGTCCTCATACCAGAGCCAACAG-3′ and reverse,
5′-GGACCAGGATGCACACAATCA-3′; and 18S ribosomal RNA forward,
5′-AGTCCCTGCCCTTTGACACA-3′ and reverse,
5′-GATCCGAGGGCCTCACTAAAC-3′. The 18S ribosomal RNA was used as an
internal control. All primers were obtained from Tiangen Biotech
Co., Ltd. (Beijing, China). A melting curve assay was performed to
determine the purity of the amplified product. Contamination with
genomic DNA was not detected in any of the analyzed samples. Each
sample was assayed in triplicate, analysis of relative gene
expression data used the 2−ΔΔCq method, as previously
described (21), and the results were
expressed as fold induction compared with the untreated group.
Western blot analysis
Detection of the CD146, AKT, p-AKT, P-4E-BP1,
p-S6K1, p-GSK-3β and β-actin by SDS-PAGE was performed as
previously described (21).
Soft agar colony-forming assay
Cells were treated with 10 µg/ml AA98, 2.5 µmol/l
vorinostat or vorinostat + AA98 for 24 h. DMSO-treated cells were
used as a negative control. A total of 1×103 cells were
then plated in 60-mm culture plates in medium containing 0.3% agar
overlying a 0.5% agar layer. The cells were subsequently incubated
for 14 days at 37°C and colonies were stained with 0.5 ml of
0.0005% crystal violet solution for 1 h and counted using a
dissecting microscope (×50 magnification). The results are
expressed as a percentage of colonies in the DMSO-treated
group.
Animal experiments
In total, 120 female athymic BALB/c nude mice were
obtained from the Animal Center of the Chinese Academy of Medical
Science (Beijing, China). The 6-week-old mice used were maintained
in a laminar-flow cabinet under specific pathogen free conditions.
In tumor xenograft models, 1×107 SKOV3 cells were
injected subcutaneously. Once tumors had grown between 5 and 6 mm,
the mice were grouped (n=10) and administered intraperitoneally
with 8 mg/kg of AA98 or 20 mg/kg of vorinostat or vorinostat + AA98
twice a week until the mice were sacrificed (tumor volume >1,000
mm3 or 42 days subsequent to treatment). PBS served as a
control. Tumor size was determined twice a week and tumor volume
was determined according to the equation: Tumor size
(cm3)=width2xlengthx(π/6).
Laser scanning cytometry (LSC)
LSC slides were scanned using an LSC instrument
equipped with argon (Ar; 488 nm) and helium-neon (HeNe; 633 nm)
laser and iCys3.3.4 software (CompuCyte; Beckman Coulter, Inc.,
Brea, CA, USA). DNA staining based on hematoxylin served as the
trigger/contouring parameter. The following channels and settings
were used for data collection: Argon green photomultiplier tube
(PMT, 15–25%; offset, 0.2; gain, 13%) and HeNe LongRed (LR; PMT,
14–22%; offset, 0–0.3; gain, 13%). The present study analyzed the
immunohistochemical tissue samples in phantom mode. Argon green and
HeNe LongRed parameters were collected with aberration
compensation. Statistical analysis was performed on the results of
3 independent experiments using the paired Student's t-test.
Statistical analysis
The statistical significance of differences between
experimental and control groups was determined by one-way analysis
of variance followed by the Student-Newman-Keuls test using SPSS
software version 13.0 (SPSS, Inc., Chicago, IL, USA). All
statistical tests were two sided, and P<0.05 was considered to
indicate a statistically significant difference. Statistical
analysis of LSC findings was performed on the results of 3
independent experiments using a paired Student's t test.
Results
Induction of adhesion molecular CD146
is a common phenomenon in vorinostat-treated ovarian cancer cells
in vitro and in vivo
In previous studies, adhesion molecule CD146 was
observed to be significantly upregulated following HDACi treatment
in ovarian cancer cells (13). In
addition, previous studies have linked CD146 with apoptosis
resistance in cancer cells (21,22). To
additionally verify whether expression of CD146 is induced by
vorinostat, the present study investigated the effects of
vorinostat on mRNA and protein expression of CD146 in ovarian
cancer cells. A2780 and SKOV3 cells were treated with 2.5 µmol/l
vorinostat for 12 h. As shown in Fig.
1A, subsequent to treatment with vorinostat, transcriptional
induction of CD146 reached an extremely high level, 468.5 fold for
A2780 and 450.3 fold for SKOV3 (P<0.001), compared with the
basal transcriptional level.
