Introduction
Colon cancer is the third most prevalent type of
human cancer, which is cured primarily via surgical excision and
chemotherapy (1). Therapeutic methods
utilizing molecular targets to alter cancer cell properties possess
more powerful antitumor functions and fewer toxic effects towards
healthy cells (2). As a result, small
molecule target therapeutics for cancer has been the subject of
numerous investigations (2). The heat
shock protein (hsp) family, particularly hsp90, is associated with
the stability of a number of key oncogenic target proteins,
including receptor and non-receptor tyrosine kinases, and is
therefore a promising molecular target for cancer treatment
(3). Increased levels of hsp90
expression affects the early-stage of carcinogenesis and the
morphology of cancer cells, which is associated with the
therapeutic effects of hsp90 inhibitors (4). As a potent anti-cancer treatment, a
number of hsp90 inhibitors have been the subjects of several
studies (5,6). The overexpression of hsp90 has been
demonstrated in colon cancer and is associated with the
proliferation of colon cancer cells.
17-allylamino-17-demethoxygeldanamycin (17-AAG) is a
derivative of geldanamycin (an hsp90 inhibitor), which specifically
interacts with the N-terminal ATP binding domain of hsp90,
inhibiting the intrinsic ATPase activity that is important for its
chaperone function (7). Subsequently,
hsp90 substrate proteins are degraded via the ubiquitin-proteasome
pathway. The majority of these substrate proteins are signaling
molecules involved in numerous signaling pathways that influence
tumor development (8). The present
study aimed to elucidate the effects of 17-AAG on the cell cycle,
proliferation and apoptosis of HCT-116 cells, and investigate the
molecular mechanisms underlying the effect of 17-AAG treatment on
colon cancer cells.
Materials and methods
Cell culture
The HCT-116 human colon carcinoma cell line was
provided by Professor Zheng Huachuan of The First Affiliated
Hospital of Liaoning Medical University (Jinzhou, China). The cells
were cultured in RPMI-1640 supplemented with 10% fetal bovine serum
(FBS) and 100 U/ml penicillin (all from Hyclone; GE Healthcare Life
Sciences, Shanghai, China) and incubated at 37°C with 5%
CO2. Cells were monitored daily and the medium was
replaced every 2–3 days as required. Cells in the logarithmic
growth phase were selected and transferred to 96-well plates
(5×104 cells/ml). Upon cell adherence, five
concentrations of 17-AAG (1.25, 2.5, 1.5, 1.10 and 1.20 mg/l) were
added to the experimental groups and an untreated control group was
included. The control group was only cultured with RPMI-1640
supplemented with 10% FBS and 10 µl RPMI-1640 was added when 10 µl
17-AAG was added to the other groups. MTT solution was added to the
wells after 24 or 48 h, and the medium was discarded after 4 h.
Subsequently, 150 µl/well dimethyl sulfoxide was added and the
plate was agitated for 15 min. Optical density (OD) was evaluated
at a wavelength of 490 nm using a microplate reader (ELX808;
BioTek, China, Beijing, China). Finally, the growth inhibition
rates at various doses of 17-AAG were calculated using the
equation: Growth inhibition rate = [(OD control group) - (OD17 -
AAG group)]/(OD control group - OD blank group).
Cell cycle and apoptosis assays
HCT-116 cells (5×104 cells/ml) in the
logarithmic phase of growth were added to petri dishes to establish
a control group (RPMI-1640, 10% FBS) and experimental groups with
various concentrations (1.25, 2.5 and 5 mg/l) of 17-AAG. Following
48 h the cells were collected by centrifugation at 1,000 × g at 4°C
for 5 min. Subsequently, 2 ml PBS was added to each group to
resuspend the cells in order to wash them. Cells for the apoptosis
assay were then resuspended in 500 µl binding buffer (Hangzhou
Biosci Biotech Co., Ltd., Hangzhou, China) and stained with 5 µl
propidium iodide (PI) and 5 µl Annexin V (Hangzhou Biosci Biotech
Co., Ltd.) for 15 min at room temperature in dark. Subsequently,
the cells were analyzed by Flow cytometry (BD FACSVerse; BD
Biosciences, Franklin Lakes, NJ, USA). The cells for cell cycle
assay were collected after 48 h and incubated in 70% ethanol for 12
h at −20°C. Subsequently, the cells (106) were
resuspended in 500 µl binding buffer (Hangzhou Biosci Biotech Co.,
Ltd.) following centrifugation at 1,000 × g and 4°C for 5 min.
Ribonuclease A (125 mg/ml, 1 µl; Beyotime Institute of
Biotechnology, Jiangsu, China) was added to the buffer and the cell
suspension was incubated at 37°C for 30 min. Following incubation,
5 µl PI (Hangzhou Biosci Biotech Co., Ltd.) was added to each cell
sample and incubated for 30 min at 20°C in dark. Flow cytometry (BD
FACSVerse; BD Biosciences) was used to detect the cell cycle stage
of HCT-116 cells and apoptotic cells.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-PCR)
analysis
HCT-116 cells at the logarithmic phase of growth
were added to petri dishes to establish a control group (RPMI-1640,
10% FBS) and experimental groups with various concentrations of
17-AAG (1.25, 2.5 and 5 mg/l). All the cells were collected after
48 h and added to Eppendorf tubes, following which 1 ml TRIzol®
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) was added and
tubes were incubated on ice for 10 min. Subsequently, 200 µl
chloroform was added, and the cells were placed in a shaker for 15
sec at room temperature and centrifuged for 15 min at 12,000 × g
and 4°C. Following centrifugation, the upper layer of supernatant
(~200 µl) was collected and ~200 µl isopropyl alcohol was added to
each tube. The tubes were kept on ice for 10 min and centrifuged
for 15 min at 12,000 × g and 4°C. Subsequently, all the liquid was
discarded and cells were washed with 70% ethanol diluted with
diethylpyrocarbonate-treated water, and the RNA concentration of
each sample was evaluated using an ultraviolet
spectrophotometer.
RT-PCR was performed using the TaKaRa RNA PCR (avian
myeloblastosis virus) kit (Takara Biotechnology Co., Ltd., Dalian,
China) according to the manufacturer's protocol (9). Primer sequences for signal transducer
and activator of transcription (STAT)3, cyclin D1, cyt-c,
caspase 9 and caspase 3 are provided in Table I, and were used for DNA amplification
following RT.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene name | Sequence,
5′-3′ | Annealing
temperature, °C | Number of
cycles | Product size,
bp |
|---|
| STAT3 | F:
CCTCCTCAGCATCTTATCCG | 59 | 30 | 499 |
|
| R:
CAGCCTGGGCATCCTTG |
|
|
|
| Caspase 3 | F:
TGGACCTGTTGACCTGA | 57 | 30 | 269 |
|
| R:
CACAAAGCGACTGGATG |
|
|
|
| Caspase 9 | F:
TGGACCTGTTGACCTGA | 57 | 30 | 885 |
|
| R:
CACAAAGCGACTGGATG |
|
|
|
| Cyt-c | F:
GTCCGGTTGCGCTTTCCTT | 60 | 40 | 156 |
|
| R:
CGCAGTTTCCTCAAATTCTTTCTTC |
|
|
|
| Cyclin D1 | F:
TGTCCTACTACCGCCTCACACGCTTCCTCTCCG | 63 | 35 | 160 |
|
| R:
TCCTCTTCCTCCTCCTCGGCGGCCTTG |
|
|
|
| GAPDH | F:
CAATGACCCCTTCATTGACC | 60 | 30 | 135 |
|
| R:
TGGAAGATGGTGATGGGATT |
|
|
|
Western blot analysis
HCT-116 cells at the logarithmic phase of growth
were added to petri dishes, including a control group (RPMI-1640,
10% FBS) and experimental groups with various concentrations of
17-AAG (1.25 and 2.5 mg/l). The cells were collected after 48 h.
