Introduction
Epithelial-mesenchymal transition (EMT) is a
biological process observed in embryo neural crest formation
(1). In order to migrate easily to
distant locations, embryonic epithelial cells undergo EMT to become
mesenchymal cells (2). In addition to
embryonic cells, cancer cells also undergo EMT (3). This phenomenon was proposed as a cancer
metastasis hypothesis, in which epithelial cancer cells
downregulate E-cadherin to detach from the primary tumour (4). E-cadherin and vimentin, expressed in
epithelial and mesenchymal cells, respectively, have been
considered as key markers for EMT (5). Reports suggest that several factors,
including Snail and Twist, are able to regulate E-cadherin
expression (6,7). It was believed that epithelial-type
cancer cells underwent EMT, changed to mesenchymal-type cancer
cells, moved to a secondary organ and subsequently underwent
mesenchymal-epithelial transition (MET) to form a secondary tumour
mass (8). However, to the best of our
knowledge, very little information has been reported regarding the
MET process. A previous study reported that paired box 2, bone
morphogenetic protein 7 and Wilms tumour 1 were associated with MET
during kidney formation in the embryo (9). The cancer stem cell model is a
hypothesis associated with cancer metastasis. Cancer stem cells
possess similarities with stem cells, sharing properties including
self-renewal, differentiation and chemoresistance ability (10). With the aforementioned
characteristics, it is believed that only cancer stem cells have an
ability to develop a tumour mass in distant organs. There have been
studies combining cancer stem cell and EMT theories in the analysis
of mammary and lung cancer (11,12).
Numerous markers for cancer stem cells have been identified in
various types of solid and blood cancer. For example, cluster of
differentiation (CD)133, CD44 and CD24 have been widely used to
identify cancer types displaying stem cell properties (13). Lung cancer may be divided into
non-small cell lung cancer (NSCLC) and small cell lung cancer
(14). The incidence of NSCLC is 4
times higher, and it is more resistant to chemotherapy compared
with small cell lung cancer (14).
The prognosis of NSCLC patients is poor, and the 5-year survival
rate was reported to be 5–15% (14).
The A549 cancer cell line is an NSCLC cell line (15). A number of studies have been performed
to correlate the association between EMT and cancer stem cells
using the A549 cell line (15,16).
However, to the best of our knowledge, little is known about MET,
the reverse of the EMT process, and the association between MET and
cancer stem cells following transforming growth factor β1
(TGF-β1)-induced EMT. Thus, the aim of the current study was to
investigate the TGF-β1 microenvironment conditions affecting EMT
and MET in the A549 lung cancer cell line. Additionally, properties
associated with cancer stem cells were measured to reveal the
association between EMT, MET and cancer stem cells.
Materials and methods
Cells, chemicals and reagents
A549 cell line was supplied by the Korean Cell Line
Bank (Seoul, South Korea). Cisplatin, phosphate-buffered saline
(PBS) and ribonuclease A were purchased from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany). RPMI-1640 medium, fetal bovine serum
(FBS) and penicillin/streptomycin were obtained from Gibco (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). For flow cytometry,
monoclonal antibodies for CD24 (cat. no. 130-095-953), CD44 (cat.
no. 130-095-195) and CD133 (cat. no. 130-090-853) were purchased
from Miltenyi Biotec GmbH (Bergisch Gladbach, Germany). Protease
inhibitor cocktail was obtained from Intron Biotechnology, Inc.
(Seongnam, Korea). For western blotting, TGF-β1,
radioimmunoprecipitation assay (RIPA) lysis buffer 3 and the
primary polyclonal antibody against caspase-3 (cat. no.
ADI-AAP-113) were supplied by Enzo Life Sciences, Inc.
(Farmingdale, NY, USA). Primary antibodies against E-cadherin (cat.
no. BS1097) (polyclonal), SMAD3 (cat. no. AP0446) (polyclonal), and
phosphorylated-SMAD3 (cat. no. BS64037) (polyclonal) were supplied
by Bioworld Technology, Inc. (St. Louis Park, MN, USA). Primary
antibodies against vimentin (cat. no. A301-620A) (polyclonal) and
GAPDH (cat. no. A300-641A-M) (polyclonal) were provided by Bethyl
Laboratories Inc. (Montgomery, TX, USA). Primary antibodies against
signal transducer and activator of transcription 3 (STAT3;
polyclonal), phosphorylated-STAT3 (monoclonal), extracellular
signal-regulated kinase (ERK; polyclonal), phosphorylated-ERK
(monoclonal), nuclear factor κB (NF-κB; polyclonal) and
phosphorylated-NF-κB (polyclonal) were obtained from EMD Millipore
(Billerica, MA, USA).
Cell culture
Cells were cultured in RPMI-1640 medium with 10%
heat-inactivated FBS and 1% penicillin/streptomycin. Cells were
maintained at 37°C in an atmosphere of 5% CO2 in a
humidified incubator. The experiments were divided into 3 groups:
Group 1 (control), TGF(−) received no treatment; group 2, TGF(+)
treated with 10 ng/ml (17) TGF-β1
daily for 3 days; and group 3 (MET/return), TGF-β1-treated cells
(group 2) were incubated in media for an additional 3 days
following removal of TGF-β1.
