Introduction
Prostate cancer, the most common non-cutaneous
malignancy, is the second leading cause of cancer-associated
mortality for males in the US, behind lung cancer (1). As localized prostate cancer can be cured
by surgery or radiotherapy, the major public health burden comes
from the metastatic stage of prostate cancer, for which there are
currently limited curative options. A typical first-line treatment
for metastatic prostate cancer is androgen deprivation therapy.
However, the initial response to hormone therapy is not maintained
for a long time; hormone therapy resistance, which leads to a
disease state termed castrate-resistant prostate cancer (CRPC),
emerges within a median time of 1.5 years (2). A previous study of patients with CRPC
revealed that ~67% of patients responded to long-term androgen
hormone blockade; the median survival time of non-responder
patients was <1 year (2).
Docetaxel was approved by the US Food and Drug
Administration in 2004 and has been evaluated in randomized Phase
III trials for patients with metastatic CRPC to overcome the
limitations of hormone therapy (3).
Docetaxel chemotherapy prolonged median overall survival by ~3
months when compared with mitoxantrone and prednisone, and
exhibited palliative benefits for a number of patients with
metastatic CRPC (3). Docetaxel exerts
its effect by targeting microtubules, which consist of filamentous
polymers of α- and β-tubulin heterodimers and are critical for cell
division (4). Docetaxel binds to
β-tubulin and stabilizes microtubule structures, which inhibits the
mitotic spindle apparatus. Thus, docetaxel-susceptible cells
exhibit mitotic arrest, leading to apoptosis (5).
Although docetaxel was the first cytotoxic therapy
demonstrated to exhibit a survival benefit in patients with CRPC,
the median time to prostate-specific antigen progression is limited
to 6–8 months, and additional chemotherapy options at progression
are required (6). Furthermore, ~50%
of patients with CRPC do not respond to docetaxel therapy,
presenting a significant clinical problem (7). Furthermore, initial responders to
docetaxel treatment ultimately develop docetaxel resistance.
Proposed mechanisms for this resistance include the inhibition of
drug accumulation into cancer cells, circumventing the cytotoxic
effect via the upregulation of alternative growth pathways and the
development of apoptosis resistance (5). However, the exact mechanism for
docetaxel resistance has yet to be elucidated. De novo and
acquired resistance to docetaxel chemotherapy are likely to be the
main limitations to its efficacy.
A treatment regimen to overcome docetaxel resistance
may be a viable alternative therapeutic strategy as there are
currently a limited number of treatment options available to
patients with CRPC. In our previous study, a cisplatin-resistant
bladder cancer cell line was established to identify the genes
associated with cisplatin resistance in bladder cancer (8). A docetaxel-resistant prostate cell line
(PC3DR2) was subsequently established using the same method
(9); resistance-associated genes in
this cell line were examined in the present study through DNA
microarray, western blot analysis and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR).
In the present study, PC3DR2 cells were identified
to exhibit a 3-fold increase in the expression of insulin-like
growth factor 1 receptor (IGF1R), DBF4 homolog (DBF4), sterile α
motif and leucine zipper-containing kinase AZK (ZAK), patched 1
(PTCH1), serpin peptidase inhibitor, clade E, member 1 (SERPINE1)
and breast cancer 2 (BRCA2) from cancer-associated pathways
compared with a docetaxel-sensitive cell line (PC3). BRCA2 small
interfering (si)RNA knockdown restored docetaxel sensitivity in
PC3DR2 cells, suggesting that BRCA2 may be associated with
docetaxel resistance in human prostate cancer cells.
Materials and methods
Cell lines and chemicals
PC3 CRPC cells were obtained from the American Type
Culture Collection (Manassas, VA, USA) and cultured in Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum
(MediaTech, Inc.; Corning Incorporated, Corning, NY, USA), 100 U/ml
penicillin and 100 mg/l streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) with 5% CO2 at 37°C. A
docetaxel-resistant CRPC cell line (designated PC3DR2) was
generated by serial desensitization of PC3 cells as previously
described (8,9). Docetaxel was obtained from
Sanofi-Aventis Korea Co., Ltd. (Seoul, South Korea).
Cytotoxicity assay
The cytotoxic effect of docetaxel was determined
using a Cell Counting Kit-8 (CCK-8) assay (Dojindo Molecular
Technologies, Inc., Rockville, MD, USA). Approximately 4,000 cells
were seeded in 96-well plates with 100 µl DMEM, and the cells were
treated with increasing doses of docetaxel (0, 0.005, 0.01, 0.1,
0.5, 1, 2.5, 5, 10, 20 or 50 µM diluted with DMEM) for 72 h under
the aforementioned conditions. Following incubation, 10 µl CCK-8
solution was added and the absorbance at 450 nm was determined 3 h
after further incubation under the same conditions.
Apoptosis and survival-associated
protein expression
Total protein was extracted from PC3DR2 cells with
or without 2 µg/ml docetaxel treatment using
radioimmunoprecipitation assay lysis buffer [containing 50 mM
tris-HCl (pH 8.0), 150 mM sodium chloride, 1.0% NP-40, 0.5% sodium
doxycholate, 0.1% sodium dodecyl sulfate and 1 mM
phenylmethylsulfonyl fluoride]. Protein concentrations were
determined using the Pierce BCA Protein Assay kit (Thermo Fisher
Scientific, Inc.). Equal amounts of protein (20 µg) were separated
by 8–12% SDS-PAGE and transferred onto polyvinylidene fluoride
membranes (EMD Millipore, Billerica, MA, USA) blocked with
Tris-buffered saline with Tween-20 containing 5% skimmed milk for 1
h at room temperature.
Membranes were incubated overnight at 4°C with
primary antibodies diluted to 1:1,000 against Akt (cat. no.,
#4060), phosphorylated (p)-Akt (cat. no., #4685), phosphoinositide
3-kinase (PI3K; cat. no., #4257), p-PI3K (cat. no., #4228),
mechanistic target of rapamycin (mTOR, #2983), p-mTOR (cat. no.,
#2971), p70 ribosomal S6 kinase (p70S6; cat. no., #2708), p-p70S6
(cat. no., #9205), glycogen synthase kinase-β (GSK-β, cat. no.,
#9315), p-GSK-β (cat. no., #9323), p-eukaryotic translation
initiation factor 4E-binding protein 1 (P-4E-BP1; cat. no., #2855),
p-inhibitor of nuclear factor κB kinase α (p-IKKα; cat. no.,
#2694), cyclin A (cat. no., #4656), B1 (cat. no., #4138) and D1
(cat. no., #2978), cell division cycle 2C (CDC2C; cat. no., #9112),
p-CDC2C (cat. no., #9111), retinoblastoma protein (pRb; cat. no.,
#9308), p21 (cat. no., #2946), caspases 3 (cat. no., #9664) and 8
(cat. no., #9496), poly (ADP-ribose) polymerase (PARP; cat. no.,
#9542) cellular inhibitor of apoptosis (cIAP) 1 (cat. no., #4952)
and 2 (cat. no., #3130), β-actin (cat. no., #4970; all Cell
Signaling Technology, Inc., Danvers, MA, USA), B-cell lymphoma 2
(Bcl-2; cat. no., #sc-7382) and Bcl-2-associated agonist of cell
death (Bad; cat. no., #sc-8044) and X-apoptosis regulator (Bax;
cat. no., #sc-70405; all Santa Cruz Biotechnology, Inc., Dallas,
TX, USA). Following incubation with secondary antibodies
(anti-mouse, cat. no., sc-2055; dilution, 1:1,000; anti-rabbit,
cat. no., sc-2004, dilution, 1:5,000, Santa Cruz Biotechnology,
Inc.) for 1 h at room temperature, protein expression was detected
using an enhanced chemiluminescence western blot substrate kit
(Pierce™ ECL Western Blotting Substrate kit; Thermo Fisher
Scientific, Inc.).