Lysis buffer (M-PER™ Mammalian Protein Extraction reagent; Thermo
Fisher Scientific, Inc.) was added, and the cells were incubated on
ice for 20 min. Subsequently, the cells were centrifuged for 20 min
at 1,000 × g and 4°C. The upper layer of supernatant was collected,
and the protein concentration was evaluated by using the Pierce™
BCA Protein Assay kit (Thermo Fisher Scientific, Inc). Total cell
lysates (30 µg) were separated by 12% SDS-PAGE and transferred to a
polyvinylidene fluoride membrane (Merck Millipore, Darmstadt,
Germany), and the blots were blocked with 5% skimmed milk. The
membranes were washed three times by using 1 ml phosphate-buffered
saline (PBS; Thermo Fisher Scientific, Inc.) for 5 min. The blots
were probed with antibodies against STAT3, cyclin D1, cyclin B1,
cyt-c, caspase 9 and caspase 3. Antibodies (1 ml) against
STAT3 against STAT3 (cat. no. ab119352), cyclin D1 (cat. no.
ab134175), cyclin B1 (cat. no. ab32053), cyt-c (cat. no.
ab13575), caspase 9 (cat. no. ab32539) and caspase 3 (cat. no.
ab32042) were added to the membranes at dilutions of 1:1,000. The
membranes were placed on a shaker for 1 h at 20°C. The membranes
were subsequently washed three times using 1 ml PBS for 5 min. The
secondary antibodies horseradish peroxidase (HRP)-conjugated
anti-mouse immunoglobulin G (IgG; cat. no. ab131368) and
HRP-conjugated anti-rabbit IgG variable domain of heavy chain
single domain (cat. no. ab191866) were added to the membranes. All
primary and secondary antibodies were purchased from Abcam
(Shanghai, China). The membranes were placed in a shaker with the
secondary antibody for 1 h at 20°C, and subsequently washed 3 times
with PBS. Pierce™ enhanced chemiluminescence western
blotting substrate (Thermo Fisher Scientific, Inc.) was added to
the membranes for 3 min, and the membranes were captured with the
ChemiDoc XRS system (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Immunofluorescence assay
HCT-116 cells at the logarithmic growth phase were
added to 6-well plates on a cover glass to form a control group
(RPMI-1640, 10% FBS) and experimental groups with various
concentrations of 17-AAG (1.25, 2.5 and 5 mg/l). The cells were
collected after 48 h and washed once with PBS. Subsequently, 4%
paraformaldehyde was added to the wells, and the cells were
incubated at room temperature for 15 min prior to 3 washes with
PBS. The cells were subsequently incubated with 1% Triton X-100 for
20 min at 20°C and washed with PBS three times. Bovine serum
albumin (1%; Beyotime Institute of Biotechnology) was added to the
wells, which were then incubated for 30 min at room temperature.
STAT3 primary antibody (1:200) was added to the wells and incubated
overnight at 4°C. The secondary antibody goat anti-mouse IgG (heavy
chain and light chain; 1:400; cat. no. ab96879; Abcam) was added to
the wells and incubated for 2 h at room temperature. The cells were
washed three times with PBS. Following washing, DAPI was added to
the wells and incubated for 5 min in the dark. The cells were
observed under a fluorescence microscope and images were
captured.
Statistical analysis
Statistical analysis was performed with SPSS
(version 19.0; IBM SPSS, Armonk, NY, USA). The data were presented
as the mean ± standard deviation. Data comparisons among groups
were performed using one-way analysis of variance, and Turdey post
hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
HCT-116 cell proliferation is
inhibited by 17-AAG treatment
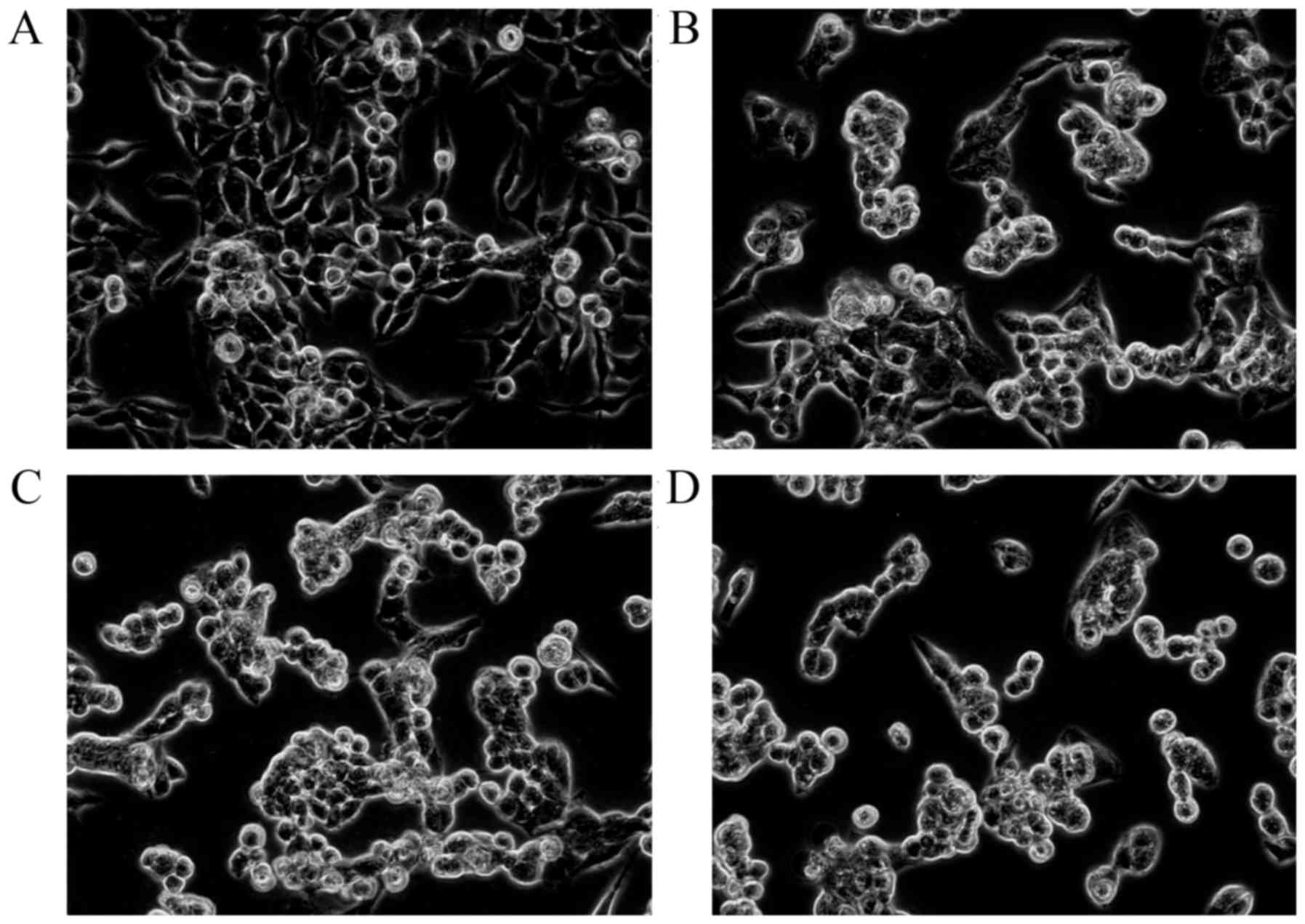
The MTT assay results revealed that 1.25–20 mg/l of
17-AAG exhibited significant inhibitory effects (P<0.01) on the
proliferation of HCT-116 cells in a concentration-dependent manner.