Morphological analysis
The cells were grown in a coated cell culture dish
(SPL Life Science, Pocheon, South Korea) and visualized without
stain using an Olympus CKX41 optical microscope and Tomoro AcquCAM
3 (both Olympus Corporation, Tokyo, Japan) digital camera.
Flow cytometry
Immunostaining of A549 cells was performed as
follows. Cells were blocked with 2% FBS reconstituted in PBS, and
subsequently incubated with antibodies against CD24, CD44 and CD133
conjugated to allophycocyanin, fluorescein isothiocyanate and
phycoerythrin (mouse anti-human, 1:11). The stained samples were
analysed using a flow cytometer equipped with
fluorescence-activated cell sorting BD CellQuest™ Pro software
version 6.0 (BD Biosciences, Franklin Lakes, NJ, USA). The flow
cytometric analysis was performed using isotype control antibodies
and single colour stained samples in multivariate flow cytometry
(18).
Cell cycle analysis
Cell cycle analysis was performed as described
previously (19). A549 cells were
seeded at a density of 1×105 cells/well in 6-well
plates. Following incubation, the cells were collected, washed with
PBS, fixed with 70% ethanol and stored at 4°C. To remove ethanol,
stored cells were washed with PBS, ribonuclease A (50 µg/ml) was
added and cells were incubated at room temperature for 5 min.
Subsequently, the cells were stained with 10 µg/ml propidium iodide
(PI), incubated at 37°C for 10 min and counted using flow
cytometry.
Cisplatin resistance test
TGF(−), TGF(+) and MET/return cells were incubated
in 48-well cell culture plates in 400 µl medium. Cells were
subsequently treated with cisplatin at a concentration of 100 ng/ml
and incubated at 37°C for 48 h. Subsequently, the cell viability
was measured using WST-1 solution (EZ-Cytox kit; Daeillab Service
Co., Ltd., Seoul, Korea) and the optical density at 450 nm was
measured using an ELISA plate reader with Magellan™
Tracker software version 3.0.0.12 (Tecan Group Ltd., Männedorf,
Switzerland) (19).
In vitro cell proliferation and
migration assay
A549 cells were incubated in a gelatin-coated 6-well
plate to form a 100% confluent monolayer prior to wounding. A wound
was made (using a 1 ml pipette tip) by scraping across the
monolayer, and the cells migrated in medium supplemented with 10%
FBS (20).
Protein extraction and western blot
analysis
Western blotting was performed as previously
described (19). The whole-cell
lysates were extracted with a mixture of RIPA lysis buffer 3,
phosphatase inhibitor cocktail and protease inhibitor cocktail,
according to the manufacturer's instructions. Lysate protein was
centrifuged at 4°C at 16,000 × g for 20 min. Lysate protein level
was measured using the Bradford protein assay (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Aliquots of 10 µg cell
lysate protein were resolved on 8% (v/v) SDS-PAGE gel, and
transferred onto a nitrocellulose membrane (Bio-Rad Laboratories,
Inc.) for 90 min at 300 V. The protein-attached membrane was
incubated with blocking buffer (TTBS, 20 mM Tris-HCl, pH 7.6, 137
mM NaCl and 0.05% Tween-20) supplemented with 5% (w/v) non-fat dry
milk for 1 h at room temperature. The membrane was washed 5 times
for 5 min each with TTBS and then incubated (overnight) with
primary antibodies (rabbit anti-human; 1:1,000). Subsequently, the
membrane was incubated (for 1 h) with horseradish peroxidase
(HRP)-conjugated secondary antibody (goat anti-rabbit; 1:5,000).
Visualization of the protein bands was performed using enhanced
chemiluminescence western blotting substrate (Chemiluminescent
Sensitive Plus HRP Microwell and/or Membrane Substrate; SurModics,
Inc., Eden Prairie, MN, USA).
Statistical analysis
The results are presented as the mean ± standard
deviation. Comparison of the groups was performed using one-way
analysis of variance. P<0.05 was considered to indicate a
statistically significant difference. Statistical analyses were
performed using SPSS version 22 (IBM SPSS, Armonk, NY, USA).