Microarray analysis
Total RNA was extracted from PC3 and PC3DR2 cells
using the RNeasy® Protect Mini kit (Qiagen, Inc.,
Valencia, CA, USA) according to the manufacturer's protocol. RNA
samples with high RNA integrity numbers (RIN>9.0). Agilent 2100
Bioanalyzer System and RNA kits developed by Agilent Technologies
(Santa Clara, CA, USA) with A260/280 ratios of 1.8–2.1 were used
for cDNA synthesis. Amplification cycles of RNA to cDNA and cDNA to
biotin-labeled RNA were performed with the GeneChip IVT Express kit
(Affymetrix; Thermo Fisher Scientific, Inc.). RNA was hybridized to
a GeneChip Human Genome HG-U133 Plus 2.0 array (Applied Biosystems;
Thermo Fisher Scientific, Inc.); all microarray steps were
performed according to the manufacturer's protocol. The MAS5
algorithm in GenPlex software ver. 3.0 (Istech Corp., Seoul, Korea)
was used for analyzing the CEL file data. The Affymetrix Microarray
Suite, MicroDB, and Data Mining Tool software v. 5.0 (Thermo Fisher
Scientific, Inc.) were used to annotate 54,120 probe sets with
17,084 genes from the UniGene database. Following global scaling
regression normalization, the data was log transformed to base 2.
Gene expression levels in PC3 and PC3DR2 cells compared using the
n-fold method. Differentially expressed gene clusters were analyzed
using GeneCluster 1.0 (MIT, Cambridge, MA, USA). GenMAPP was used
to analyze the functional pathways associated with differentially
expressed genes. (http://www.genmapp.org).
RT-qPCR
Total RNA was extracted from PC3 and PC3DR2 cells
using the RNeasy® Protect Mini kit as aforementioned.
cDNA was produced from 1 µg total RNA using oligo(dT) primers and
Omniscript reverse transcriptase enzyme (both Qiagen, Inc.)
according to the manufacturer's protocol. qPCR was performed with
the cDNA produced from 10 ng RNA with the FastStart Universal SYBR
Green Master mix (Roche Diagnostics, Indianapolis, IN, USA) using a
7500 Real-Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The sample was incubated at 95°C for 15 min,
followed by 40 cycles of 95°C for 15 sec, annealing at 58°C for 30
sec and extension at 72°C for 30 sec. GAPDH was used as the
reference gene. Fold change in gene expression was calculated
following the 2−ΔΔCq method (10). Primer sequences are presented in
Table I.
 | Table I.Primer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Gene symbol | Gene name | Direction | Primer sequence
(5′-3′) |
|---|
| IGF1R | Insulin-like growth
factor 1 receptor | Forward |
GTCCTCCTTGATGGTGGAAT |
|
|
| Reverse |
GTTCAAAACCTTGCCCACAT |
| BRCA2 | Breast cancer 2 | Forward |
ATGCAAATGCATACCCACAA |
|
|
| Reverse |
AGGTGGCCCTACCTCAAAAT |
| DBF4 | DBF4 homolog | Forward |
GGGTAACTGGAAGCCATGAA |
|
|
| Reverse |
CATGAGCCACAGGAGAGTCA |
| ZAK | Sterile α motif and
leucine zipper-containing kinase AZK | Forward |
GCTGCCTTCCTTTGAGATTG |
|
|
| Reverse |
CCGCTTCCCTGTAATGTTGT |
| PTCH1 | Patched 1 | Forward |
AGGGATTCCAAGGTGGAAGT |
|
|
| Reverse |
TGGCCTCTTTGCTTCAGATT |
| SERPINE1 | Serpin peptidase
inhibitor, clade E, member 1 | Forward |
TATCCTTGCCCTTGAGTGCT |
|
|
| Reverse |
AGTGGCTGGACTTCCTGAGA |
| CDKN2C | Cyclin-dependent
kinase inhibitor 2C | Forward |
ACGTCAATGCACAAAATGGA |
|
|
| Reverse |
TCATGAATGACAGCCAAACC |
| CDC6 | Cell division cycle
6 homolog | Forward |
TCTGATTCCCAAGAGGGTTG |
|
|
| Reverse |
TTCTGCTGAAGAGGGAAGGA |
| CDC25C | Cell division cycle
25 homolog C | Forward |
TGGGGAGATAACTGCCACTC |
|
|
| Reverse |
AAGCTGTGCTGGGCTACATT |
| CCNE2 | Cyclin E2 | Forward |
CCGAAGAGCACTGAAAAACC |
|
|
| Reverse |
GAATTGGCTAGGGCAATCAA |
| WNT3 | Wingless-type MMTV
integration site family, member 3 | Forward |
CGCCTCGGAGATGGTAGTAG |
|
|
| Reverse |
AAAGTTGGGGGAGTTCTCGT |
| FLI1 | Friend leukemia
virus integration 1 | Forward |
TGCACTCAGCTGACCACTCT |
|
|
| Reverse |
TTTCCAAGTTCTGGGACCAC |
| CUL2 | Cullin 2 | Forward |
GCATAGGACTGCATTCAGCA |
|
|
| Reverse |
GCGATGTCTGTGGAGTAGCA |
siRNA preparation and
transfection
si-BRCA2 specific for long-form BRCA2 was
synthesized by Invitrogen; Thermo Fisher Scientific, Inc.,
according to published sequences (11,12).
Scrambled siRNA (si-SCR) was obtained from Dharmacon (cat. no.,
#D-001210-01; GE Healthcare, Chicago, IL, USA). For transfection,
10 nM siRNA was mixed with DharmaFECT® 1 transfection
reagent (Dharmacon; GE Healthcare) and used according to the
manufacturer's protocol.