The cell numbers in the 17-AAG treated groups were significantly
reduced (P<0.01), compared with those observed in the control
group, with an abnormal cell morphology exhibited by the
17-AAG-treated cells (Fig. 1). The
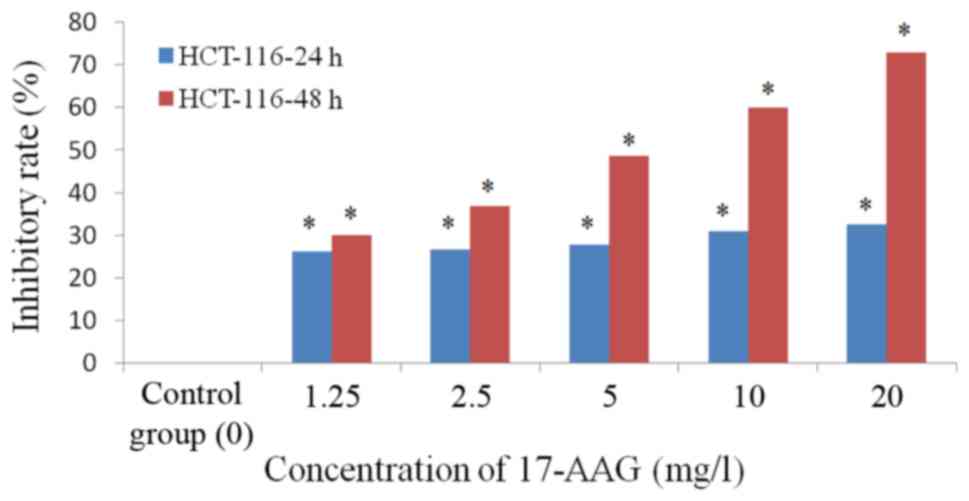
proliferation inhibition rate of 17-AAG-treated cells (1.25, 2.5,
5, 10 and 20 mg/l) at 48 h (IC50, 1.71 mg/l) was
increased, compared with that observed at 24 h (IC50,
23.24 mg/l; Table II; Fig. 2).
| Table II.Inhibitory effects of 17-AAG on the
proliferation of HCT-116 colon carcinoma cells (mean ± standard
deviation; n=6). |
Table II.
Inhibitory effects of 17-AAG on the
proliferation of HCT-116 colon carcinoma cells (mean ± standard
deviation; n=6).
|
| 24 h | 48 h |
|---|
|
|
|
|
|---|
| 17-AAG, mg/l | Absorbance |
| Inhibitory rate,
% | Absorbance |
| Inhibitory rate,
% |
|---|
| Control | 0.450±0.09 |
| 0 | 0.569±0.01 |
| 0 |
| 1.25 | 0.399±0.01 |
| 26.17a | 0.472±0.05 |
| 30.14a |
| 2.5 | 0.397±0.04 |
| 26.61a | 0.434±0.08 |
| 36.82a |
| 5 | 0.392±0.004 |
| 27.87a | 0.366±0.05 |
| 48.65a |
| 10 | 0.378±0.019 |
| 30.91a | 0.302±0.04 |
| 60.01a |
| 20 | 0.343±0.017 |
| 32.45a | 0.229±0.003 |
| 72.73a |
| IC50 |
| 60.08 |
|
| 5.24 |
|
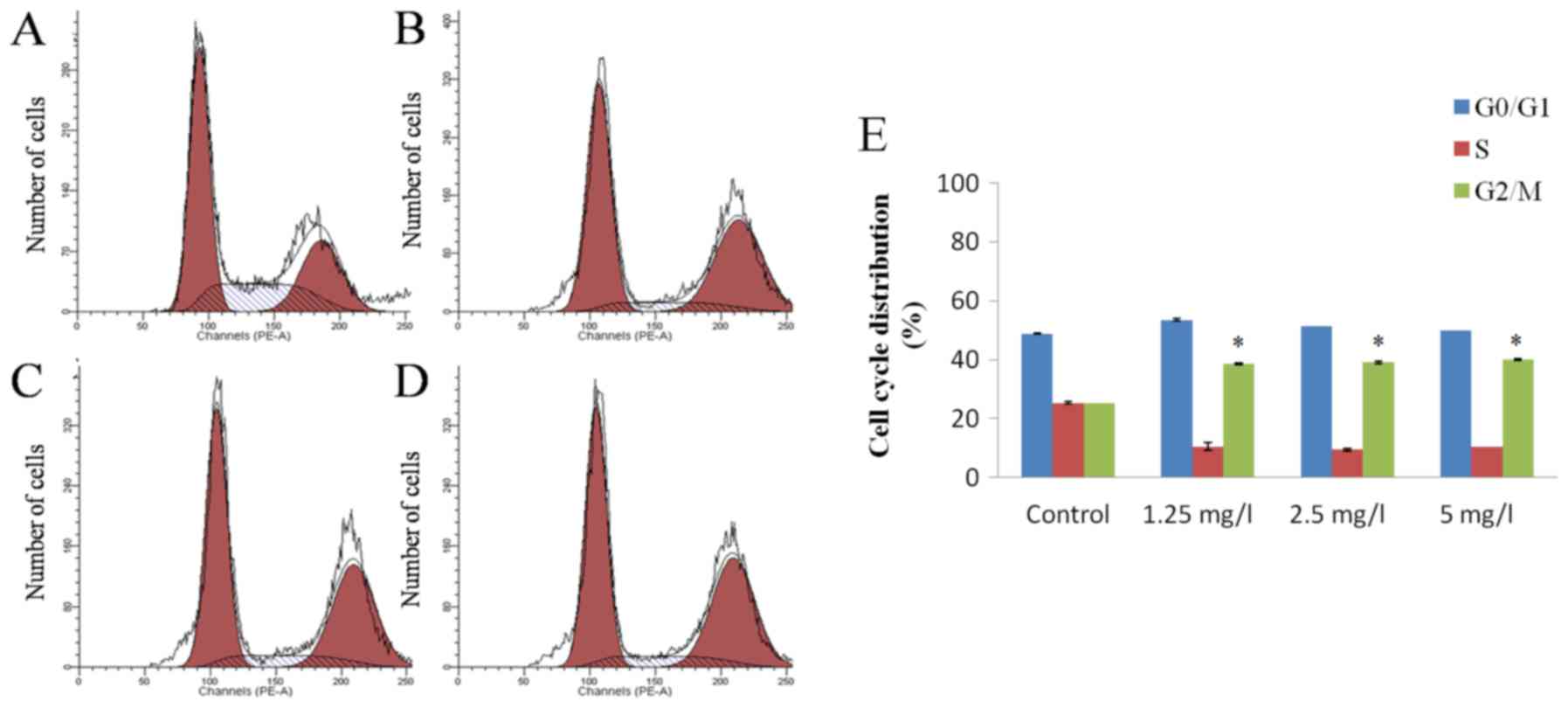
17-AAG induces G2 stage
cell cycle arrest in HCT-116 cells
PI staining detection results revealed that various
concentrations (1.25, 2.5 and 5 mg/l) of 17-AAG were able to cause
a significant arrest in cell cycle progression of HCT-116 cells at
the G2 stage after 48 h. However, this effect did not
appear to occur in a concentration-dependent manner (Fig. 3).