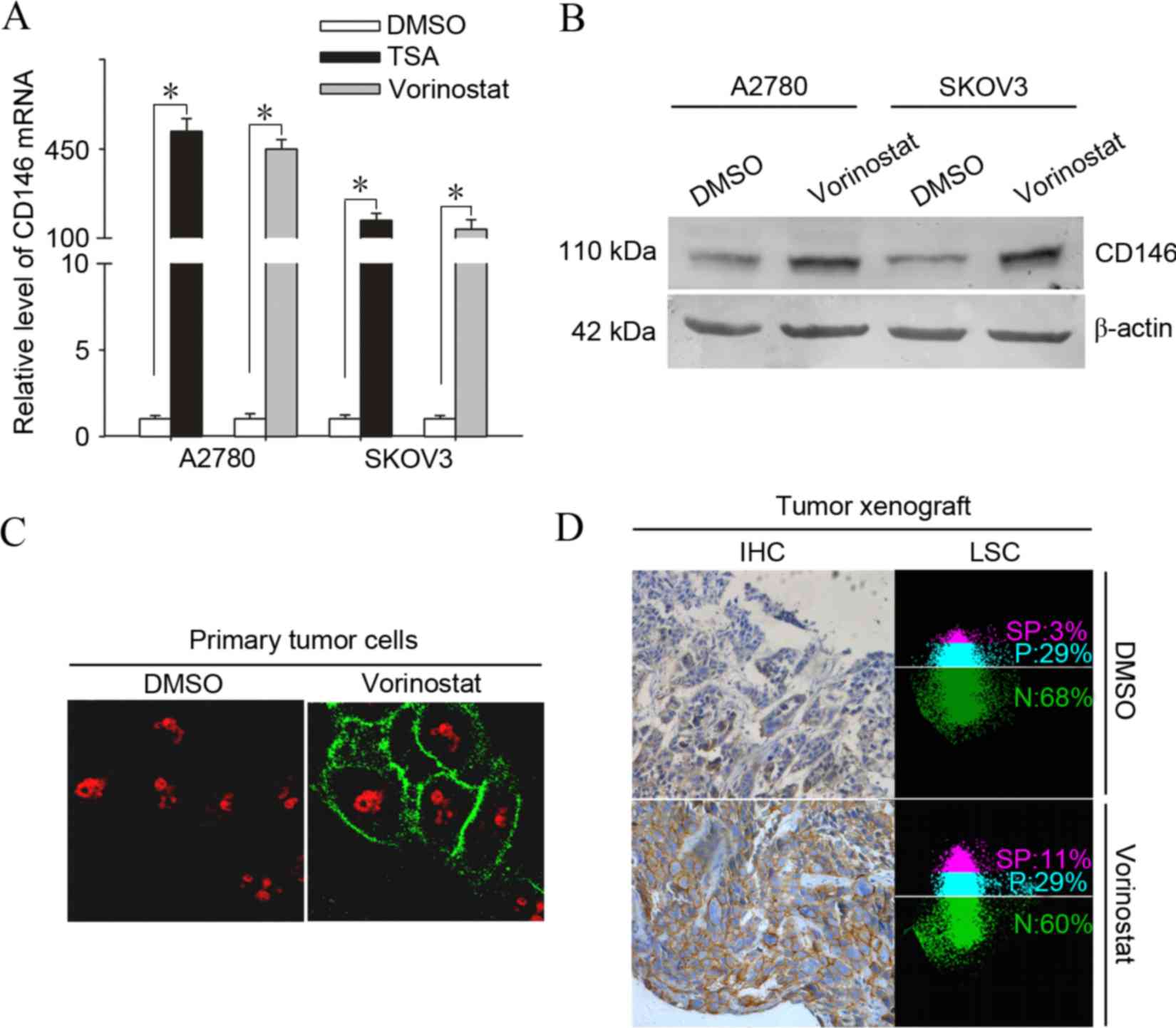 | Figure 1.Induction of the adhesion molecule
CD146 is a common phenomenon in vorinostat-treated ovarian cancer
cells in vitro and in vivo. (A) A2780 and SKOV3 cells
were treated with 2.5 µmol/l vorinostat or 500 nmol/l TSA for 12 h
and subjected to analysis of quantitative polymerase chain reaction
for the mRNA levels of CD146. Results are normalized to those of
18s RNA and expressed as the fold induction compared with the
DMSO-treated group (*P<0.05). (B) A2780 and SKOV3 cells were
treated with 2.5 µmol/l vorinostat for 24 h and analyzed for the
protein levels of CD146 by western blot analysis. (C) A2780 cells
were treated with 2.5 µmol/l vorinostat or DMSO for 12 h and were
then analyzed by immunofluorescent analysis for staining of CD146,
obtaining representative images under a confocal microscope
(magnification, ×600). (D) SKOV3 tumor-bearing mice were treated
with 25 mg/kg of vorinostat or DMSO for 24 h and CD146 expression
was determined by immunohistochemistry and quantified by laser
scanning cytometry. Images represent typical data (Total positive
rate for CD146 (SP plus P) is the mean ± standard deviation (n=10).
CD146, cluster of differentiation 146; DMSO, dimethyl sulfoxide;
TSA, trichostatin A; SP, strong positive; P, positive; N, negative;
IHC, immunohistochemistry; mRNA, messenger RNA; LSC, laser scanning
cytometry. |
Furthermore, another HDACi, TSA, significantly
induced the expression of CD146, indicating that the induction of
CD146 expression may be a common action shared by HDACi. To
determine whether the vorinostat-induced expression of CD146 occurs
in primary ovarian cancer cells, 8 primary tumor samples from
patients with ovarian cancer were treated with vorinostat.
