Introduction
Lung cancer remains the leading cause of
cancer-associated mortality worldwide, due to poor prognosis, high
resistance to therapy and a low survival rate (1). Human non-small cell lung cancer (NSCLC)
constitutes ≥80% of patients with lung cancer and includes
adenocarcinoma, squamous cell carcinoma and large-cell carcinoma
(2). Despite the abundance of
available chemotherapeutic approaches, current treatments often
lead to severe resistance and side effects (3). Therefore, it is necessary to develop a
novel and alternative small molecule drug to provide effective
chemotherapy with good efficacy and low toxicity.
Compounds composed of epoxy groups have become an
attractive target for chemotherapy development (4). Previously, a triepoxide derivative
called teroxirone, proved an effective treatment for patients
recovering from leukemia and lymphoma (4,5).
Furthermore, a previous study has demonstrated that the tumor
suppressor p53 regulates teroxirone-induced apoptosis in human
NSCLC cells by damaging cellular DNA (6). Despite the distinctive inhibitory
effects demonstrated in cell and animal models, the targets and
underlying mechanisms of teroxirone, which lead to p53 activation
and final cell growth inhibition, are not yet understood (6). As a regulatory mediator, apoptosis
serves to eliminate damaged cells without injuring the surrounding
cells (7). Thus, to further
understand the detailed mechanisms regarding the onset of
apoptosis, the present study assessed mitochondrial functions
during p53-dependent apoptosis. The results of the present study
demonstrated that teroxirone activated reactive oxygen species
(ROS), and that mitochondrial function was impaired as a result of
teroxirone treatment, which contributed to cytotoxic effects.
Pretreatment of ROS scavengers recovered cell viability by
restoring mitochondrial function, reducing DNA damage and
attenuating apoptotic characteristics. Thus, the efficacy of
teroxirone by apoptotic cell death depended on the generated ROS
following the disruption of the membrane potential cascade. The
present study also provided further information on the apoptotic
mechanisms that may lead to a novel perspective of triepoxides.
Materials and methods
Cell culture
H460 (HTB177™), H1299 (CRL5803™) and A549 (CCL185™)
human NSCLC cells were purchased from the American Type Culture
Collection (Manassas, VA, USA) and maintained in Dulbecco's
modified Eagle's medium (DMEM; Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany). All cultured cells were supplemented with
L-glutamine, sodium pyruvate and cultured with 10% heat-inactivated
fetal bovine serum (FBS; Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) in 5% CO2 at 37°C. All cell
lines were periodically examined using a MycoTect™ kit (Invitrogen;
Thermo Fisher Scientific, Inc.), to ensure the absence of
mycoplasma contamination.
Chemicals
Dichlorodihydrofluorescein diacetate (DCFH-DA),
N-acetylcysteine (NAC), dimethyl sulfoxide (DMSO) and MTT were
purchased from Sigma-Aldrich (Merck Millipore). The stock solutions
of 10 mM DCFH-DA and NAC as dissolved in DMSO, were passed through
a filter with a 0.22 µm pore size (Immobilon; EMD Millipore,
Billerica, MA, USA). The synthetic teroxirone, as used previously
(6), has a purity of >98%. A stock
solution of 10 mM DMSO was stored at −20°C and freshly dissolved in
media prior to use. The penicillin and streptomycin antibiotic
mixture, sodium pyruvate and the L-glutamine supplements were
supplied by Sigma-Aldrich (Merck Millipore).
Cell viability determination
Cell viability was determined using the trypan blue
dye exclusion-staining assay. Briefly, 3×105 cells/well were seeded
on a 6-cm dish in DMEM supplemented with 2% FBS. The suspended
cells were incubated at 37°C for 24 h to allow for attachment.
Following the indicated duration of exposure time teroxirone, the
media was removed and trypsin-EDTA was added in order to suspend
the adherent cells. The numbers of cells stained with 0.4% trypan
blue were presented as the mean ± standard deviation. The
experiments were repeated independently ≥3 times, revealing similar
results.
Detection of mitochondrial membrane
potential (MMP)
Human H460 and A549 NSCLC cells were plated at a
density of 3×105 in 6-well plates, allowed to attach overnight and
pre-treated with NAC (10 µM) for 2 h at 37°C, prior to being
treated with dimethyl sulfoxide (vehicle control), 2 or 5 µM
teroxirone for 12 h at 37°C. The treated cells were reacted with 2
µM JC-1 for 15 min at 37°C in a 5% CO2-supplemented
incubator, prior to being washed with PBS and analyzed by flow
cytometry (FACSCalibur™; BD Biosciences, Franklin Lakes, NJ, USA)
using a 488-nm excitation and data collected at 585 nm wavelength
emissions.