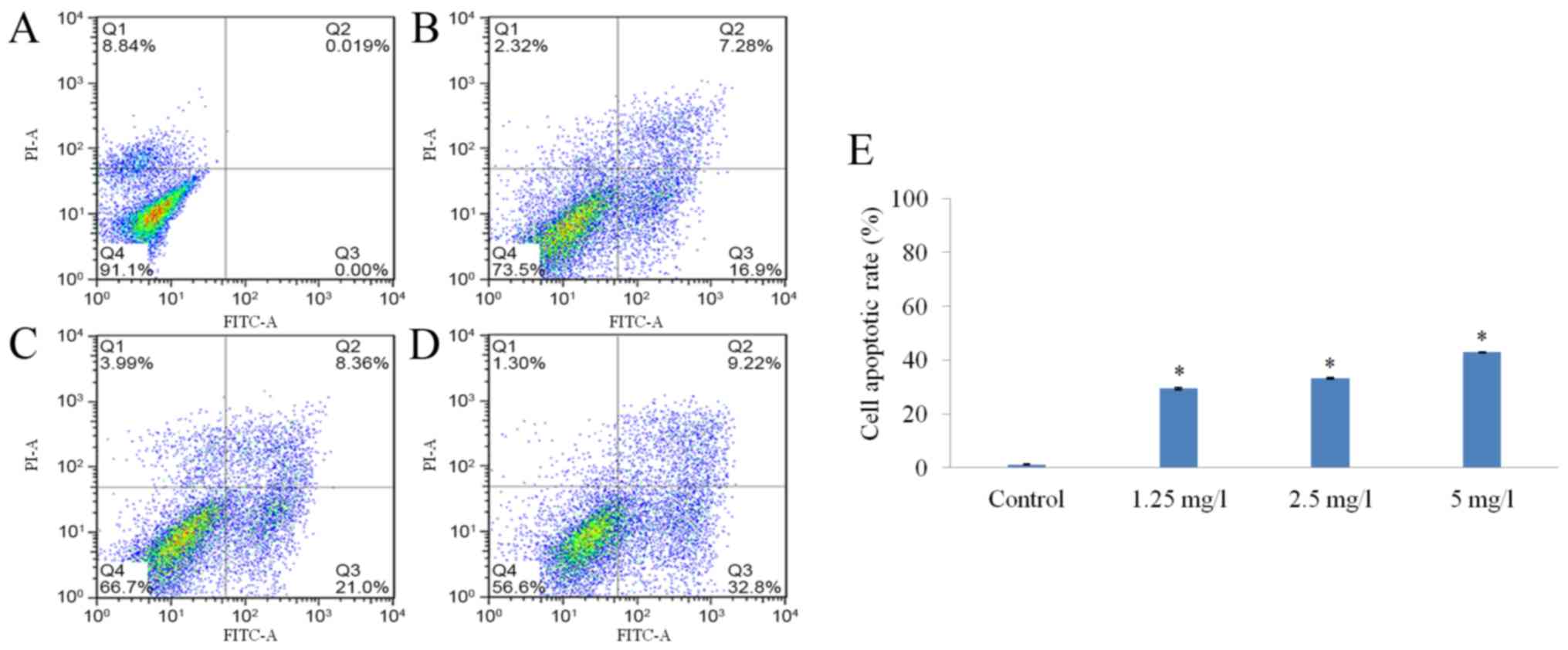
17-AAG promotes HCT-116
cell-apoptosis
The apoptotic rates of the cells in the
17-AAG-treated groups (1.25, 2.5 and 5 mg/l) were markedly
increased compared with cells in the control group. Additionally,
17-AAG appeared to increase the apoptosis rate of HCT-116 cells in
a dose-dependent manner (Fig. 4).
17-AAG-treatment affects the mRNA and
protein expression of STAT3, cyclin D1, cyclin B1 and caspase
3
RT-qPCR and western blot analysis results revealed
that the expression levels of STAT3, cyclin D1 and cyclin B1 in the
treated groups (1.25 and 2.5 mg/l) were significantly reduced
(P<0.01), compared with the control group. The expression levels
of cyt-c, caspase 9 and caspase 3 in the 17-AAG-treated
groups were significantly increased (P<0.01) compared with the
control, in a concentration-dependent manner (Fig. 5).