Vorinostat significantly induced the expression of CD146 as early
as 3 h subsequent to treatment and the increase lasted up to 12 h
in all of the samples examined (Table
I). To test whether the induction of CD146 transcription
upregulated the level of CD146 protein, cultured A2780 cells were
treated with vorinostat or DMSO and examined for CD146 protein
expression using immunofluorescence and western blotting. As
expected, treatment with vorinostat significantly enhanced the
positive immunoreactivity and protein level of CD146 in A2780 cells
(Fig. 1B and C).
 | Table I.Effect of vorinostat on the expression
of CD146 in clinical tumor samples. |
Table I.
Effect of vorinostat on the expression
of CD146 in clinical tumor samples.
|
|
|
| Time course, h |
|---|
|
|
|
|
|
|---|
| Patients | Clinical
diagnosis | Classification | 0 | 3 | 6 | 12 |
|---|
| Patient 1 | Ovarian cancer | Serous | 1 | 76.40±7.25 | 16.33±2.78 | 15.97±1.64 |
| Patient 2 | Ovarian cancer | Mucinous | 1 | 77.13±6.65 | 14.02±2.45 | 13.23±1.76 |
| Patient 3 | Ovarian cancer | Serous | 1 | 80.52±7.81 | 18.59±2.97 | 17.13±2.35 |
| Patient 4 | Ovarian cancer | Serous | 1 | 54.21±4.36 | 15.47±3.75 | 11.24±1.88 |
| Patient 5 | Ovarian cancer | Mucinous | 1 | 73.26±6.82 | 21.67±1.98 | 20.57±1.18 |
| Patient 6 | Ovarian cancer | Serous | 1 | 103.42±8.93 | 46.15±2.60 | 37.22±2.71 |
| Patient 7 | Ovarian cancer | Serous | 1 | 66.57±5.33 | 50.14±3.08 | 26.89±3.43 |
| Patient 8 | Ovarian cancer | Serous | 1 | 90.36±7.47 | 65.11±4.23 | 30.78±3.59 |
To address whether the induction of CD146 occurs
in vivo, SKOV3 tumor-bearing mice (n=10) were treated with
vorinostat at 20 mg/kg based on earlier studies (23). Similarly, CD146 expression was
markedly elevated in the tumor cell membrane 24 h subsequent to
treatment with vorinostat, as determined by immunohistochemistry
and quantified by LSC (Fig. 1D); the
total positive rate for CD146 in SKOV3 tumors treated with
vorinostat compared with those treated with DMSO was 40±2 vs.
30±1%, (P=0.001).
