Introduction
Cell growth and differentiation require accurate
control of the mechanism regulating the entry into, passage
through, and withdrawal from the cell cycle. Progress through the
G1 phase of the cell cycle is mediated by the D-type cyclin family,
which are key regulators of the cyclin-dependent kinases (CDKs) 4
and 6 (1,2). D-type cyclins/CDK complexes are key
cell-cycle regulators, which phosphorylate critical substrates for
cell-cycle progression, including the retinoblastoma (pRb) family
of proteins (3–6). Therefore, CDK4, CDK6 and D-type cyclins
serve as master integrators in the G1 phase and couple with cell
cycle mitogenic signals to mediate their oncogenic properties in
cancer cells (6–8). The highly conserved sequence among the
three D-type cyclins suggests that they serve redundant roles.
However, each member is expressed in a tissue-specific manner
(1,9).
Consistent with their role in cell proliferation, abnormal levels
of D-type cyclins have been associated in the development of
various types of human cancer. The rearrangement and/or
amplification of the cyclin D1 gene has been widely reported in a
range of human cancers, including carcinoma of the uterine cervix,
breast carcinomas, and head and neck squamous cell carcinomas
(10–12). Likewise, overexpression of the cyclin
D2 gene have been documented in testicular tumors (13,14) and
B-cell malignancies (15). The
deregulated expression of cyclin D3 has also been described in
several types of human cancer (16–19),
including malignancies of the thymus (20). Consistent with these results, Sicinska
et al (21) reported that mice
deficient in cyclin D3 exhibited specific defects in T-lymphocyte
development. Previous studies have employed the mouse skin model to
study the role of D-type cyclins in normal and neoplastic
proliferation (22–26). These studies revealed that cyclin D1
and D2 are overexpressed in mouse skin papillomas and squamous-cell
carcinomas (SCC), whereas levels of cyclin D3 protein remain
constant in skin tumors (27).
Previous studies have demonstrated that Ras-mediated skin
tumorigenesis is dependent on pathways that signal preferentially
through cyclin D1 and D2. Consistent with this idea, it was
demonstrated that ablation of cyclin D1 or cyclin D2 led to the
developmental inhibition of mouse skin papilloma (28,29).
Currently, the role of cyclin D3 in skin tumorigenesis remains
poorly understood.
The present study investigated the effect of the
genetic modification of cyclin D3 and CDK6 in skin carcinogenesis
and normal keratinocyte proliferation. A number of previous studies
by the present authors and the others have used the two-stage skin
carcinogenesis protocol, which is a well-suited model for
understanding the multistage nature of tumor progression in order
to validate the importance of tumor suppressors and oncogenes in
skin cancer formation and transformation (28–41). In
this model of chemically induced skin tumorigenesis, tumor
initiation is accomplished via a single topical application of a
genotoxic carcinogen, including 7,12-dimethylbenz(a)anthracene
(DMBA), which produces a heritable mutation in the Ha-Ras oncogene.
Tumor promotion is induce via multiple applications of a tumor
promoter, usually 12-O-tetradecanoylphorbol-13-acetate (TPA),
leading to the expansion of the initiated cells. TPA induces the
hyperproliferation of cells, which promotes the generation of
benign tumors, or papillomas, which can, in certain cases, progress
to SCC (malignant progression). In the mouse epidermis, cyclin D3
preferentially binds to CDK6 and, although counterintuitive,
overexpression of CDK6 leads to inhibition of mouse skin
tumorigenesis (27,36). The authors have hypothesized that the
tumor-inhibitory role of CDK6 depends largely on binding to cyclin
D3. In the present study, cyclin D3-deficient mice were used to
investigate the role of cyclin D3 in skin tumorigenesis. Compound
mice overexpressing CDK6 in the absence of cyclin D3 were also
assessed to determine the effect of cyclin D3 ablation in
CDK6-dependent skin tumor development. The present study
demonstrated that cyclin D3 deficiency reduces the number of benign
skin tumors (papillomas) due to increased apoptosis in the hair
follicle but does not alter normal keratinocyte proliferation.
Although cyclin D3 binds preferentially to CDK6 (27), the tumor inhibition mediated by CDK6
is independent of cyclin D3. Finally, the present study revealed
that modification in the D-type cyclin and CDK levels resulted in
unbalanced formation of CDK/cyclin complexes leading to reduced
papilloma development, but to a more aggressive tumor
phenotype.
Materials and methods
Mouse models
The cyclin D3-knockout mice were provided by Dr
Peter Sicinski, Department of Genetics, Harvard Medical School
(Boston, MA, USA) (21), and the
generation of K5CDK6 transgenic mice was as previously reported
(36). Cyclin D3+/− mice were
backcrossed with the FVB genetic background (The Jackson Lab, Bar
Harbor, ME, USA) for three generations to reduce the influence of
the genetic background. K5CDK6 mice were mated with cyclin D3+/−
mice to obtain K5CDK6/cyclin D3+/− mice and backcrossed with cyclin
D3+/− mice to acquire K5CDK6/cyclin D3-/− mice. The mice were
housing at the animal facility of the College of Veterinary
Medicine, NC State University. Housing conditions include a 12 h
light/dark cycle, 20–23°C and water and food were accessible at all
times. The genotype of the mice was confirmed by polymerase chain
reaction (PCR) as previously described (21,36) using
KAPA2G fast PCR kit (Kapa Biosystems, Inc., Wilmington, MA,
USA).
Mouse experiments
The animal research protocols were approved by the
Institutional Animal Care and Use Committee at North Carolina State
University (Raleigh, NC, USA). The end-point of the two-stage
carcinogenesis experiments was the quantification of the number of
skin tumors developed during a 20–25 weeks period or an individual
skin tumor size of 1,800 mm3, at which point the animal would have
reached the study end-point. Tumor burden may exceed 1,800 mm3 when
multiple tumors are present but no individual tumor exceeded this
dimension (tumor burden did not exceed 10% of mouse body weight).
The effect of lack of cyclin D3 in skin tumorigenesis was
determined with 15 female mice from each group (wild-type vs.
cyclin D3-/−). No phenotypical differences were observed between
wild type and cyclin D3-/− mice. Three-weeks-old mice were
administered with a single topical application of 200 nmol DMBA
(Cat D3254; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in 200
µl acetone on the dorsal surface of the mice. After 2 weeks, the
mice were dosed topically twice a week with 4 µg TPA (Cat P1680;
Sigma-Aldrich; Merck KGaA) in 200 µl acetone for 20 weeks.
Papilloma development was traced weekly until the end of the
experiment at 20 weeks. Tumors ≥1 mm in size were counted weekly,
and the multiplicity (mean number of tumors per mouse) and
incidence of tumor-bearing mice were compared between the two
groups using Fisher's exact test.
To determine the effect of cyclin D3 ablation on
K5CDK6 transgenic mice, four groups of 10 mice each were utilized
(wild-type, K5CDK6, cyclin D3−/− and K5CDK6/cyclin
D3−/− mice). K5CDK6 mice were generated in C57BL/6
background (36) and crossed with
cyclin D3−/− as aforementioned. Tumorigenesis in newborn
mice was initiated at day 3 after birth with a single application
of 50 µg DMBA in 50 µl acetone on the dorsal skin. After 3 weeks,
the mice were administered twice a week with 4 µg TPA in 200 µl
acetone for 25 weeks. Papilloma development was traced weekly until
the end of the experiment at 25 weeks after the first application
of the tumor promoter TPA. Tumors ≥1 mm in size were counted weekly
and the multiplicity and incidence of tumor-bearing mice were
compared between the groups using Fisher's exact test.
The mice were sacrificed by CO2
asphyxiation, and the dorsal skins were treated with a commercial
available depilatory agent (Nair) for 1 min.
Western blotting and
co-immunoprecipitation assays
The epidermal tissue was scraped off with a razor
blade, placed into the homogenization buffer (50 mmol/l HEPES, pH
7.5, 150 mmol/l NaCl, 2.5 mmol/l EGTA, 1 mmol/l
ethylenediaminetetraacetic acid, 0.1% Tween-20, 1 mmol/l
dithiothreitol, 0.1 mmol/l phennylmethyl sulfonyl fluoride, 0.2
U/ml aprotinin, 10 mmol/l b-glycerophosphate, 0.1 mmol/l sodium
vanadate and 1 mmol NaF), and homogenized using a manual
homogenizer. The epidermal homogenate was centrifuged at 11,000 × g
for 20 min at 4°C to collect the supernatant, which was used
directly for western blot analysis or stored at −80°C. Protein
concentration was measured using the Bio-Rad Protein Assay system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). Protein lysates
(25 µg from each sample) were electrophoresed on 10% acrylamide
gels and electrophoretically transferred onto nitrocellulose
membranes. After being blocked with 5% nonfat powdered milk in
Dulbecco phosphate-buffered saline, the membranes were incubated
with 1 µg/ml specific antibodies. The following antibodies were
used: Polyclonal antibodies against cyclin D1 (sc-717), cyclin D2
(sc-593), CDK4 (sc-260), CDK6 (sc-177) and β-actin (sc-1616), (all
from Santa Cruz Biotechnology, Inc., Dallas, TX, USA; all 1:100)
and mouse monoclonal antibody against cyclin D3 (DCS-22)
(NeoMarkers; Thermo Fisher Scientific Inc., Waltham, MA, USA). The
membranes were washed 3-times on PBS and Triton X-100
(Sigma-Aldrich; Merck KGaA) and incubated with secondary
antibodies: Goat anti-rabbit-HRP (horseradish peroxidase) (cat. no.
sc-2020) (Santa Cruz Biotechnology, Inc.) or goat anti-mouse-HRP
(cat. no. 31430) (Pierce; Thermo Fisher Scientific Inc.) and
visualized using enhanced chemiluminescence (ECL detection kit; GE
Healthcare Bio-Sciences, Pittsburgh, PA, USA).