Results
Induction of epithelial-mesenchymal
transition
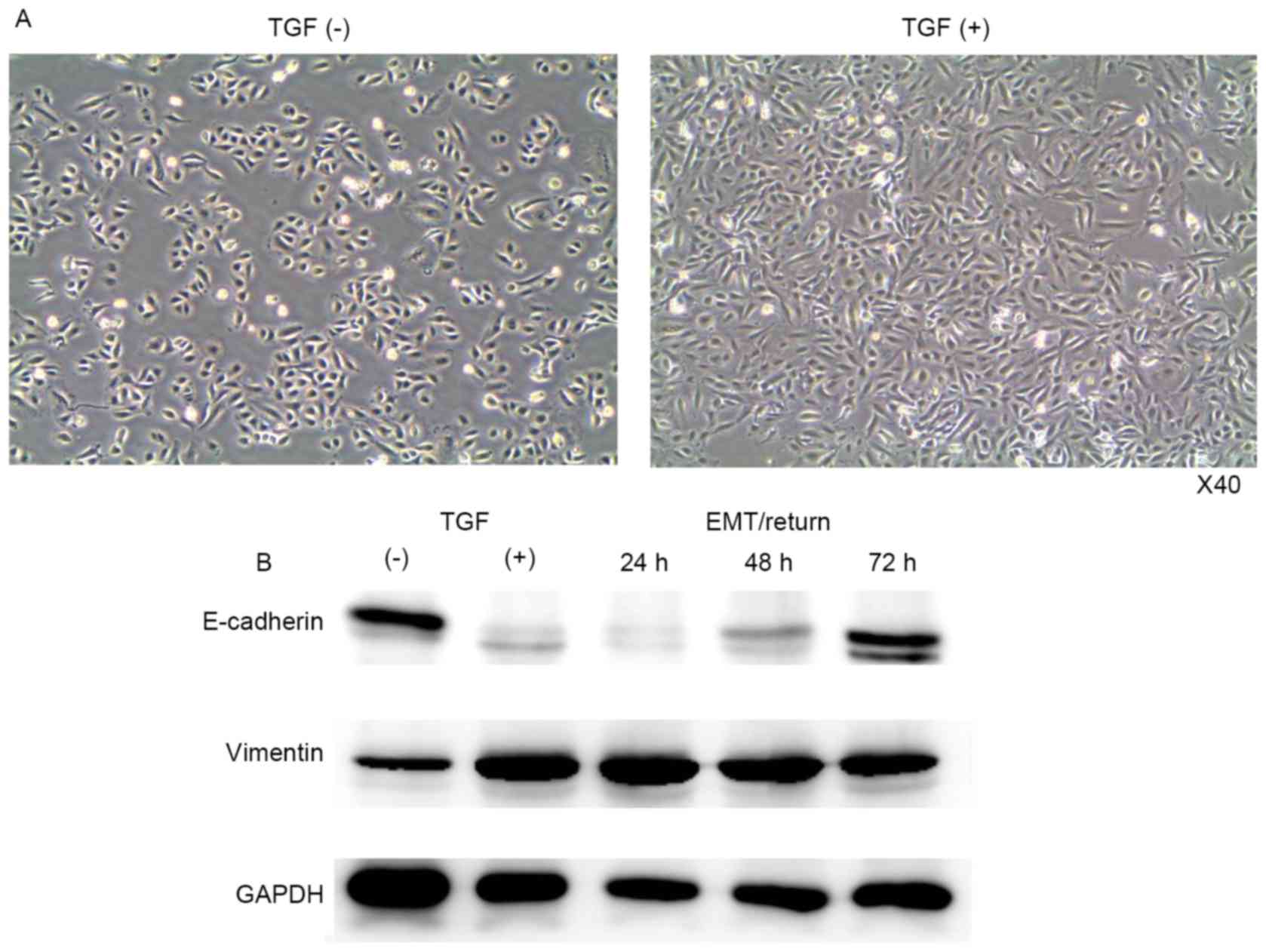
A549 cells were treated with TGF-β1 every 24 h for 3
days, at a concentration of 10 ng/ml. Morphological changes were
observed daily by microscopy and images were recorded using a
camera. After 72 h of treatment, the morphology of the A549 cells
became fibroblast-like, with an elongated shape (Fig. 1A). To confirm EMT induction, western
blotting was performed. E-cadherin (epithelial cell marker) was
decreased, whereas vimentin (mesenchymal cell marker) was
increased, following TGF-β1 treatment (Fig. 1B). In A549 cells, EMT was not fully
induced until day 2 of treatment (data not shown).
Changes of mesenchymal-epithelial
transition
Following induction of EMT, the TGF-β1 was washed
from the cell culture media (group 3) to investigate the return of
mesenchymal-type cells back to epithelial cells. Mesenchymal-type
A549 cells were converted to epithelial-type cells after 3 days of
incubation without TGF-β1 (Fig. 1B).
The level of E-cadherin returned to normal, and the vimentin level
was slightly reduced after 72 h.