Detection of intracellular ROS
production
H460 and A549 cells were plated at 3×105 cells/well,
attached overnight and treated with 2 or 5 µM teroxirone at 37°C,
followed by staining with 10 µM DCFH-DA for 30 min. The formation
of fluorescent-oxidized DCF was monitored using a FACSCalibur™ flow
cytometer (excitation at 485 nm, emission at 535 nm). The generated
ROS were quantified by evaluating the fluorescence intensity of
10,000 cells using ImageJ software (version 1.45; National
Institutes of Health, Bethesda, MD, USA).
Release of cytochrome c
Following the induced release of cytochrome c
from mitochondria in H460 and A549 cells by treatment with 2 or 5
µM teroxirone, the cells were fixed with 4% formaldehyde,
permeabilized and stained with an anti-cytochrome c
monoclonal antibody (dilution, 1:200; catalog no. 556432; BD
Pharmingen; BD Biosciences) at 4°C for 18 h. Subsequent to washing
with PBS, cells were stained with 10 mM Mitotracker Green
(mitochondrial staining; Invitrogen; Thermo Fisher Scientific,
Inc.) for 30 min at room temperature, and a secondary antibody
conjugated with tetramethylrhodamine (dilution, 1:500; catalog no.
T2402, Sigma-Aldrich; Merck KGaA) for cytochrome c for 48 h
at 4°C. The slides were counter-stained with 1:2,000 DAPI
(Sigma-Aldrich; Merck KGaA) at room temperature for 15 min. The
release of cytochrome c punctae in cells was quantified
using the ImageJ software (version 1.45; National Institutes of
Health).
Western blot analysis
Cells treated with teroxirone were washed with PBS
and scraped in a lysate buffer substituted with 1% Triton X-100,
150 mM NaCl, 5 mM EDTA, 1% aprotonin, 5 mM phenylmethylsulfonyl
fluoride and 10 µg/ml leupeptin as dissolved in 20 mM sodium
phosphate buffer. The protein concentrations were determined by a
bicinchoninic acid assay (Pierce; Thermo Fisher Scientific, Inc.)
and used for western blot analysis. Protein lysates were separated
by 10% SDS-PAGE gels and transferred onto nitrocellulose membranes.
The blots were blocked for 1 h with 1% skimmed dried milk in
Tris-buffered saline (pH 7.6). All antibodies, including secondary
antibodies, were used at a 1:2,000 dilution. The primary antibodies
used included anti-p53 (catalog no. sc-6243), anti-B-cell lymphoma
(Bcl)-2-associated X-protein (Bax; catalog no. sc-0526) (both from
Santa Cruz Biotechnology, Dallas, TX, USA), anti-caspase-3 (catalog
no. 19677; Proteintech Rosemont, IL, USA), anti-phosphorylated
protein kinase B (Akt; catalog no. GTX128414); and anti-Akt
(catalog no. GTX121937), anti-poly (ADP-ribose) polymerase (PARP;
catalog no. GTX112864), anti-Bcl-2 (catalog no. GTX100064), and
anti-cytochrome c (catalog no. GTX108585) (all from GeneTex,
Irvine, CA, USA). Membranes were then incubated with horseradish
peroxidase-conjugated anti-mouse (dilution, 1:3,000; catalog no.
F5393) or anti-rabbit IgG (dilution; 1:3,000; catalog no. F0382)
(both from Sigma-Aldrich; Merck KGaA) for 1 h at room temperature.
Control of protein loading was obtained by probing with an
anti-GAPDH antibody (catalog no. GTX100118; GeneTex). The blots
were visualized using an enhanced chemiluminescence system (GE
Healthcare Life Sciences, Chalfont, UK).
Flow cytometry and cell cycle
analysis
A total of 1×105 cells were plated in 12-well
plates. For sample preparation, cells were collected, were washed
twice with PBS and subsequently preserved with 70% alcohol
supplemented with PBS, for 24 h at −20°C. Immediately prior to
analysis, the sample cells were treated with 10 µg/ml propidium
iodide (PI; Sigma-Aldrich; Merck Millipore), 10 µg/ml RNase A (ICN
Pharmaceuticals, Inc., Costa Mesa, CA, USA) and substituted with
PBS, for 30 min in the dark. Data was analyzed by ModFit LT
software (version 2.0; BD Biosciences).