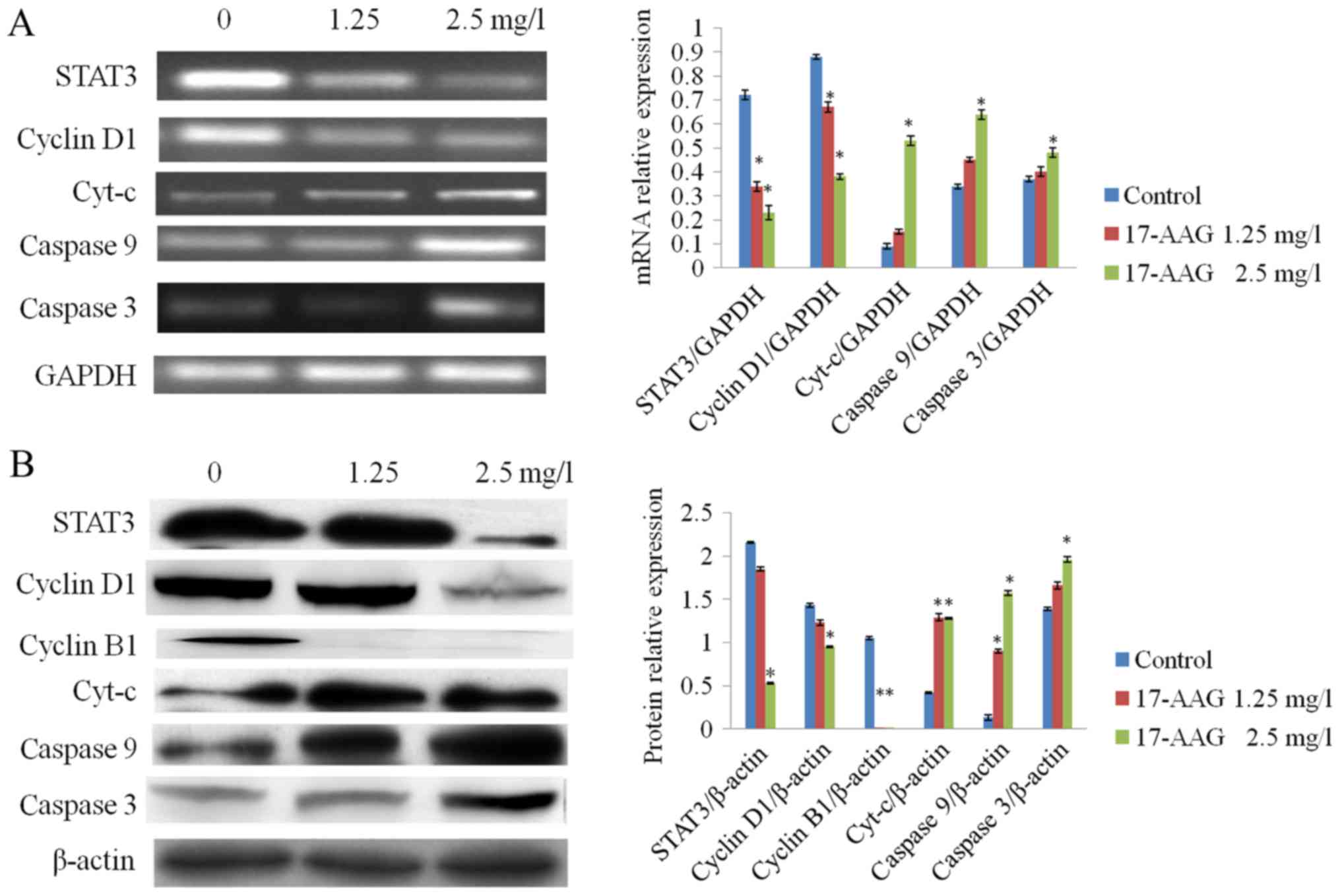 | Figure 5.Effect of 17-AAG on the expression
levels of various cellular factors. (A) Following treatment with
17-AAG, cyt-c, caspase 9 and caspase 3 mRNA expression
levels were increased in HCT-116 cells in a concentration-dependent
manner compared with the control. By contrast, STAT3 and cyclin D1
mRNA expression levels were decreased in a concentration-dependent
manner, compared with the control. (B) Following 17-AAG-treatment,
cyt-c, caspase 9 and caspase 3 protein expression was
significantly increased in a concentration-dependent manner,
compared with the control, whereas stat3, cyclin D1 and cyclin B1
expression was significantly decreased in a concentration-dependent
manner, compared with the control. *P<0.05 compared with the
control. 17-AAG, 17-allylamino-17-demethoxygeldanamycin; mRNA,
messenger RNA; cyt-c, cytochrome c; STAT3, signal
transducer and activator of transcription 3. |
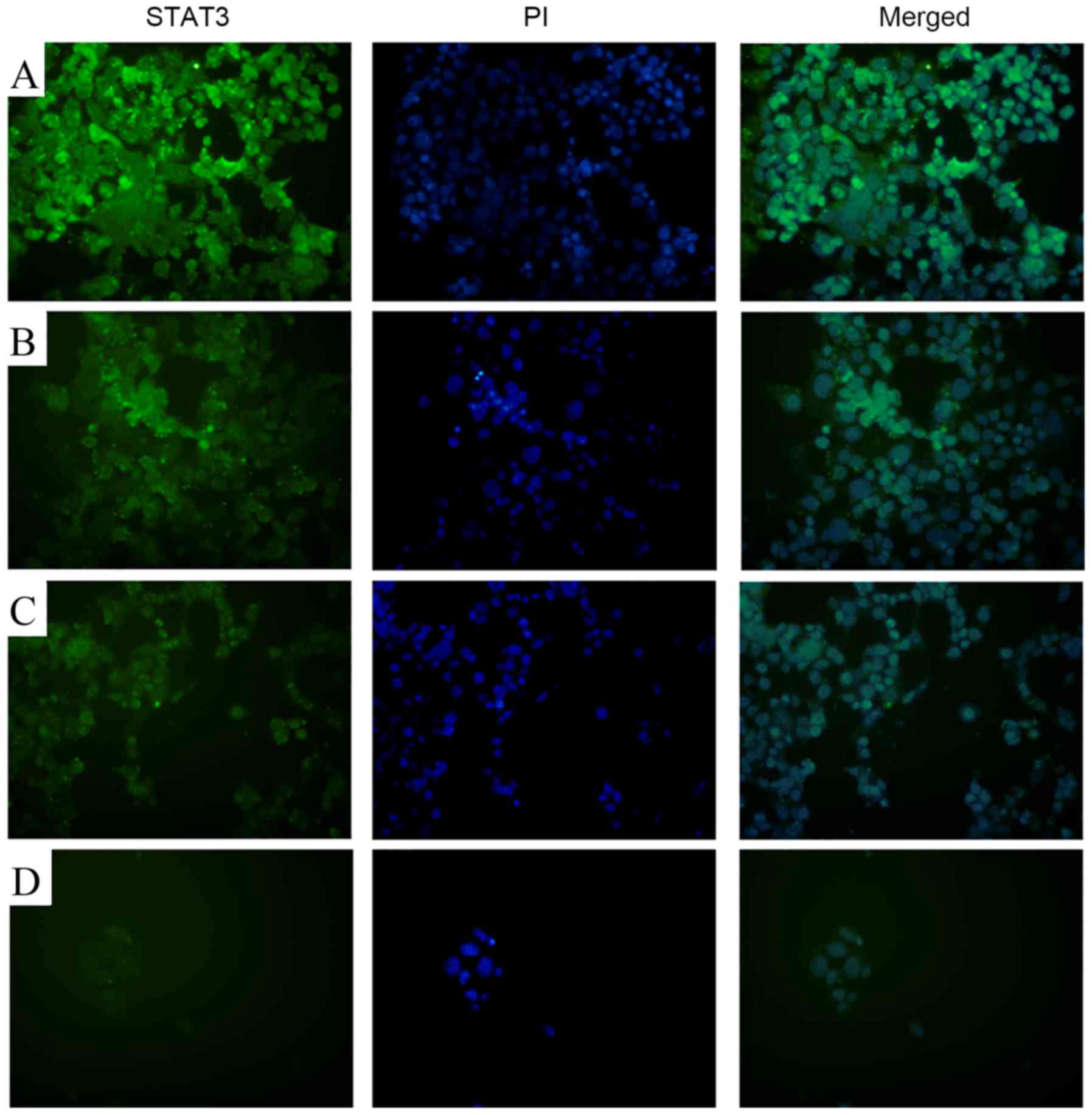
17-AAG inhibits STAT3 expression
Fluorescence microscopy revealed markedly reduced
STAT3 staining in the 17-AAG-treated groups (1.25, 2.5 and 5 mg/l),
compared with the control, in a concentration-dependent manner
(Fig. 6).