Cancer stem cell marker
properties
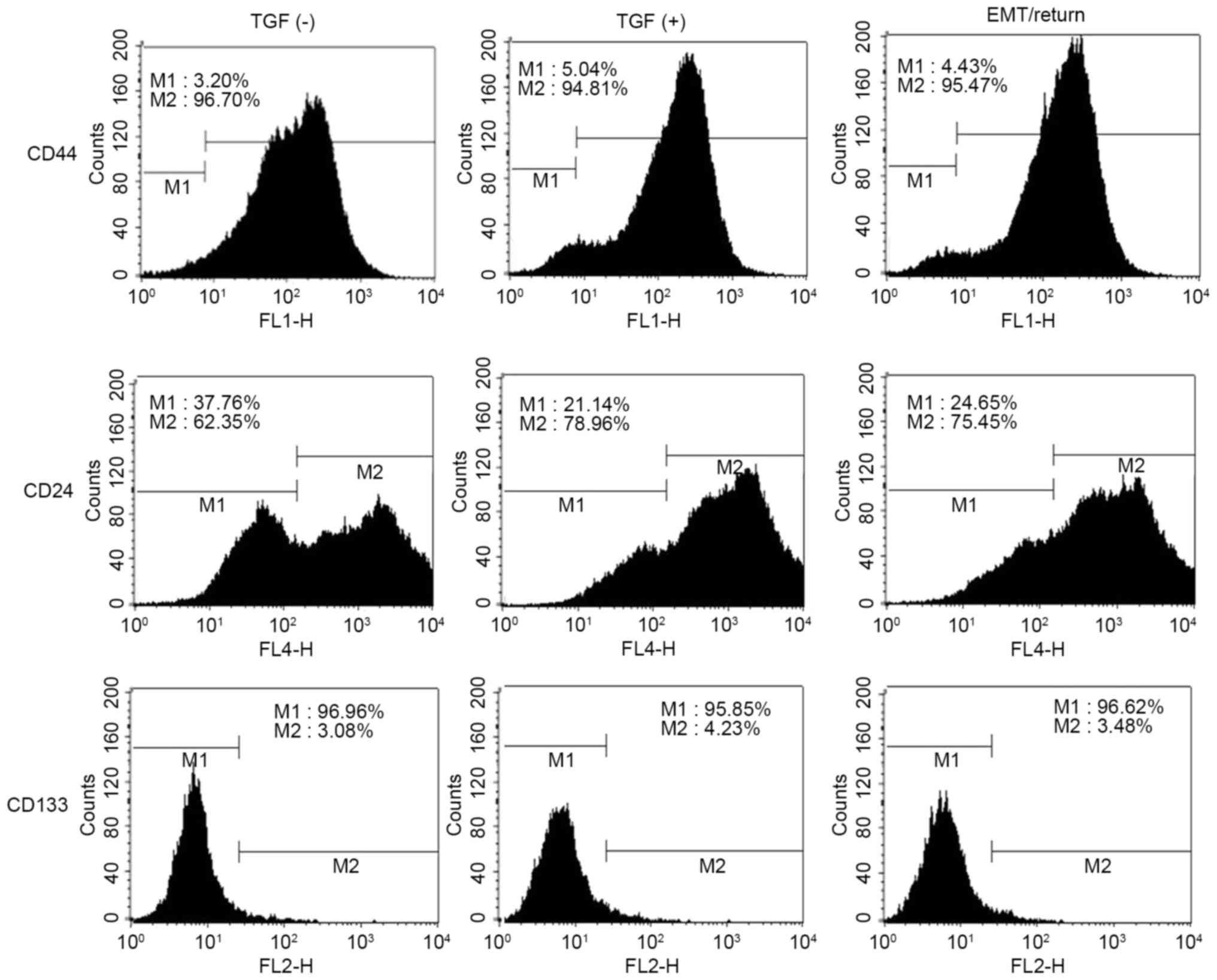
Cancer stem cell marker proteins, including CD24,
CD44 and CD133 were measured using flow cytometry. In the control
group, the population of CD44high was 96.07% (M2),
CD24low was 37.76% (M1) and CD133high was
3.08% (M2) in A549 cells (Fig. 2).
Following TGF-β1 treatment, CD44high and
CD133high were slightly changed; however, the proportion
of CD24low was reduced by ~15% (Fig. 2). Although the A549 cells in group 3
returned back to normal status, the profile of CD24low
cells remained unaltered compared to group 2 (Fig. 2).