Statistical analysis
Unless indicated otherwise, datasets consist of
>3 replicates. Data are presented as the mean ± standard
deviation. Statistical significance between groups was determined
using an unpaired Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
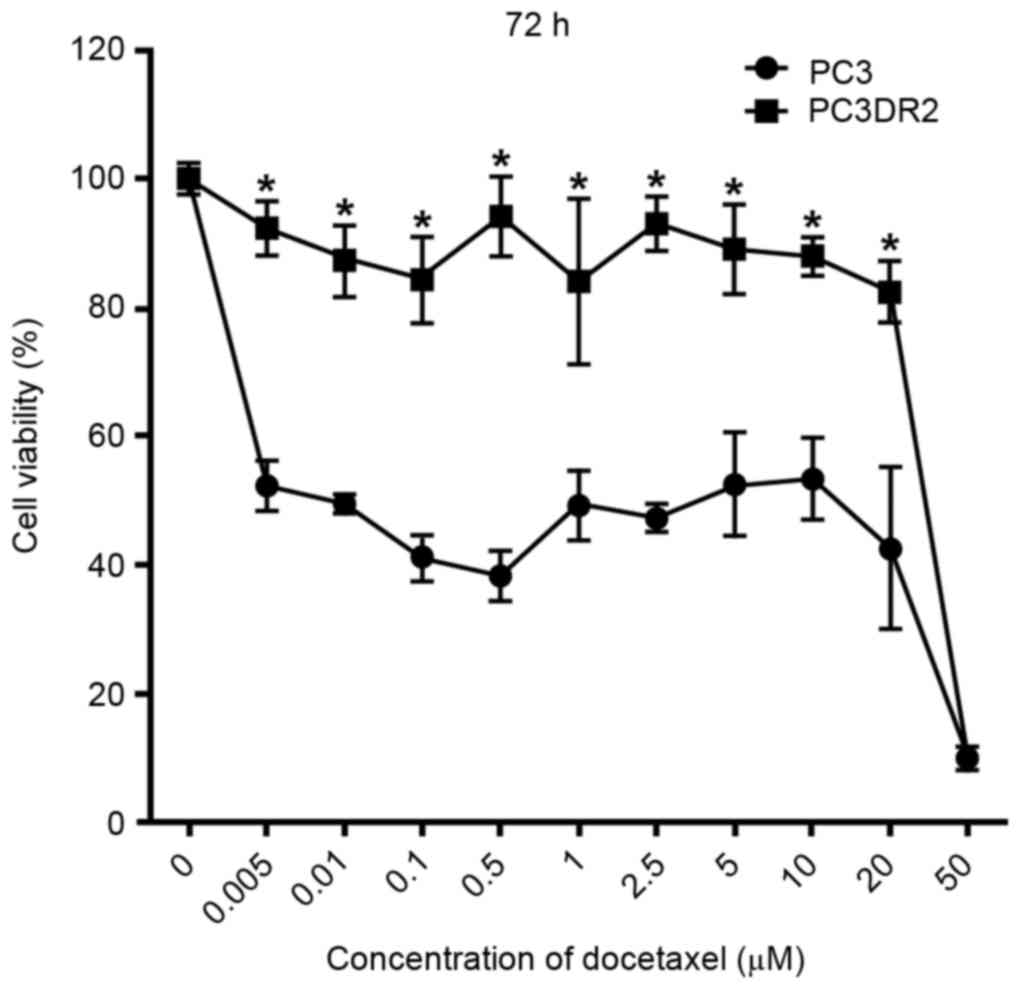
Establishment of a docetaxel-resistant
prostate cancer cell line (PC3DR2)
A docetaxel-resistant prostate cancer cell line
(PC3DR2) was generated by serial desensitization. To confirm the
docetaxel resistance, PC3 and PC3DR2 cells were exposed to
increasing doses of docetaxel (0, 0.005, 0.01, 0.1, 0.5, 1, 2.5, 5,
10, 20 or 50 µM) for 72 h. The extent of the cytotoxic effect of
docetaxel was determined using a CCK-8 assay. As presented in
Fig. 1, docetaxel decreased the
viability of PC3 cells in dose-dependent manner at 72 h; a
concentration of 0.005 µM docetaxel was sufficient to suppress
proliferation of PC3 cells by 52% at 72 h. However, PC3DR2 cells
were significantly more resistant to all concentrations of
docetaxel ≤20 µM at 72 h (P<0.05); the 20 µM PC3DR2 group
exhibited ~80% viability, whereas the 20 µM PC3 group exhibited
~50% viability. Thus, it was established that PC3DR2 cells
exhibited significant resistance to docetaxel doses ≤20 µM.
Effect of docetaxel resistance in
PC3DR2 cells on cell cycle, survival and apoptosis signaling
pathways
To investigate the characteristics of signaling
molecules and response of PC3DR2 cells to docetaxel treatment,
alterations to proteins in cell cycle-, survival-, and
apoptosis-associated signaling pathways in PC3DR2 cells were
examined following docetaxel treatment. Levels of cell
survival-(Akt, PI3K, p70S6, mTOR and GSKβ), cell cycle-(cyclin D1,
p-CDC2C and pRb) and apoptosis-(caspases 3 and 8, Bad, Bcl-2, cIAP1
and 2) associated signaling molecules were not altered in PC3DR2
cells following docetaxel treatment. Notably, 4E-BP1, a repressor
of mRNA translation, was inactivated by docetaxel treatment in
PC3DR2. However, expression of some cell cycle-associated molecules
(cyclin A, cyclin B1 and p21) and Bax was changed following
docetaxel treatment (Fig. 2).