Discussion
The over-expression of hsp90 has been detected in
various forms of carcinoma, including breast, prostate, renal,
colon, ovarian and hepatocarcinoma, multiple myeloma and leukemia
(10–12). As hsp90 is a highly expressed tumor
marker (13), hsp90 inhibitors may be
an effective anticancer therapy. Geldanamycin, an inhibitor of the
hsp90 ATP-binding domain, was the first hsp90 inhibitor to be
identified (14). This inhibitor
functions by inducing the degradation of hsp90 substrates, which
enables the suppression of various signaling pathways, in order to
impede carcinoma angiogenesis, histogenesis and cell proliferation
as well as induce cell cycle arrest and apoptosis (15). As a number of studies have revealed
the high hepatotoxicity of geldanamycin, its derivative 17-AAG was
investigated in the present study. 17-AAG has increased water
solubility and lower hepatotoxicity (16,17).
Abnormalities in cell cycle control may lead to
aberrant proliferation and cancer development (17), suggesting that induction of cell cycle
arrest may be a promising therapeutic approach for cancer (18). Numerous studies have reiterated the
importance of the cell cycle during carcinogenesis, as well as
revealing that hsp90 inhibitors are able to induce arrest at
different cell cycle stages in various tumor types (19,20).
Previous studies have suggested that treatment with 17-AAG is able
to arrest SGC-7901 gastric cancer cells and HepG2 liver cancer cell
lines at the G2/M stage (21,22) and
bladder cancer cell lines (RT4, RT112 and T24) at the G1
stage (23). In the present study
17-AAG treatment induced G2-stage arrest in CT-116 cells
in a non-concentration-dependent manner.
In order to control the cell cycle, cyclin-dependent
kinases (CDK) are primarily regulated by cyclins and CDK inhibitors
(24). High levels of CDK expression
are able to trigger anti-apoptotic mechanisms, leading to
unrestricted cell proliferation (25). As one of numerous proto-oncogenes,
cyclin D1 forms a complex with and activates CDK4, leading to the
phosphorylation of retinoblastoma protein and the release of E2
factor (26). This process induces
the initiation of S phase-associated gene transcription and the
cell cycle transition from G1 to S phase (26). The present study observed mRNA-level
and protein-level changes in cyclin D1, detecting reduced cyclin D1
expression in HCT-116 cells. As cells were arrested at the
G2 phase, it was hypothesized that the effect of 17-AAG
on the cell cycle of HCT-116 cells is not dependent on altered
cyclin D1 expression levels. The cyclin B1 subunit binds CDK1, and
this maturation-promoting factor complex promotes progression from
the G2 to M phase of the cell cycle (27). Following activation, the cyclin
B1-CDK1 complex is able to initiate the prophase of mitosis,
suggesting that an increase in its activity may lead to alterations
in the mitotic behavior of the cell (28). Therefore, the expression levels and
activity of cyclin B1 and CDK1 are able to influence cell mitosis.
During the present study, HCT-116 cells were arrested at the
G2 phase and cyclin B1 expression levels were
significantly decreased. This result is contrary to a previous
study, suggesting that the HCT-116 cell cycle is not influenced by
17-AAG treatment (29). However, the
present study concluded that 17-AAG is able to arrest HCT-116 cells
at the G2 phase, possibly due to a reduction in the
levels of cyclin B1 expression.
Under stress, cells initiate programmed cell death,
termed apoptosis, which is important for various physical and
pathological processes and is an important regulator of cell growth
(30). The hallmarks of apoptosis are
changes in cell morphology, including chromatin condensation,
nuclear fragmentation and cell shrinkage, as well as biochemical
changes, including caspase activation, degradation of DNA and
proteins and the rupture of membrane modification factors, which
target cells for phagocytosis (31).
There are two major apoptosis signaling pathways: The intrinsic
mitochondrial apoptosis pathway and the extrinsic death
receptor-mediated apoptosis pathway (32). Intracellular toxicity stimuli,
including DNA damage and growth factor deficiency, trigger an
increase in mitochondrial membrane permeability and the release of
intermembrane proteins, particularly cyt-c, initiating
intrinsic apoptosis (33). Following
its release, and aided by ATP or deoxyadenosine triphosphate,
cyt-c is able to form apoptotic bodies with caspase
regulatory factors, and activate caspase 9, and downstream caspase
3 and caspase 7 proteins, to initiate the process of cell apoptosis
(34). Abnormal levels of apoptosis
disrupt the balance between viable and dead cells to promote tumor
development (35); therefore, the
regulation of alterations in apoptosis may be a novel anticancer
therapy. This present study identified the apoptosis-inducing
ability of 17-AAG, but the underlying mechanisms require further
investigation. In the present study, an increase in the mRNA and
protein expression levels of cyt-c, caspase 9 and caspase 3
in HCT-116 cells was detected to varied extents following 17-AAG
treatment. These results demonstrated that 17-AAG may induce the
apoptosis of HCT-116 cells via the mitochondrial apoptosis
signaling pathway in a concentration-dependent manner.
The present study revealed that the expression of
STAT3 in the 17-AAG-treated groups was significantly downregulated,
indicating that a reduction in STAT3 expression levels may underlie
the inhibitory effect of 17-AAG on HCT-116 cells. Members of the
STAT family of proteins function in various physical and
pathological processes, including cell growth, proliferation and
development (36). In particular,
STAT3 is important for transcription regulation, post-translational
modification and cell functions, including apoptosis and cell death
(37). STAT3 is able to translocate
into the cell nucleus and bind with specific promoter sequences to
regulate transcription. Therefore, STAT3 is able to exert a role in
cell proliferation, differentiation and metabolism, processes that
are important during the development of inflammation and tumor
formation and progression (38).
Previous studies have detected STAT3 overexpression in numerous
forms of carcinoma, including colorectal cancer, and the loss of
control of STAT3 expression in breast, head and neck, prostate,
pancreatic, ovarian and brain cancer (39,40). STAT3
is hypothesized to be an important tumor modulatory factor
(32), and is able to induce cell
proliferation, differentiation and antiapoptotic mechanisms
following activation by carcinogenic substances, including B7H3
(41). Leptin is able to elevate
hsp90 expression levels via STAT3 regulation, to promote the
elevation of human epidermal growth factor receptor 2 expression
levels; however, 17-AAG is able to abrogate this effect and inhibit
breast cancer cell proliferation (42). This result implicates STAT3 in the
anticancer functions of 17-AAG, particularly the inhibition of cell
proliferation.