To investigate CDK/D-type cyclin complex formations,
5 µg of antibodies against CDK4 (100 µg/ml; cat. no. sc-260), CDK6
(100 µg/ml; cat. no. sc-177) and 5 µg of normal rabbit IgG (200
µg/ml; cat. no. sc-2027; Santa Cruz Biotechnology, Inc.),
conjugated with protein A-sepharose beads (Thermo Fisher
Scientific, Inc.) or Dynabeads® Protein G (Invitrogen;
Thermo Fisher Scientific, Inc.) were used. Fresh protein lysates
from epidermal tissue (250 µg) were immunoprecipitated for 1 h at
4°C with constant rotation. Following three washes with extraction
buffer, proteins that co-immunoprecipitated were analyzed by
western blot analysis as aforementioned. Protein lysate (25 µg) was
loaded as a control input.
Immunostaining
Epithelial cell proliferation was measured by
intraperitoneal injection of 60 µg/g 5-bromodeoxyuridine (BrdU) 30
min prior to the sacrifice of mice by CO2 asphyxiation.
Dorsal skin and tumor sections were fixed in 10% formalin (Cat.
HT5014; Sigma-Aldrich; Merck KGaA) at room temperature for 48 h and
embedded in paraffin prior to sectioning. Tissue sections
(thickness, 4 µm) were stained with hematoxylin and eosin (H&E)
for 5 min at room temperature and were used for histopathological
analysis.
BrdU incorporation was detected by
immunohistochemical staining of paraffin-embedded skin and tumor
sections with a mouse anti-BrdU (S7101) monoclonal antibody
(Calbiochem; EMD Millipore, Billerica, MA, USA), biotin-conjugated
anti-mouse antibody (Vector Laboratories, Inc., Burlingame, CA,
USA), and avidin-biotin Vectastain Elite peroxidase kit (Vector
Laboratories, Inc.) with diaminobenzidine as a chromogen. Apoptotic
cells were identified using terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assays with the FragEL DNA
Fragmentation Detection kit and Colorimetric-TdT enzyme (EMD
Millipore), according to the manufacturer's instructions. Briefly,
the terminal deoxynucleotidyl transferase (TdT enzyme) binds to the
exposed 3′OH ends of a DNA fragment generated in apoptosis
progression and catalyzes the addition of biotin-labeled and
unlabeled deoxynucleotides. Biotinylated nucleotides were detected
using a streptavidin-HRP conjugate. The slides were counterstained
with methyl green for quantification of normal and apoptotic cells.
The numbers of apoptotic cells in skin tumors were determined in
250 µm2 sections with a reticule grid using a light
microscope (Nikon Eclipse E400). Apoptotic keratinocytes in
interfollicular and follicular epidermis were quantified in
sections (thickness, 2 cm). To determine the incidence of
follicular apoptosis, hair follicles with ≥1 apoptotic cell in the
bulge area were counted as a positive hair follicle. In all cases,
12 fields (magnification, ×400) were counted per section in a total
of 10 sections representing 5 mice per genotype.
Statistical analysis
Statistical analysis [paired t-test, analysis of
variance (one-way ANOVA), Bonferroni's multiple comparison test and
correlation Person's test) was performed using GraphPad Prism 4
software (GraphPad Software Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Reduced sensitivity to ras-dependent
skin papilloma development in cyclin D3-null mice
The authors of the present study have previously
reported that knocking out cyclin D1- and cyclin D2 has a mild
effect on the proliferative status of mouse epidermis, but lead to
a severe reduction in the sensitivity to the development of skin
papillomas induced by chemical carcinogenesis (28,42). To
investigate the effect of cyclin D3 ablation in mouse skin, cyclin
D3-null mice developed by gene targeting in embryonic stem cells
were utilized (21). First, the
authors investigated whether ablation of cyclin D3 affected
epidermal homeostasis. To this end, formalin-fixed,
paraffin-embedded skin cross-sections of cyclin D3-/− and wild-type
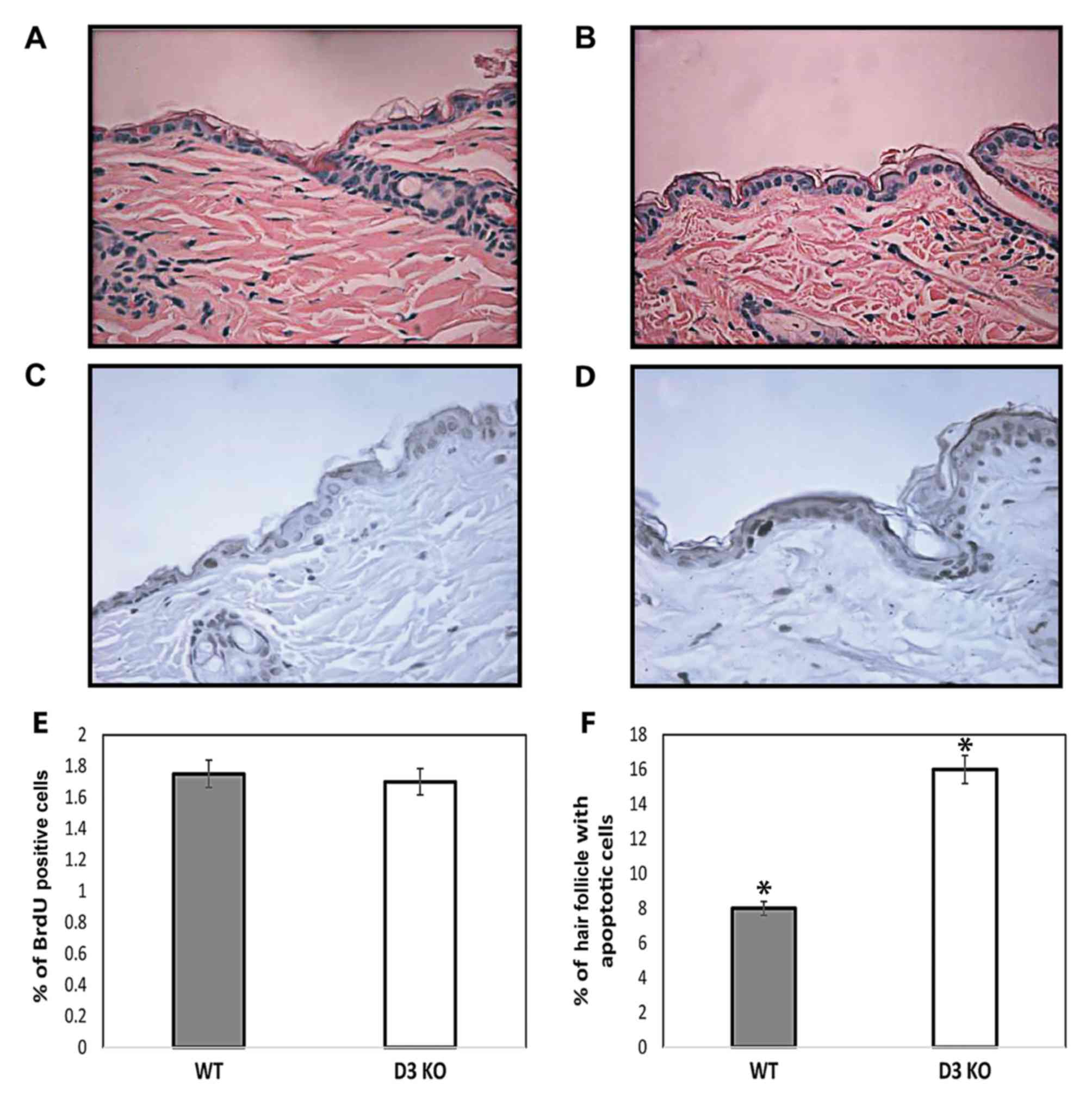
siblings were examined. H&E-stained sections revealed no
changes in the skin structure of cyclin D3-/− compared with
wild-type mice. No marked alterations in the morphology of the
follicular and interfollicular epidermis were observed between
cyclin D3-/− and wild-type siblings (Fig.
1A and B). Consistent with this data, cyclin D3-/− and
wild-type mice exhibit no differences in the total number of
nucleated keratinocytes in the interfollicular epidermis. To
determine whether loss of cyclin D3 affected epidermal homeostasis,
the rate of keratinocyte proliferation and apoptosis was examined.
Cyclin D3-/− mice did not exhibit any detectable alterations in
keratinocyte proliferation as determined by epidermal thickness
(data not shown) and the number of BrdU-positive cells in the
interfollicular epidermis compared with wild-type mice (Fig. 1C-E). The authors conclude that
epidermal keratinocytes proliferate normally in the absence of
cyclin D3, likely through compensation by other members of the
D-type cyclin family.
The authors also studied whether a lack of cyclin D3
affects apoptosis in interfollicular epidermis and hair follicles.
As previously reported, the number of apoptotic keratinocytes in
the interfollicular epidermis was barely affected by knockout of
cyclin D3 (0.2%) (42–44), and no differences were observed
between cyclin D3−/− and wild-type mice (data not
shown). Although the hair follicle stem cells do not contribute to
the homeostasis of mouse epidermis. It is known that stem cells
located in the bulge area of the hair follicle participate in
hyperproliferative conditions, including epidermis regeneration
hyperproliferation conditions (45).
Therefore, the number of hair follicles carrying apoptotic cells in
the bulge region was quantified to determine the incidence of
apoptosis in hair follicles. A two-fold increase was observed in
the number of hair follicles carrying apoptotic cells in cyclin
D3−/− mice compared with wild-type siblings (P<0.001,
t-test) (Fig. 1F). Supporting the
data of the present study, it was previously reported that
downregulation of cyclin D3 also results in increased apoptosis in
leukemia cells (46). Importantly,
multipotent stem cells localized in the bulge region of the mouse
hair follicle retain chemical carcinogens and have been
hypothesized as the origin of skin papillomas (47,48).