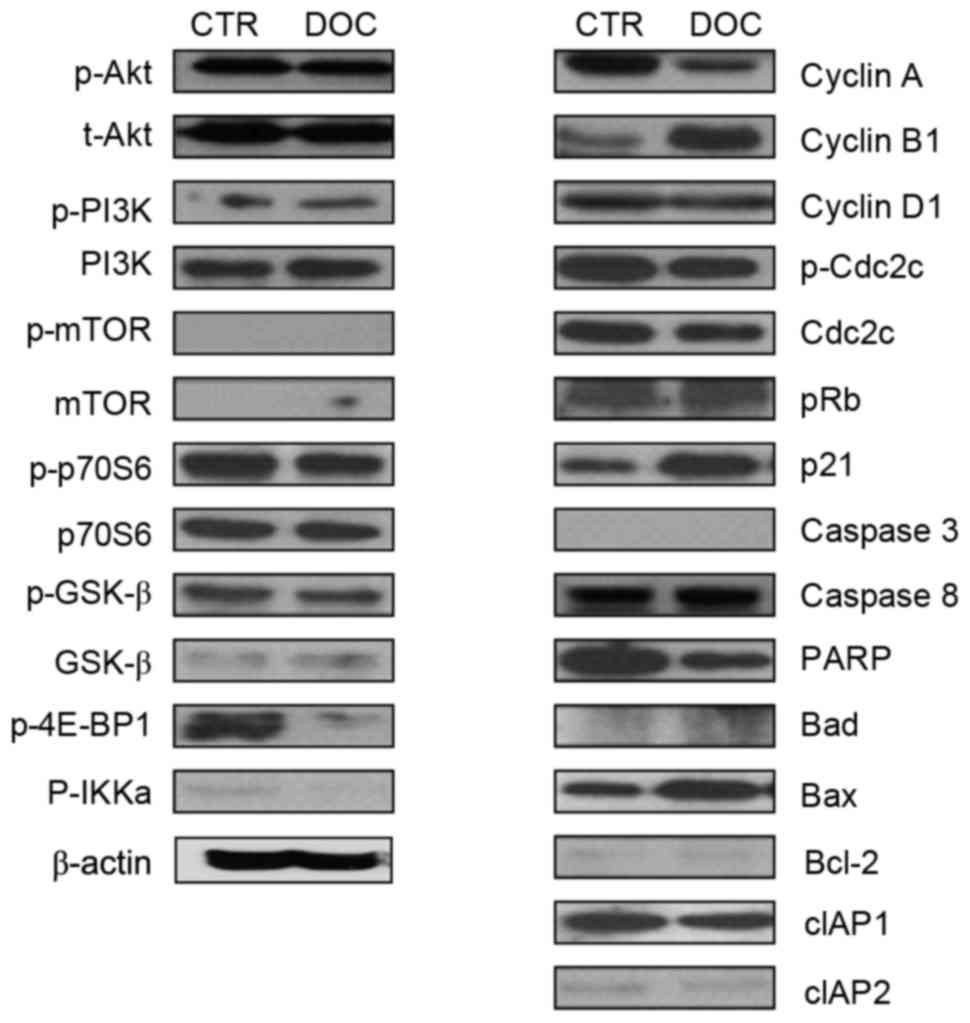 | Figure 2.Protein expression analysis with
western blotting in PC3DR2 prostate cancer cells. CTR, untreated
PC3DR2 cells; DOC, docetaxel-treated PC3DR2 cells; p,
phosphorylated; t-, total; PI3K, phosphoinositide 3-kinase; mTOR,
mechanistic target of rapamycin; p70S6, p70 ribosomal S6 kinase;
GSK-β, glycogen synthase kinase-β; 4E-BP1, eukaryotic translation
initiation factor 4E-binding protein 1; IKKa, inhibitor of nuclear
factor-κB kinase α; Cdc2C, cell division cycle 2C; pRb,
retinoblastoma protein; PARP, poly (ADP-ribose) polymerase; Bcl-2,
B-cell lymphoma 2; Bad, Bcl-2-associated agonist of cell death;
Bax, Bcl-2-associated X-apoptosis regulator; cIAP, cellular
inhibitor of apoptosis. |
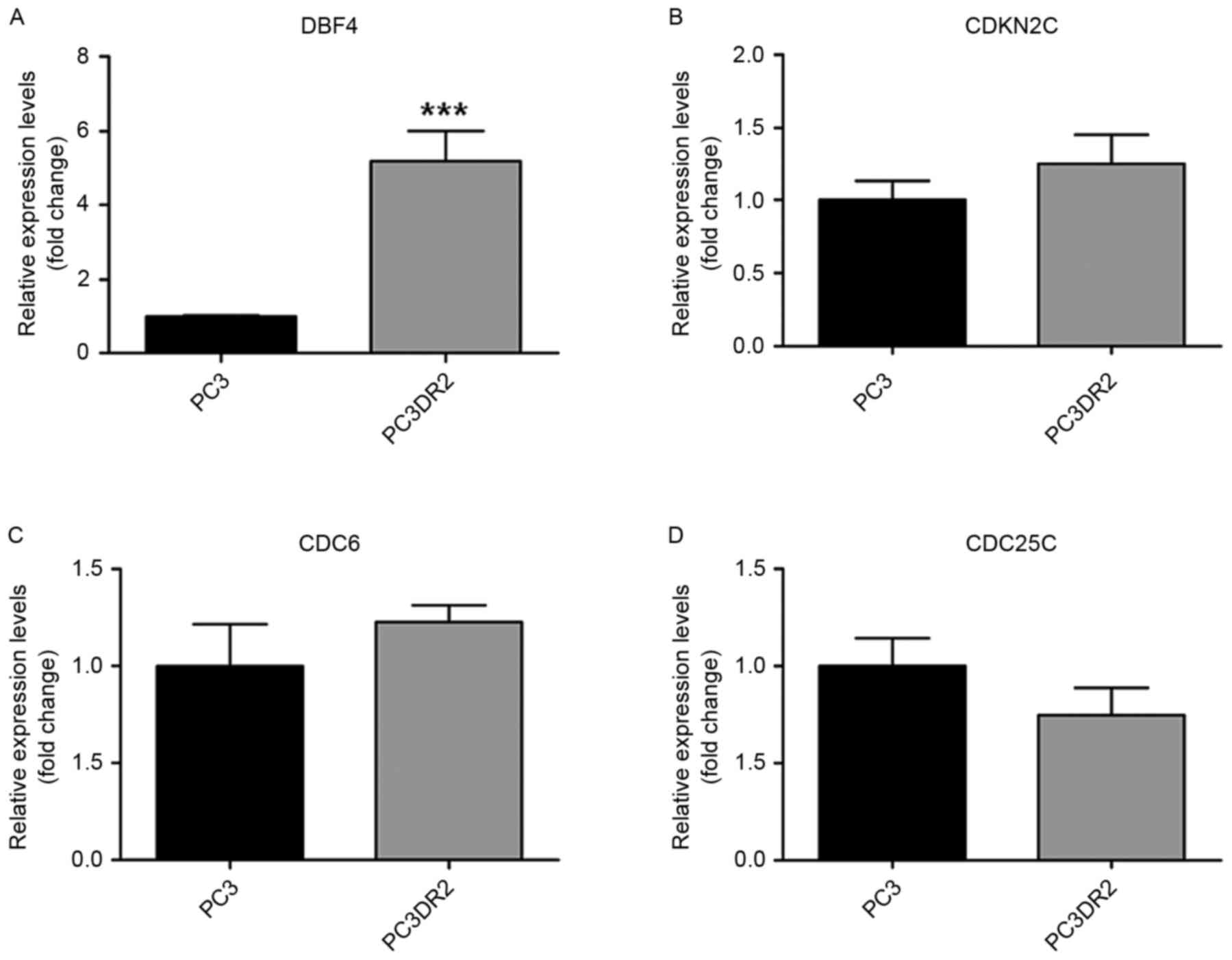
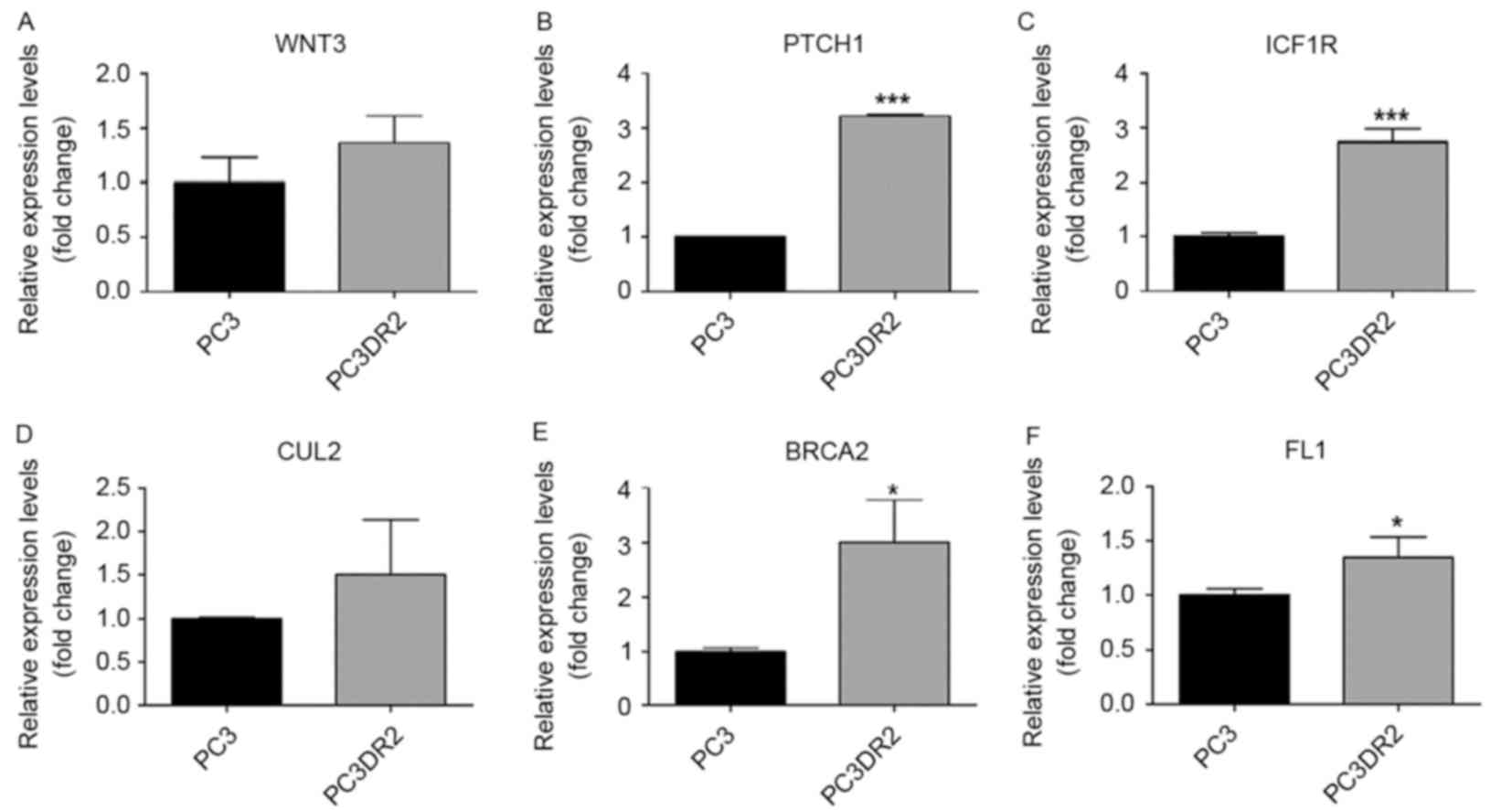
DNA microarray analysis of PC3DR2
cells
A total of 17,084 genes were analyzed; 1,227 genes
were 2-fold upregulated, whereas 1,190 genes were 2-fold
downregulated between PC3 and PC3DR2 cells. A total of 392 genes
were 3-fold upregulated, whereas 243 genes were 3-fold
downregulated (Table II). In
addition, to identify docetaxel resistance-associated genes, 13
differentially expressed genes associated with biological processes
possibly associated with docetaxel resistance were identified,
including DBF4, cyclin-dependent kinase inhibitor 2C, cell division
cycle 6 homolog, cell division cycle 25 homolog C, ZAK, SERPINE1,
cyclin E2 (CCNE2), wingless-type MMTV integration site family,
member 3, PTCH1, IGF1R, cullin 2, BRCA2 and Friend leukemia virus
integration 1 (FLI1; Table
III).v
| Table II.Number of upregulated and
downregulated genes and signaling pathways in PC3DR2 cells compared
with PC3 cells. |
Table II.
Number of upregulated and
downregulated genes and signaling pathways in PC3DR2 cells compared