Cyclin D1 is an established downstream target of
STAT3, and cyclin D1 expression is decreased upon STAT3 suppression
(43). The present study revealed
that STAT3 expression in HCT-116 cells was decreased in the
17-AAG-treated groups using RT-qPCR, western blotting and
immunofluorescence data. In addition, the current study detected
significantly reduced cyclin D1 mRNA and protein expression levels
in 17-AAG-treated cells. Therefore, it was hypothesized that 17-AAG
may downregulate the expression of cyclin D1 and cyclin B1 via
STAT3, in order to induce G2 phase arrest in HCT-116
cells.
Previous studies have suggested that, upon STAT3
inhibition, the expression levels of caspase3 may be increased
(44,45). In addition, Duan et al
(46) revealed that thymic stromal
lymphopoietin may downregulate caspase 3 expression through the
activation of the STAT3 signaling pathway, thereby suppressing the
apoptosis of decidual γδ T cells (17), indicating that caspase 3 may be a
target of STAT3. Previous studies reported that some novel
medicines, including geraniin are able to induce the intrinsic
apoptosis signaling pathway via the STAT3 pathway (47–49),
catalyzing the caspase cascade response. Therefore, it has been
suggested that STAT3 may translocate into mitochondria and regulate
apoptosis (50).
In conclusion, the present study revealed the
downregulation of STAT3 expression following 17-AAG-treatment in
HCT-116 cells, hypothesizing that 17-AAG may regulate mitochondrial
apoptosis via the STAT3 signaling pathway, leading to apoptosis
induction. However, further studies are required to elucidate the
role of STAT3 in the 17-AAG-induced apoptosis of HCT-116 cells.
Acknowledgements
The present study was supported by the Hebei
Province Education Department (grant no. ZD20140003) and University
Emphasis Subject of Hebei Province and Immunology Emphasis Subject
of Chengde Medical University
References
|
1
|
Haddock MG: Intraoperative radiation
therapy for colon and rectal cancers: A clinical review. Radiat
Oncol. 12:112017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seeber A and Gastl G: Targeted therapy of
colorectal cancer. Oncol Res Treat. 39:796–802. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen G, Yang Z, Eshleman JR, Netto GJ and
Lin MT: Molecular diagnostics for precision medicine in colorectal
cancer: Current status and future perspective. Biomed Res Int.
2016:98506902016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang C, Zhang Y, Guo K, Wang N, Jin H, Liu
Y and Qin W: Heat shock proteins in hepatocellular carcinoma:
Molecular mechanism and therapeutic potential. Int J Cancer.
138:1824–1834. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dimopoulos MA, Mitsiades CS, Anderson KC
and Richardson PG: Tanespimycin as antitumor therapy. Clin Lymphoma
Myeloma Leuk. 11:17–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Solárová Z, Mojžiš J and Solár P: Hsp90
inhibitor as a sensitizer of cancer cells to different therapies
(Review). Int J Oncol. 46:907–926. 2015.PubMed/NCBI
|
|
7
|
Hong DS, Banerji U, Tavana B, George GC,
Aaron J and Kurzrock R: Targeting the molecular chaperone heat
shock protein 90 (HSP90): Lessons learned and future directions.
Cancer Treat Rev. 39:375–387. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Soroka J, Wandinger SK, Mäusbacher N,
Schreiber T, Richter K, Daub H and Buchner J: Conformational
switching of the molecular chaperone Hsp90 via regulated
phosphorylation. Mol Cell. 45:517–528. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tian YZ, Usman T, Tian KC, Di J, Huang XX,
Xu XM, Tulafu H, Wu WW, Fu XF, Bai Y, et al: Comparative study of
13 candidate genes applying multi-reference normalization to detect
the expression of different fineness in skin tissues of wool sheep.
Genet Mol Res. 16:2017. View Article : Google Scholar
|
|
10
|
Kathiria AS, Neumann WL, Rhees J,
Hotchkiss E, Cheng Y, Genta RM, Meltzer SJ, Souza RF and Theiss AL:
Prohibitin attenuates colitis-associated tumorigenesis in mice by
modulating p53 and STAT3 apoptotic responses. Cancer Res.
72:5778–5789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pimienta G, Herbert KM and Regan L: A
compound that inhibits the HOP-Hsp90 complex formation and has
unique killing effects in breast cancer cell lines. Mol Pharm.
8:2252–2261. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sharp SY, Roe SM, Kazlauskas E, Cikotienė
I, Workman P, Matulis D and Prodromou C: Cocrystalization and in
vitro biological characterization of
5-aryl-4-(5-substituted-2-4-dihydroxyphenyl)-1,2,3-thiadiazole
hsp90 inhibitors. PLoS One. 7:e446422012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giubellino A, Sourbier C, Lee MJ,
Scroggins B, Bullova P, Landau M, Ying W, Neckers L, Trepel JB and
Pacak K: Targeting heat shock protein 90 for the treatment of
malignant pheochromocytoma. PLoS One. 8:e560832013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barrott JJ and Haystead TA: Hsp90, an
unlikely ally in the war on cancer. FEBS J. 280:1381–1396. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ye XY, Luo QQ, Xu YH, Tang NW, Niu XM, Li
ZM, Shen SP, Lu S and Chen ZW: 17-AAG suppresses growth and
invasion of lung adenocarcinoma cells via regulation of the
LATS1/YAP pathway. J Cell Mol Med. 19:651–663. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo W, Siegel D and Ross D: Stability of
the Hsp90 inhibitor 17AAG hydroquinone and prevention of
metal-catalyzed oxidation. J Pharm Sci. 97:5147–5157. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mita AC, Mita MM, Nawrocki ST and Giles
FJ: Survivin: Key regulator of mitosis and apoptosis and novel
target for cancer therapeutics. Clin Cancer Res. 14:5000–5005.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ambrosini G, Adida C and Altieri DC: A
novel anti-apoptosis gene, survivin, expressed in cancer and
lymphoma. Nat Med. 3:917–921. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yao JQ, Liu QH, Chen X, Yang Q, Xu ZY, Hu
F, Wang L and Li JM: Hsp90 inhibitor
17-allylamino-17-demethoxygeldanamycin inhibits the proliferation
of ARPE-19 cells. J Biomed Sci. 17:302010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Napper JM and Sollars VE:
17-N-Allylamino-17-demeth-oxygeldanamycin induces a diverse
response in human acute myelogenous cells. Leuk Res. 34:1493–1500.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen M, Xu J and Zhao J: Effects of HSP90
inhibitor 17-AAG on cell cycle and apoptosis of human gastric
cancer cell lines SGC-7901. Nan Fang Yi Ke Da Xue Xue Bao.