Consequently, the authors next asked whether increased apoptosis
observed throughout the bulge region of cyclin D3−/−
mice would affect skin papilloma development. To test this
hypothesis, cyclin D3−/− and wild-type littermates were
subjected to the two-stage carcinogenesis protocol. This protocol
induces skin papillomas after a single application of a carcinogen
followed by biweekly treatments with a tumor promoter that favors
the selection of cells bearing Ha-ras mutations. The dorsal skin of
cyclin D3−/− and wild-type littermates were topically
treated with a sub-carcinogenic dose of the genotoxic DMBA, and
tumor development was subsequently promoted via biweekly
applications of 12-O-tetradecanoylphorbol-13-acetate (TPA) for 20
weeks. The incidence and multiplicity of papillomas were scored in
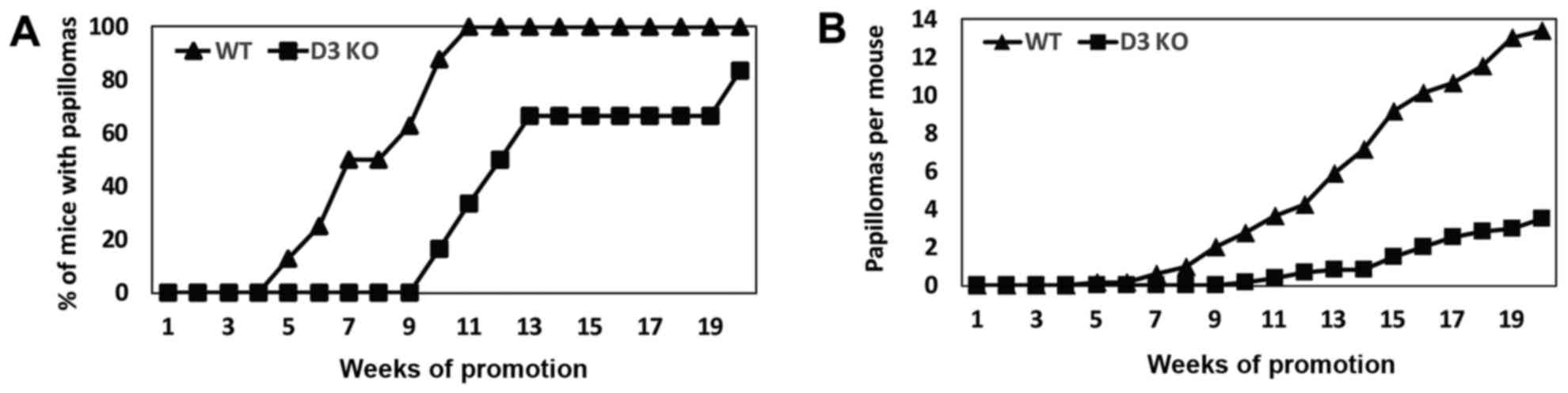
each group for 20 weeks. Papilloma development was delayed by 5
weeks in cyclin D3-null mice compared with wild-type siblings
(Fig. 2A). The incidence of papilloma
formation reached a plateau of 100% at 11 weeks in wild-type mice.
By contrast, cyclin D3−/− mice reached 83% of papilloma
incidence at 20 weeks, with ~20% of the cyclin D3−/−
mice remaining refractory to tumor development (Fig. 2A). Tumor multiplicity (mean number of
tumors per mice) shows a 74% reduction in cyclin D3−/−
mice compared with control littermates (Fig. 2B).
Collectively, these observations demonstrate that
cyclin D3 ablation does not grossly affect follicular and
interfollicular keratinocyte proliferation, but leads to increase
apoptosis in the bulge region of the hair follicle and decreased
papilloma development.
Lack of cyclin D3 does not affect the
tumor inhibitory activity of CDK6
Similar to cyclin D3 ablation, the authors have
shown previously that transgenic expression of CDK6 results in
increased apoptosis in the hair follicle and a reduced number of
skin papillomas in a two-stage carcinogenesis model (36). Notably, the authors have previously
determined a preferential binding of cyclin D3 to CDK6 and the
consequent increase in CDK6/Cyclin D3 complex formation in
hyperproliferative epidermis (49).
Likewise, forced expression of CDK6 in epidermis of K5CDK6
transgenic mice led to increased binding to cyclin D3 (36). Therefore, the authors asked whether
cyclin D3 expression is essential for the tumor inhibitory activity
of CDK6 (36). To test this
hypothesis, the authors developed the K5CDK6/cyclin D3-/− compound
mice in which overexpression of CDK6 is driven by the keratin 5
promoter (K5) to the basal cell layer of epidermis and hair
follicles in a cyclin D3-null background. Newborn animals did not
display any evident developmental abnormalities, and H&E
staining of the dorsal epidermis did not reveal alterations in the
morphology of follicular and interfollicular epidermis.
Furthermore, no significant differences in the number of nucleated
cells were observed in the interfollicular epidermis (data not
shown).
To determine whether simultaneous overexpression of
CDK6 and lack of cyclin D3 affects the expression of other
cell-cycle regulators, biochemical analysis of epidermal tissue was
performed. Protein analysis confirmed elevated CDK6 expression and
a lack of cyclin D3 in the epidermis of K5CDK6/cyclin
D3−/− mice (Fig. 3A).
Notably, a marked increase in the cyclin D1 protein level from
cyclin D3−/− and K5CDK6/cyclin D3−/−
epidermis compared with K5CDK6 and wild-type samples was observed
(Fig. 3A). A mild increase in cyclin
D2 protein expression was detected in K5CDK6 and cyclin
D3−/− epidermis. However, this effect varied between
samples.
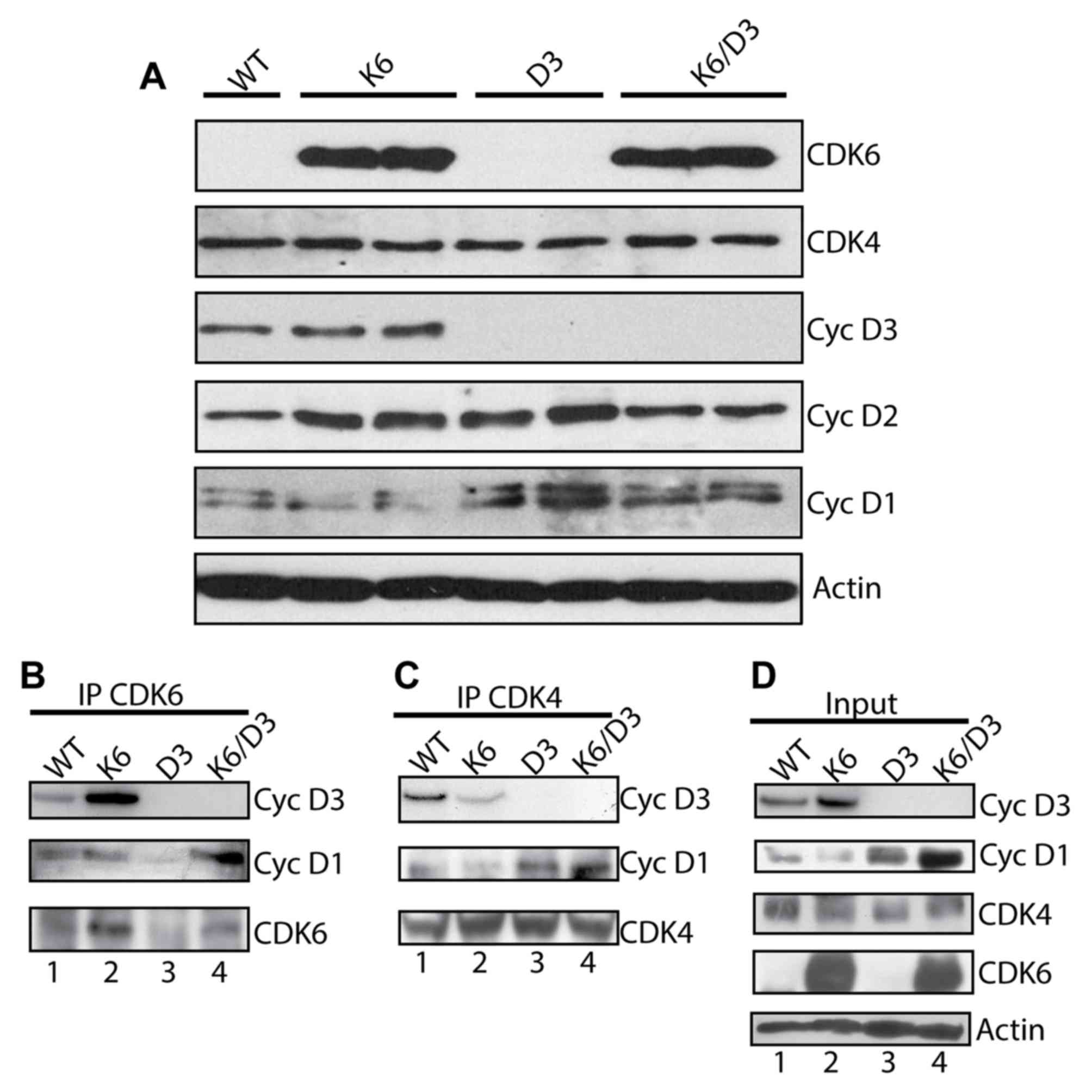 | Figure 3.Biochemical analysis of cell cycle
regulators in mouse epidermis. Epidermal lysates from WT, K6, D3
and K6/D3 mice were separated by SDS-PAGE and transferred onto a
nitrocellulose membrane. Primary antibodies against CDK6, CDK4,
cyclin D3, cyclin D2 and cyclin D1 were used for immunoblot
analysis. β-actin was used as a loading control. (A) Epidermal
lysates from WT, K6, D3 and K6/D3 mice were immunoprecipitated with
antibodies against (B) CDK6 or (C) CDK4 and blotted with antibodies
against cyclin D3, cyclin D1, CDK6 and CDK4. (D) Epidermal lysates
from WT, K6, D3 and K6/D3 mice were loaded. CDK, cyclin-dependent
kinase; K6, K5CDK6 mice; D3, cyclin D3−/− mice; K6/D3,
K5CDK6/cyclinD3−/− mice WT, wild type; IP,
immunoprecipitation. |
D-type cyclins are the limiting factors during
CDK/cyclin complex formation. Therefore, modifications in the
protein levels of cyclin D1 may change the kinetics of complex
formation in mouse epidermis (27,50). In
fact, the forced expression of cyclin D3 in mouse epidermis
resulted in elevated CDK6 and CDK4 activity, primarily associated
with increase formation of CDK4/cyclin D1, CDK6/cyclin D1 and
CDK6/cyclin D3 complexes (29).