with PC3 cells.
| Fold change
cut-off | Regulation | Genes | Significant
pathways |
|---|
| 2 | Up | 1,227 | 471 |
|
| Down | 1,190 | 362 |
| 3 | Up |
392 | 162 |
|
| Down |
243 | 67 |
| Table III.Genes associated with cell cycle and
cancer signaling pathways that were differentially expressed in
PC3DR2 cells relative to PC3 cells, as determined by DNA microarray
and RT-qPCR analyses. |
Table III.
Genes associated with cell cycle and
cancer signaling pathways that were differentially expressed in
PC3DR2 cells relative to PC3 cells, as determined by DNA microarray
and RT-qPCR analyses.
|
|
| Fold change in
PC3DR2 cells |
|---|
|
|
|
|
|---|
| Genea | Pathway | Microarray |
RT-qPCRb |
|---|
| DBF4 | Cell cycle | 24.9 |
5.2c |
| CDKN2C | Cell cycle |
3.8 | 31.2 |
| CDC6 | Cell cycle |
3.4 |
1.2 |
| CDC25C | Cell cycle |
3.2 |
0.7 |
| ZAK | MAPK signaling
pathway |
4.6 |
6.2c |
| SERPINE1 | p53 signaling
pathway |
3.7 |
3.4c |
| CCNE2 | p53 signaling
pathway |
3.7 |
1.4d |
| WNT3 | Pathways in
cancer |
3.3 |
1.4 |
| PTCH1 | Pathways in
cancer |
3.6 |
3.2c |
| IGF1R | Pathways in
cancer |
4.4 |
3.1c |
| CUL2 | Pathways in
cancer |
4.6 |
1.5 |
| BRCA2 | Pathways in
cancer |
3.2 | 3e |
| FLI1 | Transcriptional
misregulation in cancer |
9.0 |
1.4e |
Validation using RT-qPCR of candidate
genes in docetaxel resistance
The results of the microarray analysis for the
previously named genes was validated using RT-qPCR (Figs. 3–5,
Table III). Of the 13 genes, those
verified using RT-qPCR included DBF4, ZAK, SERPINE1, PTCH1, IGF1R
and BRCA2. IGF1R, PTCH1 and BRCA2 are associated with pathways in
cancer. DBF4 is associated with cell cycle. ZAK is associated with
the mitogen-activated protein kinase (MAPK) signaling pathway,
SERPINE1 and CCNE2 are associated with the p53 signaling pathway.