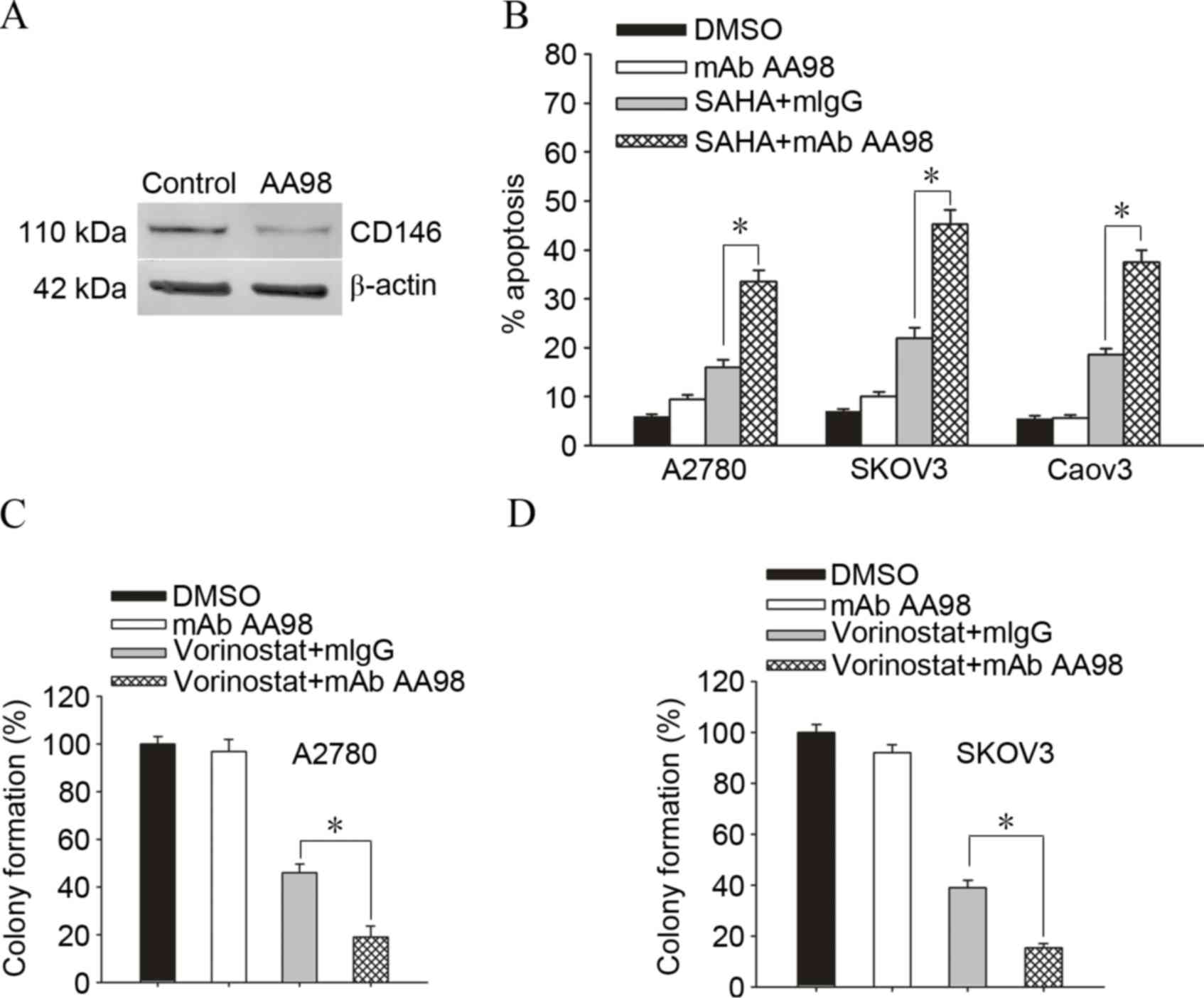
Targeting CD146 substantially enhanced
vorinostat-induced killing in ovarian cancer cells
To additionally confirm whether knockdown of CD146
enhanced vorinostat-induced cell death in ovarian cancer cells,
A2780 ovarian cancer cells were cultured with DMSO or AA98, which
has been confirmed to significantly knockdown the expression of
CD146 (Fig. 2A). A2780/SKOV3/Caov3
ovarian cancer cells are exposed to 2.5 µmol/l vorinostat for 72 h
and subjected to apoptosis assay for the determination of their
drug sensitivity. Accordingly, knockdown of CD146 increased the
sensitivity of A2780/SKOV3/Caov3 ovarian cancer cells to
vorinostat-induced apoptosis (Fig.
2B; A2780, 18.7±3.6 vs. 49.06±4.3%, P=0.001; SKOV3, 16.28±2.9
vs. 38.13±3.5%, P=0.001). Furthermore, knockdown of CD146 promoted
vorinostat-induced killing and gave rise to less survival colonies
in A2780 cells and SKOV3 cells (Fig. 2C
and D; A2780, 46.73±5.2 vs. 19.16±6.3%, P=0.004; SKOV3,
37.55±3.6 vs. 16.23±2.4%, P=0.001).
Knockdown of CD146 promotes
vorinostat-induced apoptosis via suppression of the Akt pathway in
ovarian cancer cells
Data has previously shown a link between CD146
expression and Akt activation (24);
therefore, the present study sought to determine the effects of
vorinostat/AA98 on the Akt pathway in ovarian caner cells A2780.
Vorinostat induced the phosphorylation of Akt and its downstream
targets 4E-BP1 and S6K1. AA98 co-administration with vorinostat
reverses the activation of the Akt pathway induced by vorinostat
(Fig. 3A). To additionally confirm
whether Akt had a protective effect on vorinostat/AA98-induced
apoptosis, overexpression/inhibition experiments were performed
using AAkt2 plasmid transfection or triciribine treatment. Although
transfection of AAkt2 inhibited vorinostat/AA98-induced apoptosis,
the inhibition of Akt phosphorylation by triciribine substantially
sensitized A2780 cells to vorinostat/AA98-induced killing (Fig. 3B, control vs. AAKT2, P=0.01; Fig. 3C, control vs. triciribine,
P=0.01).