Therefore, whether transgenic expression of CDK6 alters D-type
cyclins/CDKs complex formation was analyzed. Epidermal lysates from
wild type, K5CDK6, cyclin D3−/− and K5CDK6/cyclin
D3−/− mice were immunoprecipitated with antibodies
against CDK6 and CDK4 followed by western blot analysis to
determine associations between cyclin D1 and cyclin D3. In
agreement with previous findings, it was indicated that the
overexpression of CDK6 resulted in elevated levels of CDK6/cyclin
D3 complex in the epidermis of K5CDK6, compared with wild-type
littermates (Fig. 3B; Table I) (36).
Expression of CDK6 was barely visible in the epidermis of wild-type
and cyclin D3−/− mice (Fig. 3A
and B). Consistent with the elevated protein level of cyclin
D1, K5CDK6/cyclin D3−/− epidermis exhibited higher
levels of CDK6/cyclin D1 complexes compared with K5CDK6 epidermis
(Fig. 3B; Table I). In addition, epidermis from
K5CDK6/cyclin D3−/− and cyclin D3−/− mice
displayed higher levels of CDK4/cyclin D1 complex formation
compared with wild-type and K5CDK6 mice (Fig. 3C; Table
I). These results confirm that, in the absence of cyclin D3,
the compensatory increase in cyclin D1 expression leads to
increased complex formation with the two main CDKs of the G1 phase,
CDK4 and CDK6. Expression levels of CDK6, CDK4, and cyclin D1 were
quantified in protein lysates (input) and immunoprecipitated
samples from K5CDK6/cyclin D3−/− and cyclin
D3−/− mice to determine the ratio of input to
immunoprecipitation. It was established that the majority of cyclin
D1 proteins bind to CDK4 in cyclin D3−/− and
K5CDK6/cyclin D3−/− epidermis, whereas in wild-type and
K5CDK6 epidermis only 12% of cyclin D1 binds to CDK4. The
observation that CDK6 is not expressed at high levels in wild-type
and cyclin D3−/− epidermis does not allow for
verification of the ratio of cyclins to CDK6 complexes. The authors
conclude that a lack of cyclin D3 lead to changes in cyclin D1
protein level, which favors the formation of CDK4/cyclin D1 and
CDK6/cyclin D1 complexes in K5CDK6/D3−/− mice and
CDK4/cyclin D1 complexes in cyclin D3−/− mice.
Therefore, a lack of cyclin D3 is at least partially compensated by
increased cyclin D1 protein in mouse epidermis.
 | Table I.Cdk/D-type cyclin complexes in mouse
epidermis. |
Table I.
Cdk/D-type cyclin complexes in mouse
epidermis.
| Mouse genotype | CDK6/cyclin D1 | CDK6/cyclin D3 | CDK4/cyclin D1 | CDK4/cyclin D3 |
|---|
| Wild-type | + | + | + | + |
| K5CDK6 | + | +++ | + | + |
| Cyclin
D3−/− | + | − | +++ | − |
| K5CDK6/cyclin
D3-/− | +++ | − | +++ | − |
In order to define whether cyclin D3 expression is
essential for the tumor-inhibitory role of CDK6 (36), K5CDK6/cyclin D3−/−, K5CDK6,
cyclin D3−/− and wild-type littermates were subjected to
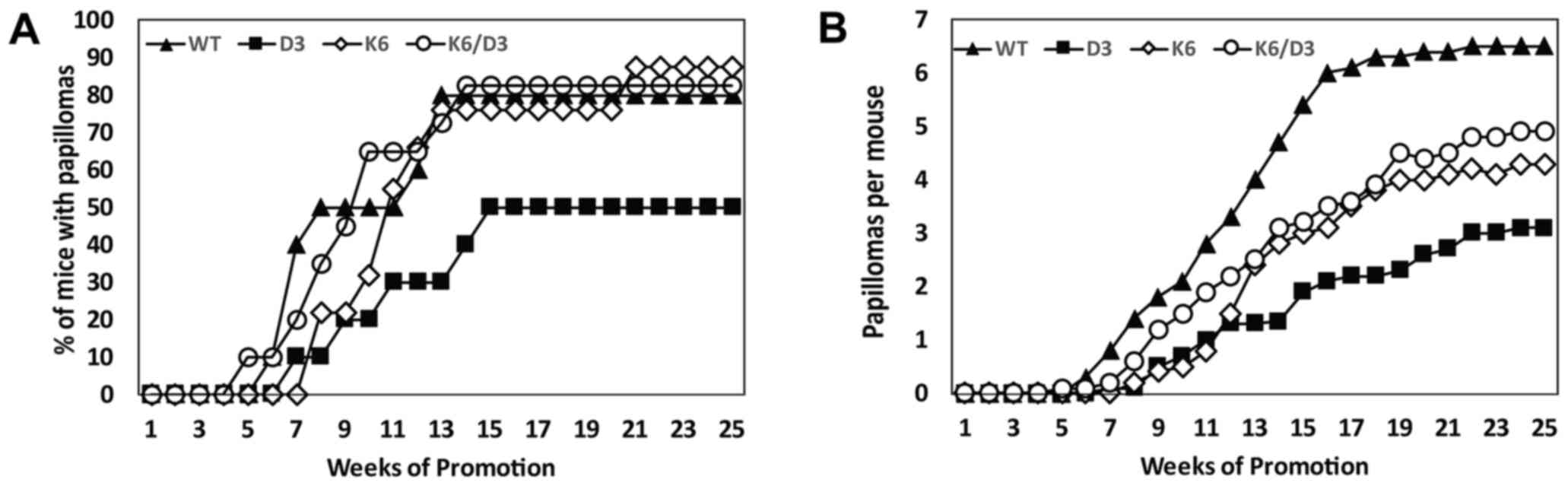
the two-stage carcinogenesis protocol for 25 weeks. The four groups
developed visible papillomas between 5–8 weeks of carcinogenic
promotion (Fig. 4A). Whereas
wild-type, K5CDK6 and K5CDK6/D3−/− mice reached 50% of
incidence by 8–11 weeks, cyclin D3−/− mice exhibited
delayed development of skin papillomas and reached 50% incidence
after 15 weeks of promotion (P=0.001, t-test), remaining at this
value until the end of the experiment. In addition, the incidence
of papilloma formation of K5CDK6, K5CDK6/D3−/− and
wild-type mice reached a plateau of ~80% at 13–14 weeks (P=0.001,
t-test; Fig. 4A). As previously
reported, K5CDK6 mice display a significantly lower number of
papillomas per mouse (multiplicity) compared with wild-type mice
(34% reduction at 25 weeks, P=0.01, t-test; Fig. 4B) (36).
In support of the findings in the present study (Fig. 2B), cyclin D3−/− mice
exhibited the strongest inhibition of papilloma multiplicity among
the four groups (52% reduction, P=0.0002, t-test; Figs. 2B and 4B). Tumor inhibition of K5CDK6/cyclin
D3−/− mice was similar to K5CDK6 mice, which displayed a
25% reduction in multiplicity compared with wild-type mice (P=0.04,
t-test; Fig. 4B). Importantly,
papilloma incidence and multiplicity between K5CDK6 and
K5CDK6/cyclin D3−/− mice showed no statistically
significant differences. Therefore, the authors confirm that
ablation of cyclin D3 causes severe papilloma repression (Figs. 2 and 4),
but the tumor inhibition driven by overexpression of CDK6 is
independent of cyclin D3 expression.
Lack of cyclin D3 reduces keratinocyte
proliferation and increases apoptosis in skin papillomas
The simultaneous overexpression of CDK6 and ablation
of cyclin D3 led to changes in the kinetics of CDK/D-type cyclin
complex formation, but did not disturb the proliferative status of
mouse keratinocytes (Fig. 3). To
investigate the biological determinants responsible for the reduced
number of papillomas observed upon variation in the levels of CDK6
and cyclin D3, the authors monitored possible alterations in
proliferation and apoptosis.