We hypothesized that these genes may be associated with docetaxel
resistance in PC3DR2 cells.
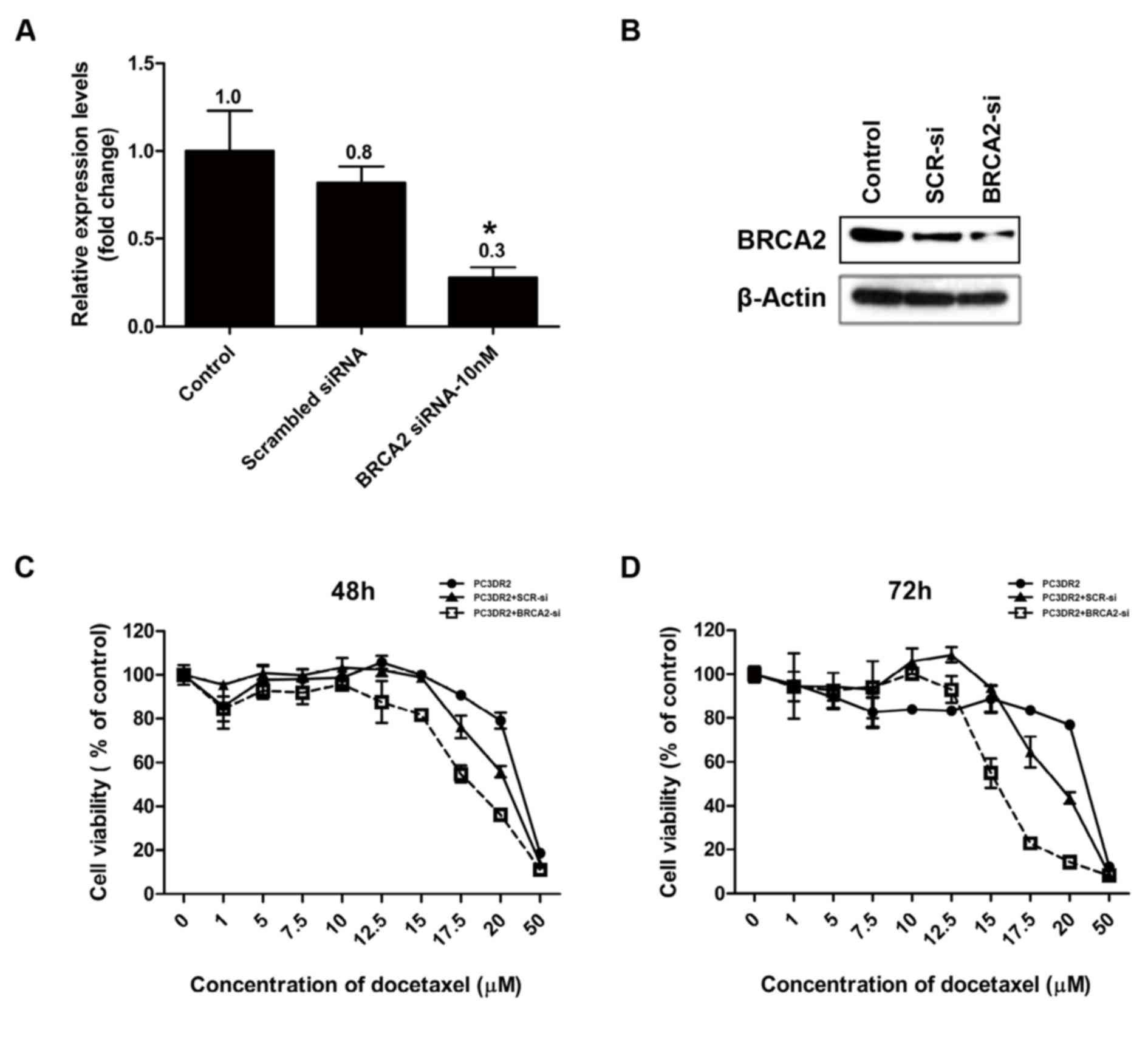
BRCA2 siRNA knockdown abolishes
docetaxel resistance in PC3DR2 cells
Following the confirmation of the upregulation of
DBF4, ZAK, SERPINE1, PTCH1, IGF1R and BRCA2 gene expression in
PC3DR2 cells, the genes were investigated for their direct
involvement in docetaxel resistance using an siRNA system. siRNA
for each gene was transfected into PC3DR2 cells and the cell
viability was determined using a CCK-8 assay following docetaxel
treatment. si-BRCA2 was confirmed by RT-qPCR and western blotting
to decrease the relative level of BRCA2 protein in PC3DR2 cells,
whereas scrambled siRNA did not significantly affect the level of
BRCA2 expression (Fig. 6A and B). The
transfection of siRNA against DBF4, ZAK, SERPINE1, PTCH1 and IGF1R
did not affect the docetaxel resistance of PC3DR2 cells (data not
shown), whereas si-BRCA2 transfection significantly reduced
docetaxel resistance at 48 and 72 h (Fig.
6C and D), suggesting that BRCA2 overexpression may be
associated with docetaxel resistance in prostate cancer cells.
Discussion
Although docetaxel represents the most effective
chemotherapeutic agent for patients with CRPC, once drug resistance
develops, there are limited effective therapeutic strategy options
for advanced CRPC. Thus, investigating the resistance mechanism of
prostate cancer cells against docetaxel is of marked urgency. In
our previous study, human docetaxel-resistance prostate cancer cell
line (PC3DR2) from docetaxel-sensitive prostate cancer cell line
(PC3) were generated by serial desensitization (9); in the present study, differential gene
expression between PC3 and PC3DR2 cells were compared with a DNA
microarray. O'Neill et al (13) also manipulated docetaxel-resistant
prostate cancer cell lines and suggested that multiple mechanisms
contribute to docetaxel resistance in partial agreement with our
results, although this study focused on the nuclear factor-κB
pathway, indicating that multiple mechanisms may be involved in
docetaxel resistance. In the present study, it was confirmed using
western blotting analysis that the expression of a number of
molecules, including those associated with the cell cycle, survival
and apoptosis, were unchanged in PC3DR2 cells subsequent to
docetaxel treatment. Using microarray analysis confirmed by
RT-qPCR, six overexpressed genes (IGF1R, DBF4, ZAK, PTCH1, SERPINE
and BRCA2) associated with cancer signaling pathways were
identified in PC3DR2 cells, exhibiting a >3-fold increase
compared with PC3 cells in the RT-qPCR data. To confirm the
association between the overexpression of these genes and docetaxel
resistance, an siRNA against each gene was transfected into PC3DR2
cells. BRCA2 knockdown abolished the docetaxel resistance in PC3DR2
cells. These results suggest the novel hypothesis that BRCA2
overexpression may be involved in docetaxel resistance.