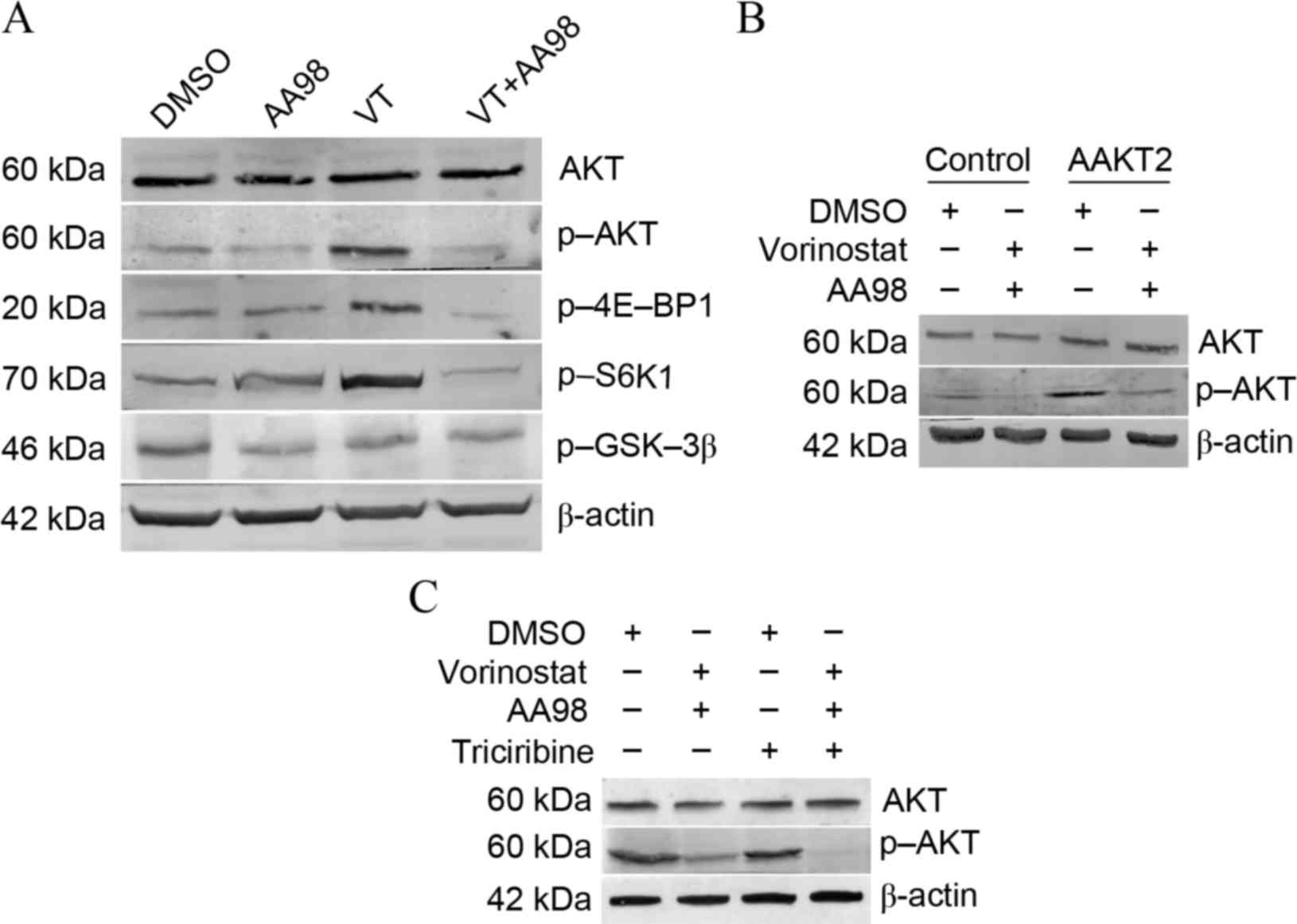 | Figure 3.Knockdown of CD146 promotes
vorinostat-induced apoptosis via suppression of the Akt pathway in
ovarian cancer cells. (A) A2780 cells were treated as depicted for
24 h (5 µmol/l vorinostat; 10 µg/ml mAb AA98) and examined for
protein levels of total Akt, p-Akt, P-4E-BP1, p-S6K1, p-GSK-3β and
β-actin. VT analysis by western blotting. (B) A2780 cells stably
transfected with the AAkt2 plasmid were treated as depicted for 24
h and examined for protein levels of total Akt and p-Akt by western
blot analysis. A2780 cells stably transfected with pcDNA3.1 plasmid
were treated as control group. (C) A2780 cells were treated with as
depicted for 24 h (5 µmol/l vorinostat; 10 µg/ml mAb AA98; 5 µmol/l
triciribine) and examined for protein levels of total Akt and p-Akt
by western blot analysis. CD146, cluster of differentiation 146;
Akt, protein kinase B; p-, phosphorylated; 4E-BP1, 4E-binding
protein 1; S6K1, ribosomal protein S6 kinase-1; GSK-3β, glycogen
synthase kinase 3β; VT, vorinostat treatment; DMSO, dimethyl
sulfoxide. |
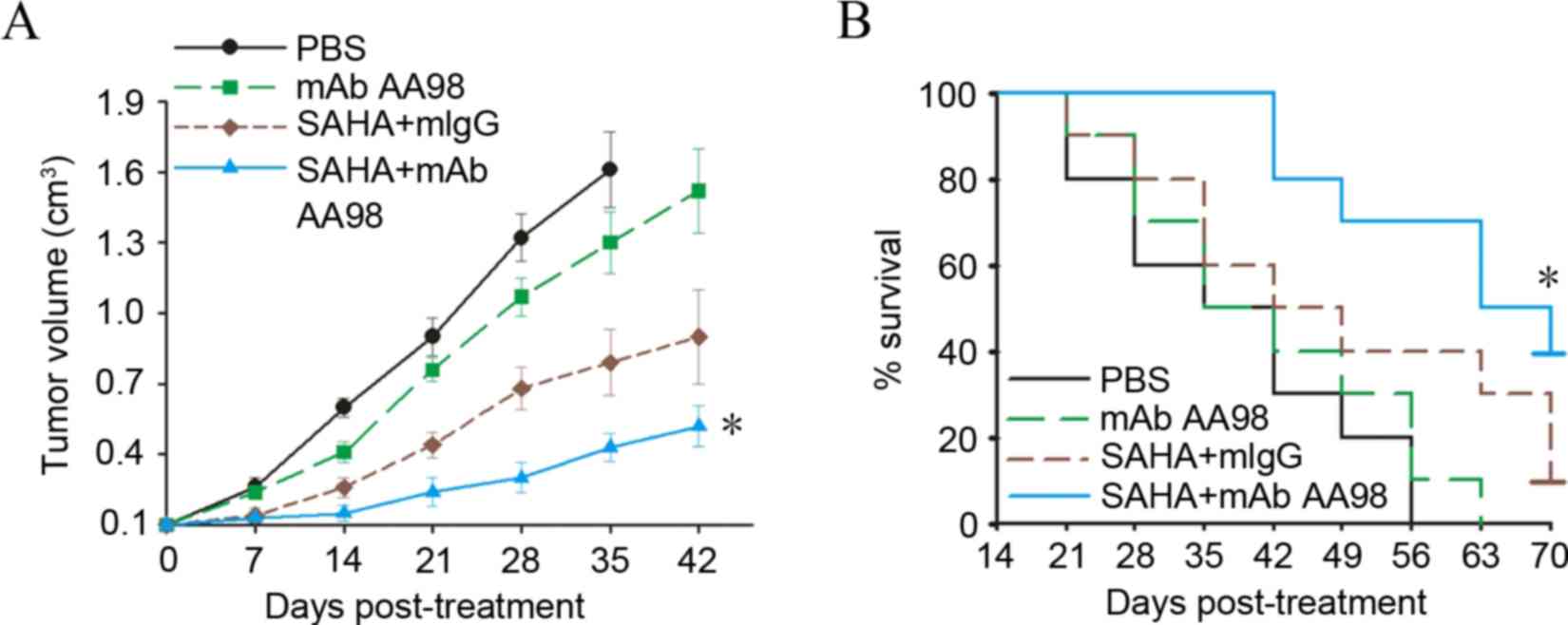
Targeting CD146 synergized with
vorinostat to substantially inhibit ovarian cancer growth
To determine the in vivo antitumor efficacy
of combined vorinostat and AA98, the present study chose lower
doses of the two agents compared with those previously reported
(13). The animal study was completed
when the tumor reached a diameter of 5–6 mm. The SKOV3
tumor-bearing mice were grouped (n=10) and administered
intraperitoneally with AA98 or vorinostat. Although no tumor
complete regression was observed in any groups with different
treatments, tumor growth was significantly retarded in the group
with combined vorinostat and AA98 treatment (P=0.02; Fig. 4A). Furthermore, combined vorinostat
and AA98 treatment significantly improved the survival rate in
SKOV3 tumor-bearing mice (P=0.001; Fig.
4B).
Discussion
The majority of patients with ovarian cancer have
progressed to advanced stages of disease by the first clinical
visit, and are therefore not eligible to be treated with surgery,