Cell cycle analysis, cisplatin
resistance and wound healing assay
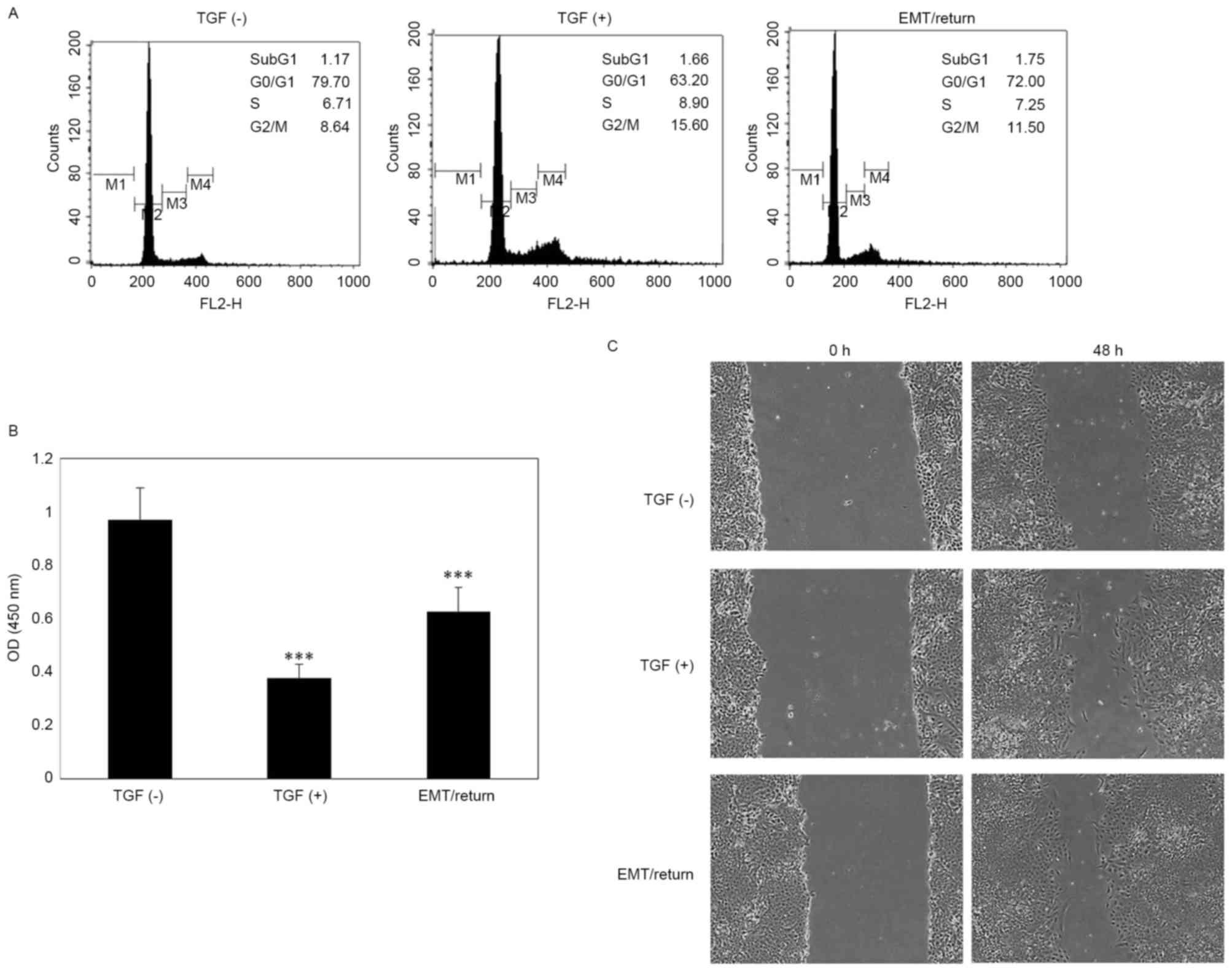
Untreated A549 cells demonstrated ~80% of the
population in G0/G1 phase and 8.64% in G2/M phase. However, in the
TGF-β1-treated group, the percentage of G2/M phase cells was almost
doubled compared with the control group. In group 3, the percentage
of cells in the G2/M phase was slightly reduced compared to group
2; however, it was still increased compared with the control
(Fig. 3A). The A549 cells of the
TGF-β1-treated and MET/return groups exhibited a significant
sensitivity (P<0.001) to cisplatin treatment compared to the
control (Fig. 3B). In the wound
healing assay, TGF-β1-treated and MET/return groups exhibited
increased proliferative and migration ability compared to the
control (Fig. 3C).
Immunoblotting assay
In screening for the mechanism of action of TGF-β1
in A549 cells, 5 different signalling factors, including SMAD3,
caspase-3, ERK, NF-κB and STAT3, were investigated. Levels of SMAD3
and the phosphorylated form of SMAD3 (known to be directly involved
in TGF-β1 signalling) were increased in TGF-β1-treated cells, and
returned to normal in the MET/return group (Fig. 4). By contrast, total caspase-3 was
increased, and the active form of caspase-3 was decreased, in the
TGF-β1-induced EMT and MET/return groups. NF-κB, STAT3 and ERK
signalling were unchanged in all groups, with the exception of
phosphorylated ERK. Phosphorylated ERK signalling was slightly
increased in the TGF-β1-treated group, and returned to normal in
the MET/return group (Fig. 4).