Docetaxel stabilizes tubulin subunits in
microtubules, leading to apoptosis (5). In addition, it has been demonstrated
that docetaxel leads to an antitumor effect by inducing the
phosphorylation of Bcl-2 (14).
Suggested docetaxel resistance mechanisms include: i)
Overexpression of the p-glycoprotein drug efflux pump; ii) mutation
of the drug-binding site; iii) Expression of another tubulin
isoform; iv) Activation of a growth factor-associated pathway; and
v) Use of an alternative metabolic pathway (5,15). In the
present study, BRCA2 overexpression was identified as an additional
possible mechanism for docetaxel resistance in PC3DR2 cells.
BRCA1 and 2 are well-known breast cancer
susceptibility genes considered to be classical tumor-suppressor
genes, since the loss of both alleles is required to promote
carcinogenesis (11,12,16,17). A
recent study demonstrated that the 12-year prostate cancer-specific
survival rate was 94.3% for patients without and 61.8% for patients
with a BRCA2 mutation, suggesting that the survival time for
patients with a BRCA2 mutation is markedly below the average for
prostate cancer (18). Mutations of
BRCA genes increase the risk of prostate cancer and are associated
with disease characteristics and therapeutic outcomes (19). It has been demonstrated that the
functional loss of BRCA2 affects the focal development of prostate
cancer (20) and the potential for
the disease to spread through upregulation of matrix
metalloproteinase-9 (21).
Conversely, the decreased expression of BRCA2 mRNA predicts a
favorable response to docetaxel in breast cancer (22). The results of the present study
revealed that BRCA2 knockdown abolished docetaxel resistance in
PC3DR2 cells. Collectively, these results may appear to be
conflicting; however, this effect is expected when considering that
the major anticancer mechanism for docetaxel is to stabilize
microtubules during mitosis to induce cell cycle arrest at G2-M
phase, leading to apoptosis (5). BRCA
proteins are also involved in the mitotic spindle assembly process.
The normal DNA repair functions of BRCA1 and BRCA2 serve a critical
function in cell cycle processes during G2-M phase. When BRCA2
expression is low, malfunction of the DNA repair system may retard
the function of the mitotic spindle to slow or arrest the G2-M
process (23). Thus, it can be
speculated that tumors with low BRCA expression may be more
sensitive to docetaxel treatment, indicating that docetaxel may
exert a greater effect on prostate cancer cells where the function
of mitotic spindles is already partially retarded due to low BRCA2
expression, in accordance the results of the present study.
Tumor suppressor genes may regulate the sensitivity
of cancer cells to chemotherapy (24). Previous studies have demonstrated that
decreased BRCA1 expression following siRNA transfection may
increase cell sensitivity to platinum compounds and topoisomerase
inhibitors (25–27). Clinical studies also indicated that
BRCA1 may be a suitable biomarker for the clinical prognosis of
ovarian, lung and breast cancer treatment after DNA-damage-based
targeted therapy, as reviewed by Stordal and Davey (28). An in vitro and in vivo
study demonstrated that low BRCA1 expression, potentially leading
to defects in the DNA damage repair mechanism, was associated with
the high sensitivity to DNA-damaging drugs including cisplatin and
PARP inhibitors (29). Preclinical
and clinical studies have revealed that the loss of BRCA1 may also
result in resistance to other types of chemotherapeutic agent,
including the anti-microtubule agents paclitaxel and docetaxel, and
molecularly targeted agents (30,31).
Therefore, BRCA1 expression affects chemosensitivity differently
depending on the type of agent.
However, in the present study, BRCA1 was not
identified to be significantly altered in PC3DR2 cells. This is
noteworthy, as BRCA1 and BRCA2 are breast cancer-susceptibility
genes that have been identified through linkage analysis of
families susceptible to breast cancer (16,17).
However, it has also been reported that sporadic breast cancer may
exhibit decreased BRCA1 mRNA levels, whereas BRCA2 mRNA levels were
variable, compared with normal breast tissue (32–34). These
results may be caused by the hypermethylation of BRCA1 promoter,
which explains the downregulation in sporadic breast cancer,
whereas the promoter for BRCA2 is not hypermethylated (34). BRCA2 may have different operating
system from BRCA1 and it may be possible for only BRCA2 to be
involved in docetaxel resistance.
In conclusion, the results of the present study
suggest the novel hypothesis that BRCA2 may be associated with
docetaxel resistance in human prostate cancer cells. To clarify
this suggestion, further study with an in vivo model is
required.
Acknowledgements
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea,
funded by the Ministry of Education (grant no.
NRF-2014R1A1A2059537), and the Seoul National University Bundang
Hospital Research Fund (grant no. 02-2014-020).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sella A, Yarom N, Zisman A and Kovel S:
Paclitaxel, estramustine and carboplatin combination chemotherapy
after initial docetaxel-based chemotherapy in castration-resistant
prostate cancer. Oncology. 76:442–446. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Berthold DR, Pond GR, Soban F, de Wit R,
Eisenberger M and Tannock IF: Docetaxel plus prednisone or
mitoxantrone plus prednisone for advanced prostate cancer: Updated
survival in the TAX 327 study. J Clin Oncol. 26:242–245. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cortes JE and Pazdur R: Docetaxel. J Clin
Oncol. 13:2643–2655. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hwang C: Overcoming docetaxel resistance
in prostate cancer: A perspective review. Ther Adv Med Oncol.
4:329–340. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Petrylak DP: New paradigms for advanced
prostate cancer. Rev Urol. 9:(Suppl 2). S3–S12. 2007.PubMed/NCBI
|
|
7
|
Petrylak DP, Tangen CM, Hussain MH, Lara
PN Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M,
et al: Docetaxel and estramustine compared with mitoxantrone and
prednisone for advanced refractory prostate cancer. N Engl J Med.