and can only receive chemotherapy, with poor results (25). Drug resistance is the primary cause of
mortality in late-stage patients. The flood of new second line
drugs in previous years has provided numerous marked improvements
in anticancer therapy (26). Thus,
the development of new therapeutic strategies and search for novel
genes with new mechanisms of action that can lead to drug
resistance of ovarian cancer cell have become the focuses of
current cancer research.
HDACis have emerged as novel second line drugs, with
their high specificity for tumor cells. However, since the targets
of HDACis are so extensive, it is not surprising that HDACis would
initiate anti-apoptotic and pro-apoptotic therapeutic responses.
HDACis usually exhibit relatively low potency when used as single
agents. The majority of the current HDACi combination strategies
are more empirical than mechanism-based applications, and
accordingly, are not optimal for this class of drugs (27,28). In
our previous study, a cDNA microarray analysis was conducted and it
was found that the expression of adhesion molecule CD146 was
significantly induced in HDACi-treated tumor cells, particularly in
ovarian cancer cells (13). In the
present findings, it was verified that the induction of CD146 is a
common phenomenon in vorinostat-treated ovarian cancer cells in
vitro and in vivo. Targeting CD146 substantially
sensitized ovarian cancer cells to vorinostat-induced killing.
Treatment with vorinostat plus AA98 also preferentially inhibits
cell proliferation, enhances apoptotic rate of ovarian cancer cells
and ablates cancer colony formation. The present findings provide
the first evidence that an undesired protective signal is initiated
by HDACi and highlight a novel molecular mechanism by which HDACi
induces the expression of CD146 as a protective response to offset
the antitumor efficacy. By contrast, the induction of CD146 may be
exploited as a novel strategy for the enhanced killing of ovarian
cancer cells. Similarly, the synergistic killing effect of
vorinostat and targeting of CD146 was observed in vivo.