On the last day of the study, remaining animals were
injected with BrdU solution, and tumors were collected and
preserved in formalin. Paraffin-embedded tumors were immunostained
to determine the number of cells in S phase by BrdU incorporation
analysis. The proliferation status of cyclin D3−/−
keratinocytes exhibited a 0.5-fold reduction compared with
wild-type tumors (P<0.05, Bonferroni's multiple comparisons
test; Fig. 5). By contrast,
keratinocyte proliferation increased by 1.5-fold in K5CDK6 and
K5CDK6/cyclin D3−/− mouse papillomas compared with the
wild-type (P<0.05, Bonferroni's multiple comparisons test;
Fig. 5). Therefore, no positive
association was observed between the reduced papilloma development
of cyclin D3−/− and K5CDK6 mice and their proliferative
status. In order to define whether other mechanisms compensate for
the changes detected in keratinocyte proliferation, the number of
apoptotic keratinocytes in papillomas was monitored by TUNEL
labeling. Notably, papillomas from genetically modified mice
exhibited elevated apoptosis compared with wild-type tumors
(Fig. 6). Consistent with a previous
study by the authors (36),
papillomas from K5CDK6 mice displayed a 2.4-fold increase in
apoptosis compared the wild-type (P<0.05, Bonferroni's multiple
comparisons test). Likewise, papillomas from cyclin
D3−/− and K5CDK6/cyclin D3−/− mice exhibited
a 2-fold increase in the number of apoptotic keratinocytes compared
with wild-type tumors (P<0.05, Bonferroni's multiple comparisons
test; Fig. 6). In agreement with
these results, a recent study demonstrated that overexpression of
CDK6 and cyclin D1 inhibits cell differentiation and causes
increased apoptosis (51). The BrdU
index and the number of apoptotic cells in K5CDK6, cyclin
D3−/− and K5CDK6/cyclin D3−/− tumors were
normalized to the corresponding values of wild-type samples
(Figs. 5 and 6), and the rate of apoptosis/proliferation
was calculated for each genotype (A/P index). A positive
correlation was observed between the reduced number of papillomas
(multiplicity) and the increased apoptotic/proliferation index
(Pearson's test, P=0.0169; Fig. 6E,
A/P index). These results suggest that changes in the levels of
CDK/D-type cyclin complexes can serve a role altering the rate of
proliferation and apoptosis leading to modifications in the kinetic
of mouse skin tumor development.
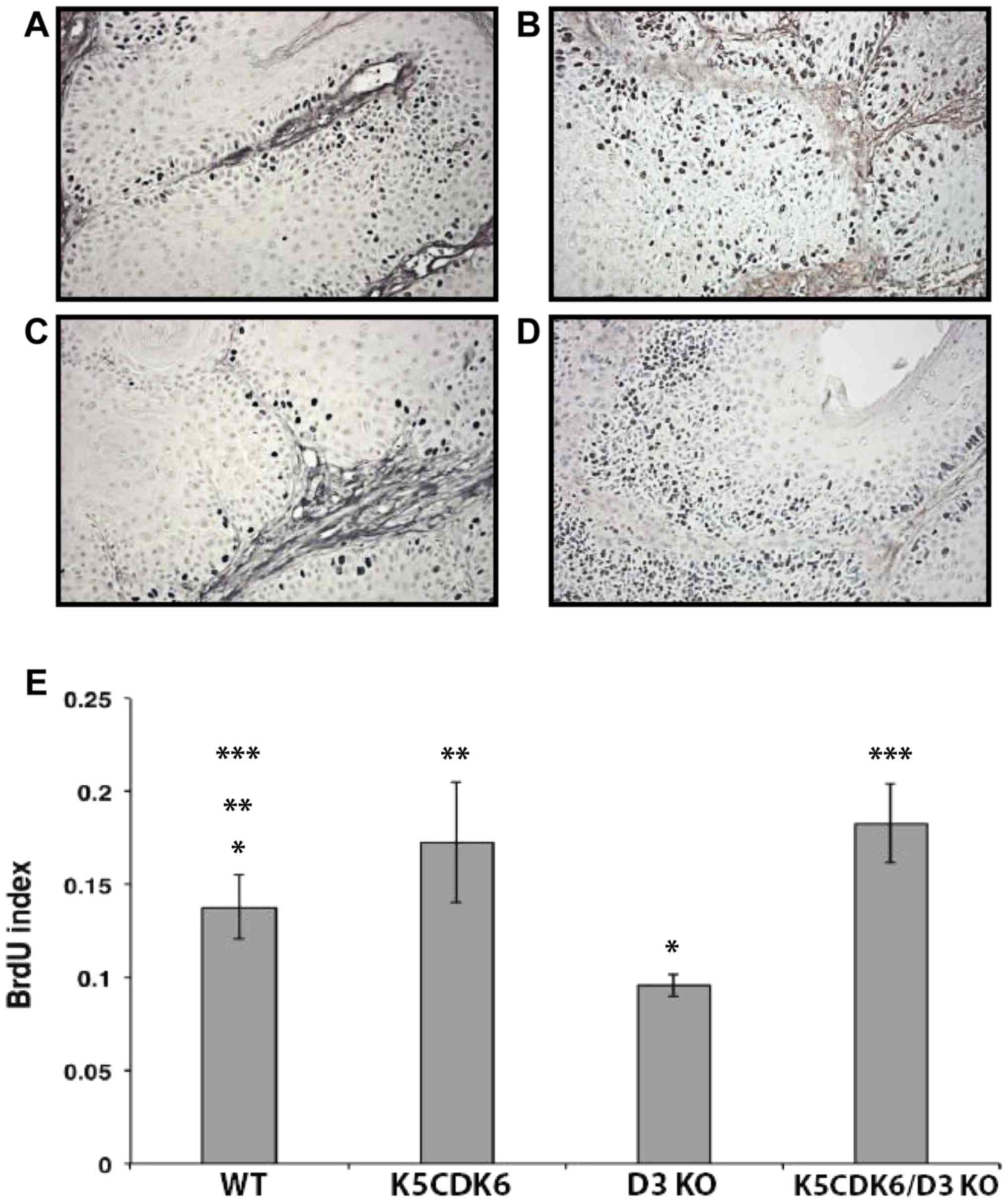 | Figure 5.Cyclin D3 ablation reduces
keratinocyte proliferation in skin papillomas. BrdU incorporation
in papillomas from (A) WT, (B) K5CDK6, (C) D3 KO, and (D) K5CDK6/D3
KO mice. Magnification, ×400. (E) Quantification of BrdU-positive
cells in skin papillomas (BrdU label index) shows reduced
proliferation in D3 KO tumors compared with WT papillomas
(*P<0.05 D3 KO vs. wild-type controls, one-way ANOVA and
Bonferroni's multiple comparisons test). Increased proliferation of
K5CDK6 and K5CDK6/D3KO tumors compared with WT papillomas
(**P<0.05, K5CDK6 vs. wild-type controls and ***P<0.05,
K5CDK6/D3KO vs. wild-type controls; one-way ANOVA and Bonferroni's
multiple comparisons test). BrdU, bromodeoxyuridine; WT, wild type;
KO, knockout. |
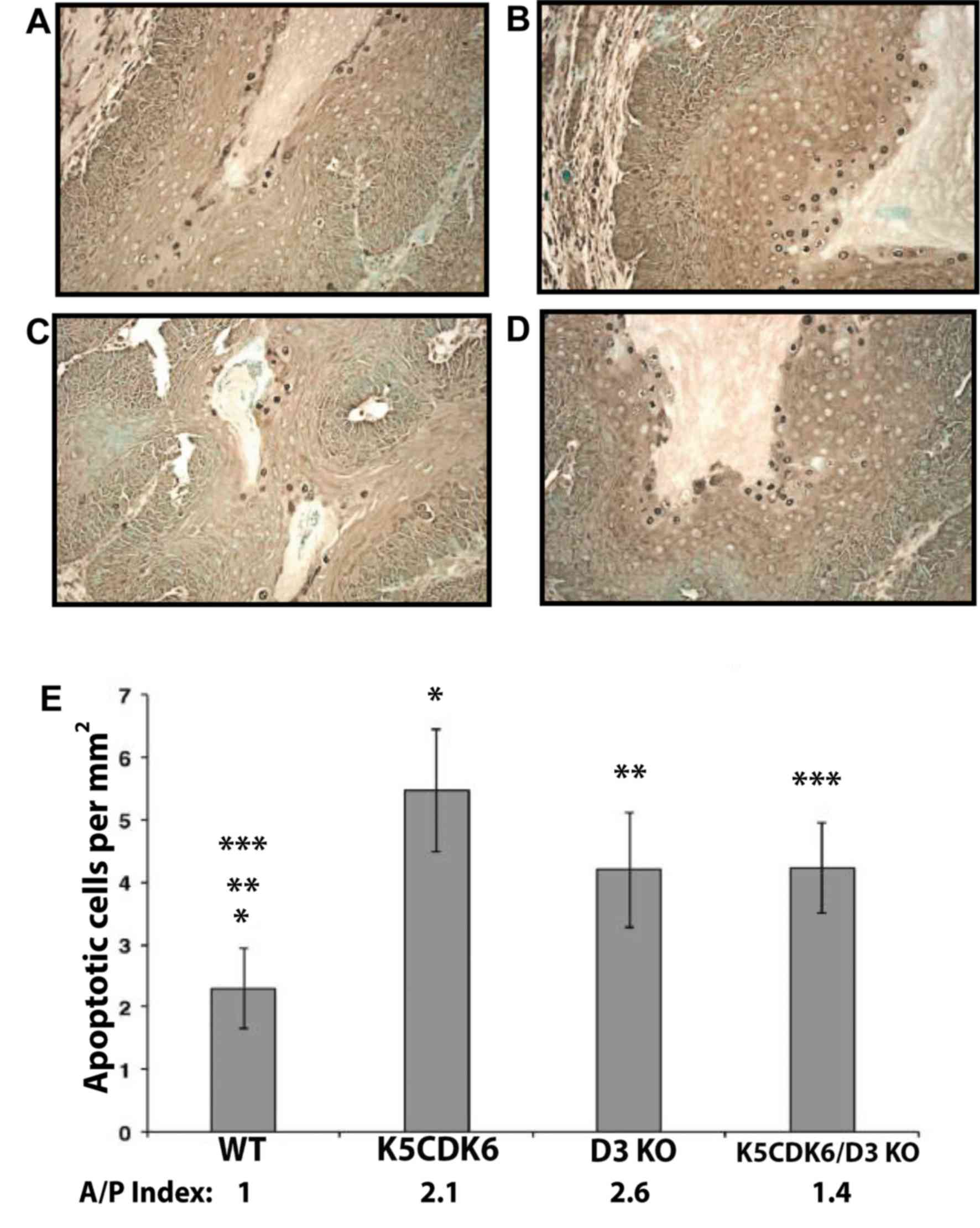 | Figure 6.Increased apoptosis in skin
papillomas from cyclin D3−/−, K5CDK6 and K5CDK6/cyclin
D3−/− mice. Apoptotic cells were visualized by terminal
deoxynucleotidyl transferase dUTP nick end labeling assay in
papillomas from (A) WT, (B) K5CDK6, (C) D3 KO, and (D) K5CDK6/D3 KO
mice. Magnification, ×400. (E) Increased apoptosis in keratinocytes
from K5CDK6, D3 KO, and K5CDK6/D3 KO tumors compared with WT
papillomas (*P<0.05, K5CDK6 vs. wild-type controls, **P<0.05,
D3 KO vs. wild-type controls, and ***P<0.05, K5CDK6/D3KO vs.