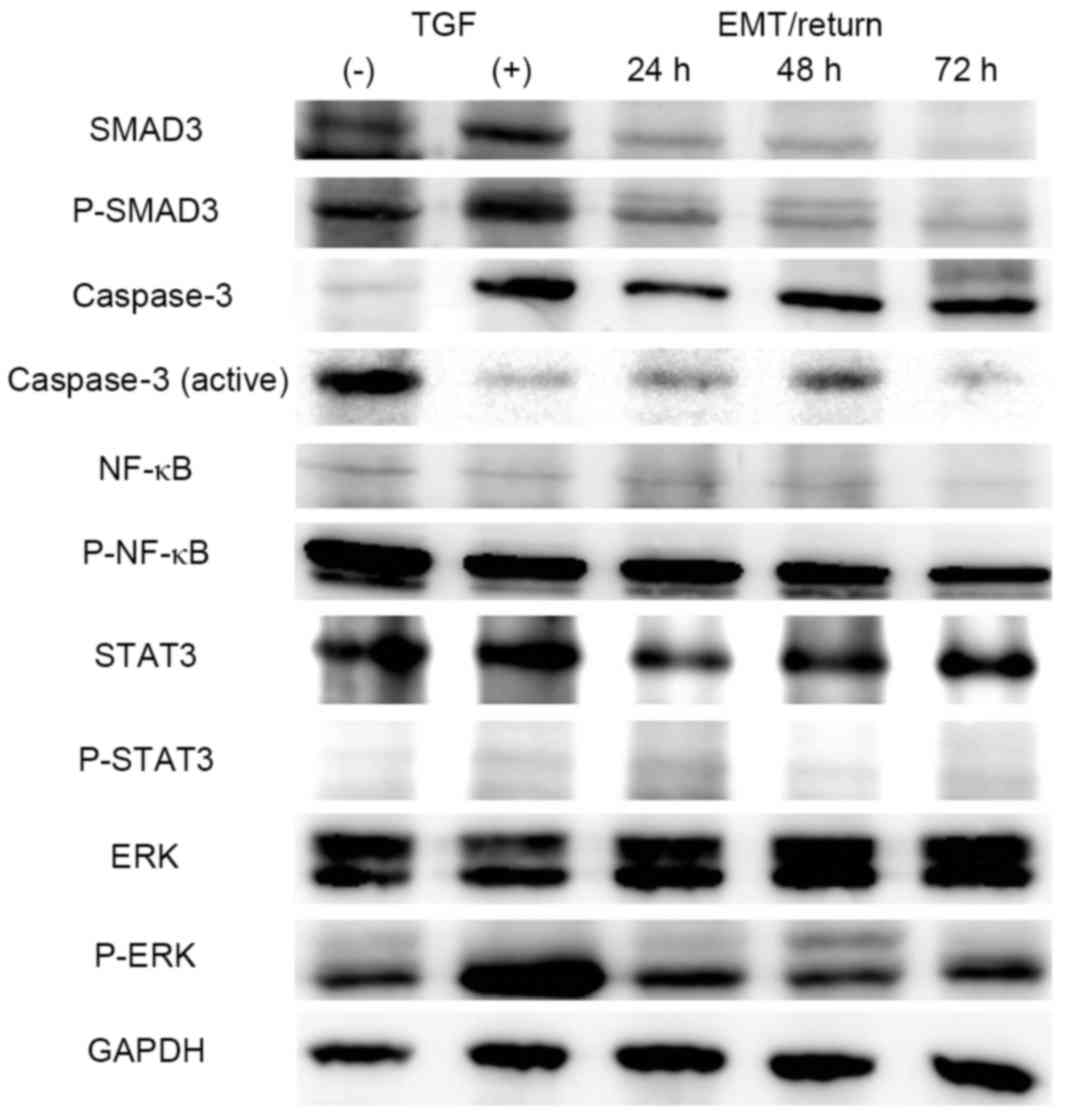 | Figure 4.Mechanism screening by western
blotting. Whole-cell lysates were immunoblotted with STAT3, ERK,
NF-κB, caspase-3, SMAD3 and GAPDH (housekeeping gene) antibodies.
STAT3, signalling transducer and activator of transcription 3; ERK,
extracellular signal-regulated kinase; NF-κB, nuclear factor-κB;
TGF, transforming growth factor; EMT, epithelial-mesenchymal
transition; P, phosphorylated. |
Discussion
The association between EMT and cancer stem cells
was originally reported in 2008 by Mani et al (11). They sorted immortalized human mammary
epithelial cells and observed that
CD44high/CD24low cells demonstrated vimentin
expression and cancer stemness properties. However, in the present
study, the portion of CD24low cells was decreased,
CD44high was unchanged and the CD133+ lung cancer stem
cell marker demonstrated negative results in TGF-β1-treated cells,
findings which contrasted to other studies (11,21).
However, the proportion of CD24low cells in the
MET/return group was unchanged compared to that in the
TGF-β1-treated group. From a hypothetical point of view regarding
the association between cancer stem cells and EMT (3), this means that CD24 may be a key marker
of cancer stemness in the A549 lung cancer cell line, a suggestion
supported by Zheng et al (22). By contrast, it was reported that CD24
and CD44 were not considered as cancer stem cell markers in the
A549 lung cancer cell line (23). In
this previous study, ~70% of normal A549 cells were recorded as
CD44high cells, and 30% were CD24low
(23). In the present study, the
percentage of CD24low cells was reduced in
TGF-β1-treated and MET/return groups. This finding indicates that
EMT induced by TGF-β1 is associated with CD24, and CD24 may be
considered as a cancer stem cell marker. The results of the present
study contrast with those of Roudi et al (23), who evaluated cancer stem cell markers
without considering the EMT. Furthermore, the CD24low
population was not significantly altered following the MET/return
period, which means that TGF-β1 associated with cancer stem cells
only during the commencement period.