Treatment of SKOV3 xenografts with vorinostat plus AA98 resulted in
a more pronounced decrease in tumor volume compared with single
drug-treated mice. Additionally, to inhibit tumor growth, it was
shown that the combined regimen of vorinostat and AA98 is able to
significantly prolong the survival rate of tumor-bearing mice.
It is well known that the sensitivity of cancer
cells to chemotherapeutic drug-induced apoptosis depends on the
balance between pro-apoptotic and anti-apoptotic signals (29). Therefore, inhibition of anti-apoptotic
signals, such as those mediated by the Akt pathway, has been
proposed as a promising strategy to enhance the efficacy of
chemotherapeutic agents (30). The
present data show that the increased sensitivity to vorinostat
caused by AA98 was strongly associated with Akt signaling in
ovarian carcinomas. The combination of vorinostat with AA98
attenuates Akt phosphorylation and 4E-BP1 expression. A similar
association has been reported between Akt activation and HDACi
sensitivity in cervical cancer cell lines (31). However, Akt kinase activity is not the
sole determinant of sensitivity to vorinostat, and certain factors,
such as S6K1, can result in sensitivity to vorinostat in the
absence of Akt activation (32). In
ovarian cancer A2780 cells, AA98 co-administration reverses the
activation of S6K1 induced by vorinostat. Furthermore, it was
confirmed that Akt had a protective effect on
vorinostat/AA98-induced apoptosis by overexpression/inhibition
experiments.
Collectively, targeting CD146 may be exploited as a
novel strategy to more effectively kill ovarian cancer cells.
However, the identification of an optimal HDACi-based regimen
requires long-term and painstaking clinical trials and suboptimal
application. The current preclinical approach may accelerate the
design of an optimal HDACi-containing regimen in the treatment of
ovarian cancer.
Acknowledgements
The present study was supported by a grant from the
Beijing Nova Program (grant no. Z141107001814015), National Natural
Science Foundation of China (grant no. 81101970) and PhD Programs
Foundation of Ministry of Education of China (grant no.
20111107120009).
References
|
1
|
Suh DH, Lee KH, Kim K, Kang S and Kim JW:
Major clinical research advances in gynecologic cancer in 2014. J
Gynecol Oncol. 26:156–167. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jayson GC, Kohn EC, Kitchener HC and
Ledermann JA: Ovarian cancer. Lancet. 384:1376–1388. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zahedi P, Yoganathan R, Piquette-Miller M
and Allen C: Recent advances in drug delivery strategies for
treatment of ovarian cancer. Expert Opin Drug Deliv. 9:567–583.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vecchione A, Belletti B, Lovat F, Volinia
S, Chiappetta G, Giglio S, Sonego M, Cirombella R, Onesti EC,
Pellegrini P, et al: A microRNA signature defines chemoresistance
in ovarian cancer through modulation of angiogenesis. Proc Natl
Acad Sci USA. 110:9845–9850. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zwergel C, Valente S, Jacob C and Mai A:
Emerging approaches for histone deacetylase inhibitor drug
discovery. Expert Opin Drug Discov. 10:599–613. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ma XL, Duan H, Liu J, Mo Q, Sun C, Ma D
and Wang J: Effect of LIV1 on the sensitivity of ovarian cancer
cells to trichostatin A. Oncol Rep. 33:893–898. 2015.PubMed/NCBI
|
|
7
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Slingerland M, Guchelaar HJ and Gelderblom
H: Histone deacetylase inhibitors: An overview of the clinical
studies in solid tumors. Anticancer Drugs. 25:140–149. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Højfeldt JW, Agger K and Helin K: Histone
lysine demethylases as targets for anticancer therapy. Nat Rev Drug
Discov. 12:917–930. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang Z, Peng S, Knoff J, Lee SY, Yang B,
Wu TC and Hung CF: Combination of proteasome and HDAC inhibitor
enhances HPV16 E7-specific CD8+ T cell immune response and
antitumor effects in a preclinical cervical cancer model. J Biomed
Sci. 22:72015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Straus DJ, Hamlin PA, Matasar MJ, Lia
Palomba M, Drullinsky PR, Zelenetz AD, Gerecitano JF, Noy A,
Hamilton AM, Elstrom R, et al: Phase I/II trial of vorinostat with
rituximab, cyclophosphamide, etoposide and prednisone as palliative
treatment for elderly patients with relapsed or refractory diffuse
large B-cell lymphoma not eligible for autologous stem cell
transplantation. Br J Haematol. 168:663–670. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rasheed WK, Johnstone RW and Prince HM:
Histone deacetylase inhibitors in cancer therapy. Expert Opin
Investig Drugs. 16:659–678. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma X, Liu J, Wu J, Yan X, Wu P, Liu Y, Li
S, Tian Y, Cao Y, Chen G, et al: Synergistic killing effect between
vorinostat and target of CD146 in malignant cells. Clin Cancer Res.