wild-type controls; one-way ANOVA and Bonferroni's multiple
comparisons test), n=10 per group. The number of apoptotic cells
and BrdU index were normalized to the wild-type samples, and the
rate of apoptosis/proliferation was calculated (A/P Index). WT,
wild type; D3 KO, cyclin D3−/− mice; K5CDK6/D3 KO,
K5CDK6/cyclin D3−/− mice. |
To determine whether modifications in CDK6 and
cyclin D3 levels and the consequent changes in the level of
CDK4/cyclin D1 complexes affect the rate of malignant progression,
histopathological analysis of skin papillomas was performed. The
dysplastic and anaplastic changes that included disturbed cell
polarity (mainly on the basal cell layer), basal cell hyperplasia,
disturbed differentiation sequence, increased number of mitosis,
mitosis in suprabasal layers, abnormal mitosis, nuclear
hyperchromatism, prominent nucleoli and increased
nuclear/cytoplasmic ratio were all taken into account. Notably, 42%
of papillomas from K5CDK6/cyclin D3−/− presented atypia
in basal and certain suprabasal layers and lack of the
differentiation pattern (intrapapilloma carcinoma and carcinoma
in situ). By contrast, 14–15% of wild-type and K5CDK6
papillomas displayed areas of intrapapilloma carcinoma, whereas all
cyclin D3−/− papillomas were regular or moderately
dysplastic (Table II). These results
suggest that the increased level of CDK4/cyclin D1 and CDK6/cyclin
D1 complexes in K5CDK6/cyclin D3−/− mice may be
associated with increased malignant progression. Supporting this
hypothesis, previous data demonstrated that increased CDK4/cyclin
D1 complexes led to increased malignant progression upon the
overexpression of CDK4 in mouse epidermis (25,33).
| Table II.Histopathological analysis of skin
tumors. |
Table II.
Histopathological analysis of skin
tumors.
|
|
| Tumor type |
|---|
|
|
|
|
|---|
| Mouse genotype | Analyzed tumors per
group, n |
Papillomaa, n (%) | SCCb, n (%) |
|---|
| Wild-type | 21 | 18 (86) | 3
(14) |
| K5CDK6 | 20 | 17 (85) | 3
(15) |
| Cyclin
D3−/− | 10 | 10
(100) | 0 (0) |
| K5CDK6/cyclin
D3-/− | 24 | 14 (58) | 10 (42) |
Discussion
Role of cyclin D3 in epidermal
proliferation
Early studies by the authors have demonstrated that
D-type cyclins are differently regulated in mouse epidermis
(22,23,27–30).
Whereas cyclin D1 was upregulated in TPA-induced proliferative
keratinocytes, cyclin D2 and cyclin D3 levels remained constant
(27). In the present report, it was
demonstrated that a lack of cyclin D3 expression does not affect
keratinocyte proliferation. Consistent with this data, cyclin D3-/−
and wild-type mice exhibited no differences in the total number of
nucleated keratinocytes in the interfollicular epidermis. Moreover,
histological analysis of skin sections revealed no changes in the
skin structure of cyclin D3-/− mice. Biochemical analysis of
epidermal lysates revealed an increased level of cyclin D1 protein
and elevated formation of CDK4/cyclin D1 complexes. Therefore,
epidermal keratinocytes proliferate normally in the absence of
cyclin D3, likely through the formation of CDK4/6-cyclin D1
complexes, as a compensatory mechanism due to the lack of
CDK4/6-cyclin D3 complexes. Although a lack of cyclin D3 expression
does not alter the apoptotic index in the interfollicular
epidermis, a relevant increase was observed in the number of
apoptotic keratinocytes in the bulge area of the hair follicles.
These results suggest that a lack of cyclin D3 expression affects
the long-lived epithelial stem cells located within the bulge
region. Fate-mapping experiments have demonstrated that these cells
do not normally contribute to epidermal homeostasis (45), which explains why a lack of cyclin D3
does not affect the normal structure of epidermis. However,
apoptosis in the bulge area of the hair follicle may affect
re-epithelialization following wounding as epithelial injury leads
to the recruitment of bulge-derived keratinocytes into the
epidermis (45). Early studies by the
authors and the results presented throughout this report
demonstrated that the molecular mechanisms leading to compensatory
changes in D-type cyclin levels respond only to modifications in
cyclin D3, but not cyclin D1 or cyclin D2 (29,50). In
fact, transgenic expression of cyclin D3 is compensated by a
reduction in levels of cyclin D2 (50), whereas a lack of cyclin D3 is
compensated by overexpression of cyclin D1 (Fig. 3). The authors conclude that changes in
the expression of cyclin D3 does not affect epidermal homeostasis,
due to compensatory mechanisms by other members of the D-type
cyclin family. However, increased levels of cyclin D1/cdk4 in
cyclin D3-/− epidermis led to hair follicle apoptosis in the bulge
area, similar to results observed in retinoblastoma-null mice
(40).
Cyclin D3 in skin tumorigenesis
The authors have previously reported that
overexpression and ablation of D-type cyclins results in varied
sensitivities to chemically induced mouse skin papillomas (29,30,52,53).
By using the two-stage carcinogenesis model, genetic evidence that
ras-mediated skin tumor development depends on signaling pathways
that act preferentially through cyclin D1 and cyclin D2 has been
provided (28,29,35).
However, the role of cyclin D3 in skin carcinogenesis has not yet
been defined. Substantial evidence has suggested that cells from
the bulge area of hair follicles have characteristics similar to
stem cells, including slow cycling, label-retaining properties and
a high proliferative capacity (54–56). In
this regard, Morris et al (47) demonstrated that bulge-area stem cells
(BuSCs) retain carcinogen-DNA adducts, which supports the idea that
these cells are targeted during chemical initiation (47,57). The
number of skin papillomas is associated with the number of
initiated cells and/or other early events, including the clonal
expansion of initiated cells. It was observed that cyclin D3
deletion and CDK6 overexpression (36) resulted in an increased number of hair
follicles bearing apoptotic cells in the bulge region. Therefore,
the authors hypothesized that cyclin D3 ablation leads to a reduced
number of initiated cells and decreased skin papilloma development.
To test this hypothesis, the two-stage carcinogenesis protocol was
used, which allowed for study of the effect of cyclin D3 ablation
during tumor initiation (an irreversible genetic alteration in a
target cell), promotion (the process by which an initiated tissue
develops focal proliferation leading to clonogenic expansion of the
initiated cells), and malignant progression to SCC (the final
transition to invasive behavior) (58). Supporting the hypothesis, a relevant
reduction in multiplicity and incidence of papillomas was observed
in cyclin D3-null mice compared with wild-type mice. Furthermore,
50% of cyclin D3-/− mice remained refractory to papilloma
development (Figs. 2 and 4). These results provide genetic evidence
that a lack of cyclin D3 expression and a further increase in
apoptosis in the bulge region of the hair follicle, affects the
number of initiated cells or, alternatively, the clonal expansion
of the initiated cells. Similarly, ablation of cyclin D1, cyclin
D2, CDK4 and Skp2 led to a reduced number of papillomas due to
failure during the initiation or early promotion stages (28,29,32–34,36).
Likewise, overexpression of CDK6 resulted in increased apoptosis in
the bulge region of the hair follicle and inhibition of papilloma
development (36).
The fact that cyclin D3 preferentially binds to CDK6
in mouse keratinocytes (27,36) led to the hypothesis that the
inhibitory action of CDK6 depends on cyclin D3. To test this
hypothesis, K5CK6/cyclin D3−/− compound mice were
generated and subjected to a two-stage carcinogenesis protocol.
Contrary to the hypothesis, the inhibition of papilloma formation
driven by CDK6 expression and a lack of cyclin D3 did not act in a
linear fashion, since a lack of cyclin D3 expression did not affect
the tumor-repressing activity of CDK6. Cyclin D3 ablation resulted
in the most substantial tumor inhibition, when compared with K5CDK6
and K5CDK6/cyclin D3−/− mice. The authors speculate that
forced expression of CDK6 and increased formation of CDK6/cyclin D1
and CDK6/cyclin D3 complexes played a positive role during the
clonal expansion of initiated cells in K5CDK6 and
K5CDK6/D3−/− mice, but not in cyclin D3−/−
mice. Supporting this view, CDK6/cyclin D3 and CDK6/cyclin D1
showed a faster kinetic of complex formation upon TPA treatment on
mouse epidermis, compared with other D-type cyclin/CDKs complexes
(27). The molecular mechanisms by
which alterations in the kinetics of CDK/D-type cyclin complex
formation affects the clonogenic expansion of the initiated cells
remains unclear and is beyond the scope of the present study.