351:1513–1520. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee S, Yoon CY, Byun SS, Lee E and Lee SE:
The role of c-FLIP in cisplatin resistance of human bladder cancer
cells. J Urol. 189:2327–2334. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Park HS, Hong SK, Oh MM, Yoon CY, Jeong
SJ, Byun SS, Cheon J, Lee SE and du Moon G: Synergistic antitumor
effect of NVP-BEZ235 and sunitinib on docetaxel-resistant human
castration-resistant prostate cancer cells. Anticancer Res.
34:3457–3468. 2014.PubMed/NCBI
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Merajver SD, Frank TS, Xu J, Pham TM,
Calzone KA, Bennett-Baker P, Chamberlain J, Boyd J, Garber JE,
Collins FS, et al: Germline BRCA1 mutations and loss of the
wild-type allele in tumors from families with early onset breast
and ovarian cancer. Clin Cancer Res. 1:539–544. 1995.PubMed/NCBI
|
|
12
|
Gudmundsson J, Johannesdottir G,
Bergthorsson JT, Arason A, Ingvarsson S, Egilsson V and
Barkardottir RB: Different tumor types from BRCA2 carriers show
wild-type chromosome deletions on 13q12-q13. Cancer Res.
55:4830–4832. 1995.PubMed/NCBI
|
|
13
|
O'Neill AJ, Prencipe M, Dowling C, Fan Y,
Mulrane L, Gallagher WM, O'Connor D, O'Connor R, Devery A, Corcoran
C, et al: Characterisation and manipulation of docetaxel resistant
prostate cancer cell lines. Mol Cancer. 10:1262011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rangel C, Niell H, Miller A and Cox C:
Taxol and taxotere in bladder cancer: In vitro activity and urine
stability. Cancer Chemother Pharmacol. 33:460–464. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gottesman MM, Fojo T and Bates SE:
Multidrug resistance in cancer: Role of ATP-dependent transporters.
Nat Rev Cancer. 2:48–58. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Miki Y, Swensen J, Shattuck-Eidens D,
Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM,
Ding W, et al: A strong candidate for the breast and ovarian cancer
susceptibility gene BRCA1. Science. 266:66–71. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wooster R, Neuhausen SL, Mangion J, Quirk
Y, Ford D, Collins N, Nguyen K, Seal S, Tran T, Averill D, et al:
Localization of a breast cancer susceptibility gene, BRCA2, to
chromosome 13q12-13. Science. 265:2088–2090. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Akbari MR, Wallis CJ, Toi A, Trachtenberg
J, Sun P, Narod SA and Nam RK: The impact of a BRCA2 mutation on
mortality from screen-detected prostate cancer. Br J Cancer.
111:1238–1240. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alanee SR, Glogowski EA, Schrader KA,
Eastham JA and Offit K: Clinical features and management of BRCA1
and BRCA2-associated prostate cancer. Front Biosci (Elite Ed).
6:15–30. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Francis JC, McCarthy A, Thomsen MK,
Ashworth A and Swain A: Brca2 and Trp53 deficiency cooperate in the
progression of mouse prostate tumourigenesis. PLoS Genet.
6:e10009952010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moro L, Arbini AA, Yao JL, di Sant'Agnese
PA, Marra E and Greco M: Loss of BRCA2 promotes prostate cancer
cell invasion through up-regulation of matrix metalloproteinase-9.
Cancer Sci. 99:553–563. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Egawa C, Miyoshi Y, Takamura Y, Taguchi T,
Tamaki Y and Noguchi S: Decreased expression of BRCA2 mRNA predicts
favorable response to docetaxel in breast cancer. Int J Cancer.
95:255–259. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mullan PB, Quinn JE, Gilmore PM,
McWilliams S, Andrews H, Gervin C, McCabe N, McKenna S, White P,
Song YH, et al: BRCA1 and GADD45 mediated G2/M cell cycle arrest in
response to antimicrotubule agents. Oncogen. 20:6123–6131. 2001.
View Article : Google Scholar
|
|
24
|
Lai D, Visser-Grieve S and Yang X: Tumour
suppressor genes in chemotherapeutic drug response. Biosci Rep.
32:361–374. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Husain A, He G, Venkatraman ES and Spriggs
DR: BRCA1 up-regulation is associated with repair-mediated
resistance to cis-diamminedichloroplatinum(II). Cancer Res.
58:1120–1123. 1998.PubMed/NCBI
|
|
26
|
Quinn JE, Kennedy RD, Mullan PB, Gilmore
PM, Carty M, Johnston PG and Harkin DP: BRCA1 functions as a
differential modulator of chemotherapy-induced apoptosis. Cancer
Res. 63:6221–6228. 2003.PubMed/NCBI
|
|
27
|
Tassone P, Tagliaferri P, Perricelli A,
Blotta S, Quaresima B, Martelli ML, Goel A, Barbieri V, Costanzo F,
Boland CR and Venuta S: BRCA1 expression modulates chemosensitivity
of BRCA1-defective HCC1937 human breast cancer cells. Br J Cancer.
88:1285–1291. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stordal B and Davey R: A systematic review
of genes involved in the inverse resistance relationship between
cisplatin and paclitaxel chemotherapy: Role of BRCA1. Curr Cancer
Drug Targets. 9:354–365. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang D, Khan S, Sun Y, Hess K, Shmulevich
I, Sood AK and Zhang W: Association of BRCA1 and BRCA2 mutations
with survival, chemotherapy sensitivity, and gene mutator phenotype
in patients with ovarian cancer. JAMA. 306:1557–1565. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chabalier C, Lamare C, Racca C, Privat M,
Valette A and Larminat F: BRCA1 downregulation leads to premature
inactivation of spindle checkpoint and confers paclitaxel
resistance. Cell Cycle. 5:1001–1007. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Papadaki C, Tsaroucha E, Kaklamanis L,
Lagoudaki E, Trypaki M, Tryfonidis K, Mavroudis D, Stathopoulos E,
Georgoulias V and Souglakos J: Correlation of BRCA1, TXR1 and TSP1
mRNA expression with treatment outcome to docetaxel-based
first-line chemotherapy in patients with advanced/metastatic
non-small-cell lung cancer. Br J Cancer. 104:316–323. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thompson ME, Jensen RA, Obermiller PS,
Page DL and Holt JT: Decreased expression of BRCA1 accelerates
growth and is often present during sporadic breast cancer
progression. Nat Genet. 9:444–450. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bièche I, Noguès C and Lidereau R:
Overexpression of BRCA2 gene in sporadic breast tumours. Oncogene.
18:5232–5238. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Collins N, Wooster R and Stratton MR:
Absence of methylation of CpG dinucleotides within the promoter of
the breast cancer susceptibility gene BRCA2 in normal tissues and
in breast and ovarian cancers. Br J Cancer. 76:1150–1156. 1997.
View Article : Google Scholar : PubMed/NCBI
|