16:5165–5176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Z and Yan X: CD146, a
multi-functional molecule beyond adhesion. Cancer Lett.
330:150–162. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xie S, Luca M, Huang S, Gutman M, Reich R,
Johnson JP and Bar-Eli M: Expression of MCAM/MUC18 by human
melanoma cells leads to increased tumor growth and metastasis.
Cancer Res. 57:2295–2303. 1997.PubMed/NCBI
|
|
16
|
Wu GJ, Wu MW, Wang SW, Liu Z, Qu P, Peng
Q, Yang H, Varma VA, Sun QC, Petros JA, et al: Isolation and
characterization of the major form of human MUC18 cDNA gene and
correlation of MUC18 over-expression in prostate cancer cell lines
and tissues with malignant progression. Gene. 279:17–31. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zabouo G, Imbert AM, Jacquemier J, Finetti
P, Moreau T, Esterni B, Birnbaum D, Bertucci F and Chabannon C:
CD146 expression is associated with a poor prognosis in human
breast tumors and with enhanced motility in breast cancer cell
lines. Breast Cancer Res. 11:R12009. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Leslie MC, Zhao YJ, Lachman LB, Hwu P, Wu
GJ and Bar-Eli M: Immunization against MUC18/MCAM, a novel antigen
that drives melanoma invasion and metastasis. Gene Ther.
14:316–323. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xing S, Luo Y, Liu Z, Bu P, Duan H, Liu D,
Wang P, Yang J, Song L, Feng J, et al: Targeting endothelial CD146
attenuates colitis and prevents colitis-associated carcinogenesis.
Am J Pathol. 184:1604–1616. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma XL, Ma QF, Liu J, Tian Y, Wang B,
Taylor KM, Wu P, Wang D, Xu G, Meng L, et al: Identification of
LIV1, a putative zinc transporter gene responsible for
HDACi-induced apoptosis, using a functional gene screen approach.
Mol Cancer Ther. 8:3108–3116. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Satyamoorthy K, Muyrers J, Meier F, Patel
D and Herlyn M: Mel-CAM-specific genetic suppressor elements
inhibit melanoma growth and invasion through loss of gap junctional
communication. Oncogene. 20:4676–4684. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lei X, Guan CW, Song Y and Wang H: The
multifaceted role of CD146/MCAM in the promotion of melanoma
progression. Cancer Cell Int. 15:32015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cooper AL, Greenberg VL, Lancaster PS, van
Nagell JR Jr, Zimmer SG and Modesitt SC: In vitro and in vivo
histone deacetylase inhibitor therapy with suberoylanilide
hydroxamic acid (SAHA) and paclitaxel in ovarian cancer. Gynecol
Oncol. 104:596–601. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeng Q, Wu Z, Duan H, Jiang X, Tu T, Lu D,
Luo Y, Wang P, Song L, Feng J, et al: Impaired tumor angiogenesis
and VEGF-induced pathway in endothelial CD146 knockout mice.
Protein Cell. 5:445–456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lheureux S, Karakasis K, Kohn EC and Oza
AM: Ovarian cancer treatment: The end of empiricism? Cancer.
121:3203–3211. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Modugno F and Edwards RP: Ovarian cancer:
Prevention, detection, and treatment of the disease and its
recurrence. molecular mechanisms and personalized medicine meeting
report. Int J Gynecol Cancer. 22:S45–S57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bots M and Johnstone RW: Rational
combinations using HDAC inhibitors. Clin Cancer Res. 15:3970–3977.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Khabele D: The therapeutic potential of
class I selective histone deacetylase inhibitors in ovarian cancer.
Front Oncol. 20:1112014.
|
|
29
|
Mohammad RM, Muqbil I, Lowe L, Yedjou C,
Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB, Dou QP, et al:
Broad targeting of resistance to apoptosis in cancer. Semin Cancer
Biol. 35:(Suppl). S78–S103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ocana A, Vera-Badillo F, Al-Mubarak M,
Templeton AJ, Corrales-Sanchez V, Diez-Gonzalez L, Cuenca-Lopez MD,
Seruga B, Pandiella A and Amir E: Activation of the PI3K/mTOR/AKT
pathway and survival in solid tumours: Systematic review and
meta-analysis. PLoS One. 9:e952192014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Feng D, Cao Z, Li C, Zhang L, Zhou Y, Ma
J, Liu R, Zhou H, Zhao W, Wei H and Ling B: Combination of valproic
acid and ATRA restores RARβ2 expression and induces differentiation
in cervical cancerthrough the PI3K/Akt pathway. Curr Mol Med.
12:342–354. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moschetta M, Reale A, Marasco C, Vacca A
and Carratù MR: Therapeutic targeting of the mTOR-signalling
pathway in cancer: Benefits and limitations. Br J Pharmacol.
171:3801–3813. 2014. View Article : Google Scholar : PubMed/NCBI
|