Similar to the cyclin D3−/− model, ablation of Rb, the
primary substrate of CDK4 and CDK6, in the mouse epidermis also
resulted in increased apoptosis and a reduction in the number of
papillomas (40). Altogether, the
results of the present study and from the epidermal-specific
Rb−/− mice (40) support a
model in which the phosphorylation/inactivation of Rb by elevated
CDK activity leads to apoptosis in BuSCs with the consequent
reduction in the number of skin papillomas. The likelihood that
CDKs and/or D-type cyclins exert their tumor inhibitory effect
through Rb-independent mechanisms cannot be ruled out. In this
regard, it has been reported that CDK6 is part of a transcription
complex that induces the expression of the tumor suppressor
p16Ink4 (59). Whether
this kinase-independent function of CDK6 is active in keratinocytes
and BuSCs is unknown. However, the authors hypothesize that
downregulation of D-type cyclins results in elevation of the
free-CDK6 and its transcription activity, leading to the blockage
of the expansion of initiated cells via p16Ink4a
transcription. In the last 5 years, a number of non-canonical
functions of D-type cyclins as transcription factors have been
described (60,61). For instance, the transcription factor
Runx1 is negatively regulated by cyclin D3 in a kinase-independent
manner (62). Runx1 promotes the
proliferation of hair follicle stem cells and induces epithelial
tumor formation in mouse skin (63).
Therefore, a lack of and/or overexpression of cyclin D3 may
unbalance the apoptotic/proliferative rate in BuSCs, with a direct
impact on the rate of tumor formation.
Histopathological analysis of skin tumors indicated
that simultaneous overexpression of CDK6 and a lack of cyclin D3
expression resulted in an increased rate of malignant progression.
The K5CDK6/D3−/− mice developed more aggressive tumors
and 42% were classified as premalignant papillomas or SCC, whereas
only 15% of K5CDK6 and 14% of wild-type tumors exhibited similar
characteristics (Table II). Cyclin
D3−/− tumors were classified as regular papillomas with
no atypia in the basal layers and no invasion of epidermis cells
into the dermis. Biochemical analysis of epidermal lysates from
K5CDK6/cyclin D3−/− mice revealed elevated levels of
CDK6/cyclin D1 and CDK4/cyclin D1 complexes, whereas cyclin
D3−/− epidermis only displayed elevated levels of
CDK4/cyclin D1 complexes (Table I).
Similarly, it has been previously reported that increased CDK4
expression in K5CDK4 transgenic mice induces malignant progression
of skin papillomas (25,44). Notably, the malignant progression
observed in K5CDK4 mice was partially mediated via
p27Kip1 sequestration by the CDK/cyclin complexes and
the indirect activation of CDK2 (25,44).
Therefore, data of the present study suggest that the simultaneous
increase in the level of CDK4/cyclin D1 and CDK6/cyclin D1
complexes observed in K5CDK6/cyclin D3−/− mice may
result in CDK2 activation, leading to increased malignant
progression.
Collectively, the results of the present study
indicate that the absence of cyclin D3 leads to decreased papilloma
formation in mouse epidermis due to increase apoptosis in the bulge
region of the hair follicle. These observations indicate that
regulation of cell survival of BuSC is a crucial mechanism for
crippling cellular defense against neoplasia. The data of the
present study suggest that cyclin D3 inhibition blocks an early
event of the multistage process of non-melanoma skin tumorigenesis.
Although cyclin D3 binds preferentially to CDK6 (27), the tumor inhibition mediated by CDK6
is independent of cyclin D3. Finally, the present study indicates
that the unbalanced formation of CDK4/6-D-type cyclin complexes
leads to reduced papilloma development and may result in a more
aggressive tumor phenotype, increasing malignant progression.
Acknowledgements
The authors thank Ms. Rima Majumdar (North Carolina
State University, College of Veterinary Medicine, Raleigh, NC, USA)
for her technical support, Dr Piotr Sicinski and Dr Ewa Sicinska
(Harvard Medical School, Department of Genetics, Boston, MA, USA)
for providing the cyclin D3-null mice, the Laboratory Animal
Resources and the Histology Core at the College of Veterinary
Medicine, North Carolina State University for aiding with the
processing and staining of skin and tumor samples. The present
study was supported by NIEHS (award no. P30ES025128) Center for
Human Health and the Environment.
References
|
1
|
Sherr CJ: D-type cyclins. Trends Biochem
Sci. 20:187–190. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weinberg RA: The retinoblastoma protein
and cell cycle control. Cell. 81:323–330. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Farkas T, Hansen K, Holm K, Lukas J and
Bartek J: Distinct phosphorylation events regulate p130- and
p107-mediated repression of E2F-4. J Biol Chem. 277:26741–26752.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leng X, Noble M, Adams PD, Qin J and
Harper JW: Reversal of growth suppression by p107 via direct
phosphorylation by cyclin D1/cyclin-dependent kinase 4. Mol Cell
Biol. 22:2242–2254. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meyerson M and Harlow E: Identification of
G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell
Biol. 14:2077–2086. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Blain SW, Montalvo E and Massague J:
Differential interaction of the cyclin-dependent kinase (Cdk)
inhibitor p27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J Biol
Chem. 272:25863–25872. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sherr CJ and McCormick F: The RB and p53
pathways in cancer. Cancer Cell. 2:103–112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ciemerych MA, Kenney AM, Sicinska E,
Kalaszczynska I, Bronson RT, Rowitch DH, Gardner H and Sicinski P:
Development of mice expressing a single D-type cyclin. Genes Dev.
16:3277–3289. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cheung TH, Yu MM, Lo KW, Yim SF, Chung TK
and Wong YF: Alteration of cyclin D1 and CDK4 gene in carcinoma of
uterine cervix. Cancer Lett. 166:199–206. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dickson C, Fantl V, Gillett C, Brookes S,
Bartek J, Smith R, Fisher C, Barnes D and Peters G: Amplification
of chromosome band 11q13 and a role for cyclin D1 in human breast
cancer. Cancer Lett. 90:43–50. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fujii M, Ishiguro R, Yamashita T and
Tashiro M: Cyclin D1 amplification correlates with early recurrence
of squamous cell carcinoma of the tongue. Cancer Lett. 172:187–192.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Houldsworth J, Reuter V, Bosl GJ and
Chaganti RS: Aberrant expression of cyclin D2 is an early event in
human male germ cell tumorigenesis. Cell Growth Differ. 8:293–299.
1997.PubMed/NCBI
|
|
14
|
Sicinski P, Donaher JL, Geng Y, Parker SB,
Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers
JD, et al: Cyclin D2 is an FSH-responsive gene involved in gonadal
cell proliferation and oncogenesis. Nature. 384:470–474. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Delmer A, Ajchenbaum-Cymbalista F, Tang R,
Ramond S, Faussat AM, Marie JP and Zittoun R: Overexpression of
cyclin D2 in chronic B-cell malignancies. Blood. 85:2870–2876.
1995.PubMed/NCBI
|
|
16
|
Hedberg Y, Roos G, Ljungberg B and
Landberg G: Cyclin D3 protein content in human renal cell carcinoma
in relation to cyclin D1 and clinico-pathological parameters. Acta
Oncol. 41:175–181. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ito Y, Takeda T, Wakasa K, Tsujimoto M and
Matsuura N: Expression and possible role of cyclin D3 in human
pancreatic adenocarcinoma. Anticancer Res. 21:1043–1048.
2001.PubMed/NCBI
|
|
18
|
Pruneri G, Pignataro L, Valentini S,
Fabris S, Maisonneuve P, Carboni N, Pece S, Capra M, Del Curto B,
Neri A and Viale G: Cyclin D3 immunoreactivity is an independent
predictor of survival in laryngeal squamous cell carcinoma. Clin
Cancer Res. 11:242–248. 2005.PubMed/NCBI
|
|
19
|
Wong SC, Chan JK, Lee KC and Hsiao WL:
Differential expression of p16/p21/p27 and cyclin D1/D3, and their
relationships to cell proliferation, apoptosis, and tumour
progression in invasive ductal carcinoma of the breast. J Pathol.
194:35–42. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Filipits M, Jaeger U, Pohl G, Stranzl T,
Simonitsch I, Kaider A, Skrabs C and Pirker R: Cyclin D3 is a
predictive and prognostic factor in diffuse large B-cell lymphoma.
Clin Cancer Res. 8:729–733. 2002.PubMed/NCBI
|
|
21
|
Sicinska E, Aifantis I, Le Cam L, Swat W,
Borowski C, Yu Q, Ferrando AA, Levin SD, Geng Y, von Boehmer H and
Sicinski P: Requirement for cyclin D3 in lymphocyte development and
T cell leukemias. Cancer Cell. 4:451–461. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rodriguez-Puebla ML, LaCava M and Conti C:
Cyclin d1 overexpression in mouse epidermis increases
cyclin-dependent kinase activity and cell proliferation in vivo but
does not affect skin tumor development. Cell Growth Differ.
10:467–472. 1999.PubMed/NCBI
|
|
23
|
Rodriguez-Puebla ML, LaCava M,
Gimenez-Conti IB, Jonhson DG and Conti CJ: Deregulated expression
of cell-cycle proteins during premalignant progression in SENCAR
mouse skin. Oncogene. 17:2251–2258. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rodriguez-Puebla ML, de Miliani Marval PL,
LaCava M, Moons DS, Kiyokawa H and Conti CJ: Cdk4 deficiency
inhibits skin tumor development but does not affect keratinocyte
proliferation. Am J Pathol. 161:405–411. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
de Miliani Marval PL, Macias E, Conti CJ
and Rodriguez-Puebla ML: Enhanced malignant tumorigenesis in Cdk4
transgenic mice. Oncogene. 23:1863–1873. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang SY, Liu SC, Goodrow T, Morris R and
Klein-Szanto AJ: Increased expression of G1 cyclins and
cyclin-dependent kinases during tumor progression of chemically
induced mouse skin neoplasms. Mol Carcinog. 18:142–152. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rodriguez-Puebla ML, Robles AI, Johnson
DG, LaCava M and Conti CJ: Synchronized proliferation induced by
12-O-tetradecanoylphorbol-13-acetate treatment of mouse skin: An in
vivo model for cell cycle regulation. Cell Growth Differ. 9:31–39.