Scheel and Weinberg (24) demonstrated that mesenchymal mammary
cancer cells demonstrate resistance to chemicals and limited
proliferation ability, findings which contrast with those of the
present study. The present study reported that the TGF-β1-treated
group exhibited sensitivity to cisplatin treatment, and high
proliferation and migration ability compared to that in the
control. The differences between studies may be attributed to
different cell lines (mammary vs. lung cancer cell line), duration
of treatment (24 h vs. 3 days) and the origin of the cells (primary
vs. immortalized).
In the present study, the control A549 cells
demonstrated higher cisplatin-resistance properties compared with
the TGF-β1-treated group. It may be hypothesised that cancer
stem-like cell properties, including self-renewal, chemoresistance
and differentiation, should be considered independently when
establishing a hypothesis connecting cancer stem cells and EMT.
Wellner et al (12) proposed
that zinc finger E-box binding homeobox 1 links EMT-activation and
stemness, which was maintained via suppression of
stemness-inhibiting microRNAs. However, this previous study focused
only on tumorigenicity, by evaluating sphere culture, a feature
known for its self-renewal ability (12). There may be a possibility that EMT
could be partially connected with cancer stem cell properties, a
suggestion supported by Xiao and He (25), who stated that neither reduced
E-cadherin, nor induced N-cadherin, are associated with poor
progression. In the present study, the MET/return group
demonstrated little change regarding cell cycle,
cisplatin-resistance, and proliferation and migration, compared
with the TGF-β1-treated group. It may be suggested that EMT induced
by TGF-β1 could be merely a trigger for cancer stemness properties,
with no further sustained effects following initiation.
TGF-β1 was reported to be involved in various
cellular physiological changes, including proliferation,
differentiation, apoptosis and EMT (26). TGF-β1 directly activated SMAD2 and
SMAD3 via the TGF-β1 receptor (27).
In the current study, SMAD3 and SMAD3 phosphorylation were
activated in the TGF-β1-treated group; however, SMAD3 signalling
disappeared within 24 h following TGF-β1 removal. This meant that
TGF-β1 was acting as an EMT inducer in the TGF-β1-treated group,
but not in the EMT/return group. It was reported that STAT3 was
activated by TGF-β1 (28); however,
this was not the case in the A549 cell line in the present study.
By contrast, ERK was considered to be involved in TGF-β1-associated
signalling and it was reported that ERK was activated by TGF-β1 in
normal murine mammary gland epithelial cells (29). In the present study, phosphorylation
of ERK was slightly increased in the TGF-β1-treated group; however,
there was no response following TGF-β1 removal. NF-κB is a
multi-transcription factor, which increases TGF-β1 transcription in
rat mesangial cells (30). In the
present study, level of NF-κB was measured to investigate the
possibility of autocrine signalling of TGF-β1. Notably, there was
no alteration in the level of NF-κB and phosphorylated NF-κB in
A549 cells. It is difficult to rationalise the inconsistency
between studies; however, the types of cell line, media and extra-
and intracellular conditions are among the variables that may
contribute to these differences. Concerning the apoptosis
mechanism, it was reported that treatment with TGF-β1 may increase
survivin, which promotes cell cycle progression and inhibits
apoptosis during EMT (31). The
results of the present study demonstrated that the active form of
caspase-3 was decreased and G2/M phase cells were increased in the
TGF-β1-treated and MET/return groups. It may be assumed that TGF-β1
is able to act as an initiator for an anti-apoptotic mechanism;
however, additional research is strongly encouraged to support this
assumption.
In conclusion, the present study revealed that
mesenchymal cells of the A549 lung cancer cell line, induced by
TGF-β1, return to epithelial cells in the absence of TGF-β1;
however, the levels of CD24, caspase-3, cell proliferation and
migration, and cisplatin sensitivity were unchanged during MET. It
may be suggested that TGF-β1 acts as an initiator, but not a
retainer, of properties associated with cancer stemness; however,
additional research is required to confirm this hypothesis.
Acknowledgements
The present study was supported by Konkuk University
(grant no. 2015-A019-0076) in 2015.
Glossary
Abbreviations
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
TGF-β1
|
transforming growth factor β1
|
|
MET
|
mesenchymal-epithelial transition
|
|
NSCLC
|
non-small cell lung cancer
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
NF-κB
|
nuclear factor-κB
|
References
|
1
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kerosuo L and Bronner-Fraser M: What is
bad in cancer is good in the embryo: Importance of EMT in neural
crest development. Semin Cell Dev Biol. 23:320–332. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: An emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zavadil J and Bättinger EP: TGF-beta and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kang Y and Massague J:
Epithelial-mesenchymal transitions: Twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hugo H, Ackland ML, Blick T, Lawrence MG,
Clements JA, Williams ED and Thompson EW: Epithelial-mesenchymal
and mesenchymal-epithelial transitions in carcinoma progression. J
Cell Physiol. 213:374–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lipschutz JH: Molecular development of the
kidney: A review of the results of gene disruption studies. Am J
Kidney Dis. 31:383–397. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sullivan JP, Minna JD and Shay JW:
Evidence for self-renewing lung cancer stem cells and their
implications in tumor initiation, progression, and targeted
therapy. Cancer Metastasis Rev. 29:61–72. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wellner U, Schubert J, Burk UC,
Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D,
zur Hausen A, et al: The EMT-activator ZEB1 promotes tumorigenicity
by repressing stemness-inhibiting microRNAs. Nat Cell Biol.