33:271–275. 2013.(In Chinese). PubMed/NCBI
|
|
22
|
Watanabe G, Behrns KE, Kim JS and Kim RD:
Heat shock protein 90 inhibition abrogates heaptocellular cancer
growth through cdc2-mediated G2/M cell cycle arrest and apoptosis.
Cancer Chemother PHarmacol. 64:433–443. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Karkoulis PK, Stravopodis DJ, Margaritis
LH and Voutsinas GE: 17-Allylamino-17-demethoxygeldanamycin induces
downregulation of critical Hsp90 protein clients and results in
cell cycle arrest and apoptosis of human urinary bladder cancer
cells. BMC Cancer. 10:4812010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mikhail S, Albanese C and Pishvaian MJ:
Cyclin-dependent kinase inhibitors and the treatment of
gastrointestinal cancers. Am J Pathol. 185:1185–1197. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Swaffer MP, Jones AW, Flynn HR, Snijders
AP and Nurse P: CDK substrate phosphorylation and ordering the cell
cycle. Cell. 167:1750–1761.e16. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bilal I, Chowdhury A, Davidson J and
Whitehead S: Phytoestrogens and prevention of breast cancer: The
contentious debate. World J Clin Oncol. 5:705–712. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Poli A, Ramazzotti G, Matteucci A, Manzoli
L, Lonetti A, Suh PG, McCubrey JA and Cocco L: A novel
DAG-dependent mechanism links PKCa and Cyclin B1 regulating cell
cycle progression. Oncotarget. 5:11526–11540. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gong D and Ferrell JE Jr: The roles of
Cyclin A2, B1, and B2 in early and late mitotic events. Mol Biol
Cell. 21:3149–3161. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Powers MV, Valenti M, Miranda S, Maloney
A, Eccles SA, Thomas G, Clarke PA and Workman P: Mode of cell death
induced by the HSP90 inhibitor 17-AAG (tanespimycin) is dependent
on the expression of pro-apoptotic BAX. Oncotarget. 4:1963–1975.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matos AJ and Santos AA: Advances in the
understanding of the clinically relevant genetic pathways and
molecular aspects of canine mammary tumours: Part 1. Proliferation,
apoptosis and DNA repair. Vet J. 205:136–143. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guicciardi ME, Malhi H, Mott JL and Gores
GJ: Apoptosis and necrosis in the liver. Compr Physiol. 3:977–1010.
2013.PubMed/NCBI
|
|
32
|
McCracken JM and Allen LA: Regulation of
human neutrophil apoptosis and lifespan in health and disease. J
Cell Death. 7:15–23. 2014.PubMed/NCBI
|
|
33
|
Apostolova N and Victor VM: Molecular
Strategies for targeting antioxidants to mitochondria: Therapeutic
implications. Antioxid Redox Signal. 22:686–729. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hilgendorf KI, Leshchiner ES, Nedelcu S,
Maynard MA, Calo E, Ianari A, Walensky LD and Lees JA: The
retinoblastoma protein induces apoptosis directly at the
mitochondria. Genes Dev. 27:1003–1015. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hassan M, Watari H, AbuAlmaaty A, Ohba Y
and Sakuragi N: Apoptosis and molecular targeting therapy in
cancer. Biomed Res Int. 2014:1508452014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hong S, Mehta KP and Laimins LA:
Suppression of STAT-1 expression by human papillomaviruses is
necessary for differentiation-dependent genome amplification and
plasmid Maintenance. J Virol. 85:9486–1994. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kamran MZ, Patil P and Gude RP: Role of
STAT3 in cancer metastasis and translational advances. Biomed Res
Int. 2013:4218212013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yuan J, Zhang F and Niu R: Multiple
regulation pathways and pivotal biological functions of STAT3 in
cancer. Sci Rep. 5:176632015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Giordano C, Vizza D, Panza S, Barone I,
Bonofiglio D, Lanzino M, Sisci D, De Amicis F, Fuqua SA, Catalano S
and Andò S: Leptin increases HER2 protein levels through a
STAT3-mediated up-regulation of Hsp90 in breast cancer cells. Mol
Oncol. 7:379–391. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang W, Dong Z, Wang F, Peng H, Liu JY
and Zhang JT: A small molecule compound targeting STAT3 DNA-binding
domain inhibits cancer cell proliferation, migration, and invasion.
ACS Chem Biol. 9:1188–1196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Y, Guo G, Song J, Cai Z, Yang J, Chen
Z, Wang Y, Huang Y and Gao Q: B7-H3 promotes the migration and
invasion of human bladder cancer cells via the PI3K/Akt/STAT3
signaling Pathway. J Cancer. 8:816–824. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chaudhari N, Talwar P, Parimisetty A,
d'Hellencourt C Lefebvre and Ravanan P: A molecular web:
Endoplasmic reticulum stress, inflammation, and oxidative stress.
Front Cell Neurosci. 8:2132014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qin A, Yu Q, Gao Y, Tan J, Huang H, Qiao Z
and Qian W: Inhibition of STAT3/cyclinD1 pathway promotes
chemotherapeutic sensitivity of colorectal caner. Biochem BiopHys
Res Commun. 457:681–687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kamran MZ, Patil P and Gude RP: Role of
STAT3 in cancer metastasis and translational advances. Biomed Res
Int. 2013:4218212013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cimica V, Chen HC, Iyer JK and Reich NC:
Dynamics of the STAT3 transcription factor: Nuclear import
dependent on ran and importin-β1. PLoS One. 6:e201882011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Duan J, Jiang XP, Li MQ, Fan DX, Wang Y,
Li DJ and Jin LP: Thymic stromal lympHopoietin suppresses the
apoptosis of decidual gamma-delta T cells via regulation of the
signal transduction and activation of transcription 3/caspase-3
signaling pathway. Am J Reprod Immunol. 70:464–471. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhu Y, Doornebal EJ, Pirtskhalava T,
Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, Niedernhofer LJ,
Robbins PD, Tchkonia T and Kirkland JL: New agents that target
senescent cells: The flavone, fisetin, and the BCL-XL inhibitors,
A1331852 and A1155463. Aging (Albany NY). 9:955–963.
2017.PubMed/NCBI
|
|
48
|
Do DV, Ueda J, Messerschmidt DM,
Lorthongpanich C, Zhou Y, Feng B, Guo G, Lin PJ, Hossain MZ, Zhang
W, et al: A genetic and developmental pathway from STAT3 to the
OCT4-NANOG circuit is essential for maintenance of ICM lineages in
vivo. Genes Dev. 27:1378–1390. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ren Z, Zou W, Cui J, Liu L, Qing Y and Li
Y: Geraniin suppresses tumor cell growth and triggers apoptosis in
human glioma via inhibition of STAT3 signaling. Cytotechnology.
2017.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Carbognin E, Betto RM, Soriano ME, Smith
AG and Martello G: Stat3 promotes mitochondrial transcription and
oxidative respiration during maintenance and induction of naive
pluripotency. EMBO J. 35:618–634. 2016. View Article : Google Scholar : PubMed/NCBI
|