1998.PubMed/NCBI
|
|
28
|
Robles AI, Rodriguez-Puebla ML, Glick AB,
Trempus C, Hansen L, Sicinski P, Tennant RW, Weinberg RA, Yuspa SH
and Conti CJ: Reduced skin tumor development in cyclin D1-deficient
mice highlights the oncogenic ras pathway in vivo. Genes Dev.
12:2469–2474. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rojas P, Cadenas MB, Lin PC, Benavides F,
Conti CJ and Rodriguez-Puebla ML: Cyclin D2 and cyclin D3 play
opposite roles in mouse skin carcinogenesis. Oncogene.
26:1723–1730. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rodriguez-Puebla ML, LaCava M and Conti
CJ: Cyclin D1 overexpression in mouse epidermis increases
cyclin-dependent kinase activity and cell proliferation in vivo but
does not affect skin tumor development. Cell Growth Differ.
10:467–472. 1999.PubMed/NCBI
|
|
31
|
Rodriguez-Puebla ML, Robles AI and Conti
CJ: ras activity and cyclin D1 expression: An essential mechanism
of mouse skin tumor development. Mol Carcinog. 24:1–6. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rodriguez-Puebla ML, de Miliani Marval PL,
LaCava M, Moons DS, Kiyokawa H and Conti CJ: Cdk4 deficiency
inhibits skin tumor development but does not affect normal
keratinocyte proliferation. Am J Pathol. 161:405–411. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Macias E, Kim Y, de Miliani Marval PL,
Klein-Szanto A and Rodriguez-Puebla ML: Cdk2 deficiency decreases
ras/CDK4-dependent malignant progression, but not myc-induced
tumorigenesis. Cancer Res. 67:9713–9720. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sistrunk C, Kim SH, Wang X, Lee SH, Kim Y,
Macias E and Rodriguez-Puebla ML: Skp2 deficiency inhibits chemical
skin tumorigenesis independent of p27(Kip1) accumulation. Am J
Pathol. 182:1854–1864. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang X, Sistrunk C, de Miliani Marval PL,
Kim Y and Rodriguez-Puebla ML: Combined effect of cyclin D3
expression and abrogation of cyclin D1 prevent mouse skin tumor
development. Cell Cycle. 11:335–342. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang X, Sistrunk C and Rodriguez-Puebla
ML: Unexpected reduction of skin tumorigenesis on expression of
cyclin-dependent kinase 6 in mouse epidermis. Am J Pathol.
178:345–354. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Philipp J, Vo K, Gurley KE, Seidel K and
Kemp CJ: Tumor suppression by p27Kip1 and p21Cip1 during chemically
induced skin carcinogenesis. Oncogene. 18:4689–4698. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kemp CJ, Donehower LA, Bradley A and
Balmain A: Reduction of p53 gene dosage does not increase
initiation or promotion but enhances malignant progression of
chemically induced skin tumors. Cell. 74:813–822. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ise K, Nakamura K, Nakao K, Shimizu S,
Harada H, Ichise T, Miyoshi J, Gondo Y, Ishikawa T, Aiba A and
Katsuki M: Targeted deletion of the H-ras gene decreases tumor
formation in mouse skin carcinogenesis. Oncogene. 19:2951–2956.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ruiz S, Santos M, Lara MF, Segrelles C,
Ballestín C and Paramio JM: Unexpected roles for pRb in mouse skin
carcinogenesis. Cancer Res. 65:9678–9686. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ruiz S, Santos M and Paramio JM: Is the
loss of pRb essential for the mouse skin carcinogenesis? Cell
Cycle. 5:625–629. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rojas P, Benavides F, Blando J, Perez C,
Cardenas K, Richie E, Knudsen ES, Johnson DG, Senderowicz AM,
Rodriguez-Puebla ML and Conti CJ: Enhanced skin carcinogenesis and
lack of thymus hyperplasia in transgenic mice expressing human
cyclin D1b (CCND1b). Mol Carcinog. 48:508–516. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rounbehler RJ, Schneider-Broussard R,
Conti CJ and Johnson DG: Myc lacks E2F1′s ability to suppress skin
carcinogenesis. Oncogene. 20:5341–5349. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Macias E, de Miliani Marval PL, De Siervi
A, Conti CJ, Senderowicz AM and Rodriguez-Puebla ML: CDK2
activation in mouse epidermis induces keratinocyte proliferation
but does not affect skin tumor development. Am J Pathol.
173:526–535. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ito M, Liu Y, Yang Z, Nguyen J, Liang F,
Morris RJ and Cotsarelis G: Stem cells in the hair follicle bulge
contribute to wound repair but not to homeostasis of the epidermis.
Nat Med. 11:1351–1354. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang P, Pavletic ZS and Joshi SS:
Increased apoptosis in B-chronic lymphocytic leukemia cells as a
result of cyclin D3 down regulation. Leuk Lymphoma. 43:1827–1835.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Morris RJ, Fischer SM and Slaga TJ:
Evidence that a slowly cycling subpopulation of adult murine
epidermal cells retains carcinogen. Cancer Res. 46:3061–3066.
1986.PubMed/NCBI
|
|
48
|
Lapouge G, Youssef KK, Vokaer B, Achouri
Y, Michaux C, Sotiropoulou PA and Blanpain C: Identifying the
cellular origin of squamous skin tumors. Proc Natl Acad Sci USA.
108:7431–7436. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rodriguez-Puebla ML, LaCava M,
Gimenez-Conti IB, Johnson DG and Conti CJ: Deregulated expression
of cell-cycle proteins during premalignant progression in SENCAR
mouse skin. Oncogene. 17:2251–2258. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rodriguez-Puebla ML, LaCava M, De Miliani
Marval PL, Jorcano JL, Richie ER and Conti CJ: Cyclin D2
overexpression in transgenic mice induces thymic and epidermal
hyperplasia whereas cyclin D3 expression results only in epidermal
hyperplasia. Am J Pathol. 157:1039–1050. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ito K, Maruyama Z, Sakai A, Izumi S,
Moriishi T, Yoshida CA, Miyazaki T, Komori H, Takada K, Kawaguchi H
and Komori T: Overexpression of Cdk6 and Ccnd1 in chondrocytes
inhibited chondrocyte maturation and caused p53-dependent apoptosis
without enhancing proliferation. Oncogene. 33:1862–1871. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yamamoto H, Ochiya T, Takeshita F,
Toriyama-Baba H, Hirai K, Sasaki H, Sasaki H, Sakamoto H, Yoshida
T, Saito I and Terada M: Enhanced skin carcinogenesis in cyclin
D1-conditional transgenic mice: Cyclin D1 alters keratinocyte
response to calcium-induced terminal differentiation. Cancer Res.
62:1641–1647. 2002.PubMed/NCBI
|
|
53
|
Robles AI, Larcher F, Whalin RB, Murillas
R, Richie E, Gimenez-Conti IB, Jorcano JL and Conti CJ: Expression
of Cyclin D1 in epithelial tissues of transgenic mice results in
epidermal hyperproliferation and severe thymic hyperplasia. Proc
Natl Acad Sci USA. 93:7634–7638. 1996. View Article : Google Scholar
|
|
54
|
Morris RJ, Tryson KA and Wu KQ: Evidence
that the epidermal targets of carcinogen action are found in the
interfollicular epidermis of infundibulum as well as in the hair
follicles. Cancer Res. 60:226–229. 2000.PubMed/NCBI
|
|
55
|
Morris RJ, Liu Y, Marles L, Yang Z,
Trempus C, Li S, Lin JS, Sawicki JA and Cotsarelis G: Capturing and
profiling adult hair follicle stem cells. Nat Biotechnol.
22:411–417. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
56
|
Morris RJ: A perspective on keratinocyte
stem cells as targets for skin carcinogenesis. Differentiation.
72:381–386. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kangsamaksin T, Park HJ, Trempus CS and
Morris RJ: A perspective on murine keratinocyte stem cells as
targets of chemically induced skin cancer. Mol Carcinog.
46:579–584. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Slaga T: Mechanism involved in two-stage
carcinogenesis in mouse skinMechanism of Tumor Promotion. Slaga T:
CRC Press; Boca Raton: pp. 1–16. 1984
|
|
59
|
Kollmann K, Heller G, Schneckenleithner C,
Warsch W, Scheicher R, Ott RG, Schäfer M, Fajmann S, Schlederer M,
Schiefer AI, et al: A Kinase-Independent Function of CDK6 Links the
Cell Cycle to Tumor Angiogenesis. Cancer Cell. 24:167–181. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hydbring P, Malumbres M and Sicinski P:
Non-canonical functions of cell cycle cyclins and cyclin-dependent
kinases. Nat Rev Mol Cell Biol. 17:280–292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Pestell RG: New roles of cyclin D1. Am J
Pathol. 183:3–9. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Peterson LF, Boyapati A, Ranganathan V,
Iwama A, Tenen DG, Tsai S and Zhang DE: The hematopoietic
transcription factor AML1 (RUNX1) is negatively regulated by the
cell cycle protein cyclin D3. Mol Cell Biol. 25:10205–10219. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hoi CS, Lee SE, Lu SY, McDermitt DJ,
Osorio KM, Piskun CM, Peters RM, Paus R and Tumbar T: Runx1
directly promotes proliferation of hair follicle stem cells and
epithelial tumor formation in mouse skin. Mol Cell Biol.
30:2518–2536. 2010. View Article : Google Scholar : PubMed/NCBI
|