11:1487–1495. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang W, Peng J, Zhang Y, Cho WC and Jin
K: The implications of cancer stem cells for cancer therapy. Int J
Mol Sci. 13:16636–16657. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lam WK and Watkins DN: Lung cancer: Future
directions. Respirology. 12:471–477. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Kim CH and Lee JC: Epithelial to mesenchymal transition
derived from repeated exposure to gefitinib determines the
sensitivity to EGFR inhibitors in A549, a non-small cell lung
cancer cell line. Lung Cancer. 63:219–226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Seo DC, Sung JM, Cho HJ, Yi H, Seo KH,
Choi IS, Kim DK, Kim JS, Abd El-Aty AM and Shin HC: Gene expression
profiling of cancer stem cell in human lung adenocarcinoma A549
cells. Mol Cancer. 6:752007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Borthwick LA, Gardner A, De Soyza A, Mann
DA and Fisher AJ: Transforming growth factor-β1 (TGF-β1) driven
epithelial to mesenchymal transition (EMT) is accentuated by tumour
necrosis factor α (TNFα) via crosstalk between the SMAD and NF-κB
pathways. Cancer Microenviron. 5:45–57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fischer KR, Durrans A, Lee S, Sheng J, Li
F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al:
Epithelial-to-mesenchymal transition is not required for lung
metastasis but contributes to chemoresistance. Nature. 527:472–476.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim NH, Kim SK, Kim DS, Zhang D, Park JA,
Yi H, Kim JS and Shin HC: Anti-proliferative action of
IL-6R-targeted antibody tocilizumab for non-small cell lung cancer
cells. Oncol Lett. 9:2283–2288. 2015.PubMed/NCBI
|
|
20
|
Chen L, Zhang JJ and Huang XY: cAMP
inhibits cell migration by interfering with Rac-induced
lamellipodium formation. J Biol Chem. 283:13799–13805. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pore MM, Buikema L, Hiltermann T and Kruyt
F: TGF beta-mediated epithelial to mesenchymal transition in non
small cell lung cancer: Effects on stemness, invasiveness and
chemotherapy sensitivity. Cancer Res. 72:24022012. View Article : Google Scholar
|
|
22
|
Zheng Y, de la Cruz CC, Sayles LC,
Alleyne-Chin C, Vaka D, Knaak TD, Bigos M, Xu Y, Hoang CD, Shrager
JB, et al: A rare population of CD24(+)ITGB4(+)Notch(hi) cells
drives tumor propagation in NSCLC and requires Notch3 for
self-renewal. Cancer Cell. 24:59–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Roudi R, Madjd Z, Ebrahimi M, Samani FS
and Samadikuchaksaraei A: CD44 and CD24 cannot act as cancer stem
cell markers in human lung adenocarcinoma cell line A549. Cell Mol
Biol Lett. 19:23–36. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Scheel C and Weinberg RA: Cancer stem
cells and epithelial-mesenchymal transition: Concepts and molecular
links. Semin Cancer Biol. 22:396–403. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiao D and He J: Epithelial mesenchymal
transition and lung cancer. J Thorac Dis. 2:154–159.
2010.PubMed/NCBI
|
|
26
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fuxe J, Vincent T and de Herreros A
Garcia: Transcriptional crosstalk between TGF-β and stem cell
pathways in tumor cell invasion: Role of EMT promoting Smad
complexes. Cell Cycle. 9:2363–2374. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu
Z, Zhao J and Zhang HT: JAK/STAT3 signaling is required for
TGF-β-induced epithelial-mesenchymal transition in lung cancer
cells. Int J Oncol. 44:1643–1651. 2014.PubMed/NCBI
|
|
29
|
Xie L, Law BK, Chytil AM, Brown KA, Aakre
ME and Moses HL: Activation of the Erk pathway is required for
TGF-beta1-induced EMT in vitro. Neoplasia. 6:603–610. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lan Y, Zhou Q and Wu ZL: NF-kappa B
involved in transcription enhancement of TGF-beta 1 induced by
Ox-LDL in rat mesangial cells. Chin Med J (Engl). 117:225–230.
2004.PubMed/NCBI
|
|
31
|
Lee J, Choi JH and Joo CK: TGF-β1
regulates cell fate during epithelial-mesenchymal transition by
upregulating survivin. Cell Death Dis. 4:e7142013. View Article : Google Scholar : PubMed/NCBI
|