Statistical analysis
The data are expressed as the mean ± standard
deviation. Statistical differences between two groups were analyzed
using one-way analysis of variance and Fisher's least significant
difference test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Teroxirone induces a decrease in MMP
and generates ROS in NSCLC cells
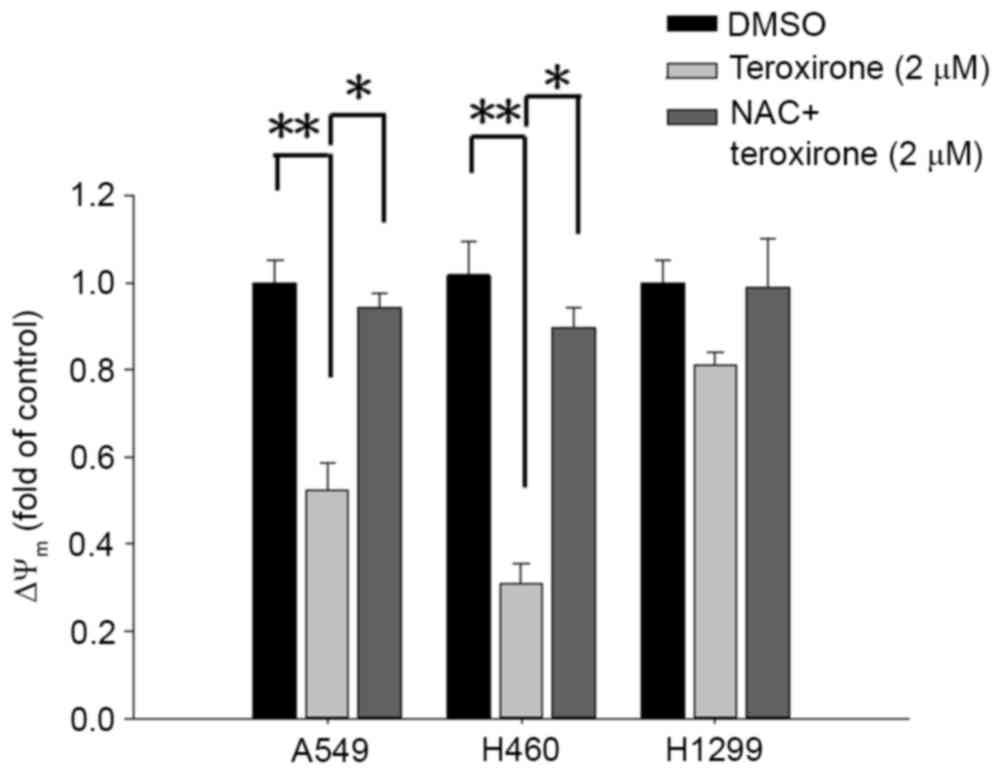
MMP variations were evaluated by incorporating the
cells with the voltage-sensitive dye JC-1. The dye aggregates when
polarized at high transmembrane potentials emit red fluorescence at
585 nm. The depolarized monomers release green fluorescence at 530
nm as measured by flow cytometry. Treatment with low concentrations
of teroxirone resulted in an MMP drop in A549 and H460 cells. The
detection of JC-1 fluorescence at higher wavelengths suggests that
the induced apoptosis began with disruption of the membrane
potential following a 12-h treatment in A549 and H460 cells,
whereas H1299 cells were unaffected. Pretreatment of cells with NAC
enables cells to recover from membrane potential collapse caused by
teroxirone (Fig. 1).
Due to its close association with the leakage of
electron transport during MMP decrease, ROS generation was a
reasonable selection for investigation in the present study. The
stained membrane-impermeable probe DCFH-DA, which was converted
into the fluorescent polar derivative DCF, indicated an increase in
the level of ROS during flow cytometry analysis. Following MMP
disruption, intracellular ROS were identified 18 h following
treatment with low concentrations of teroxirone in H460 and A549
cells. Subsequent to a 1-h pretreatment with 10 µM antioxidant NAC,
the emitted fluorescence was attenuated that signified the loss of
ROS in A549 cells (Fig. 2A). In
addition, pretreatment with antioxidant blocked the inhibitory
effect on H460 cell growth rate induced by teroxirone (Fig. 2B).
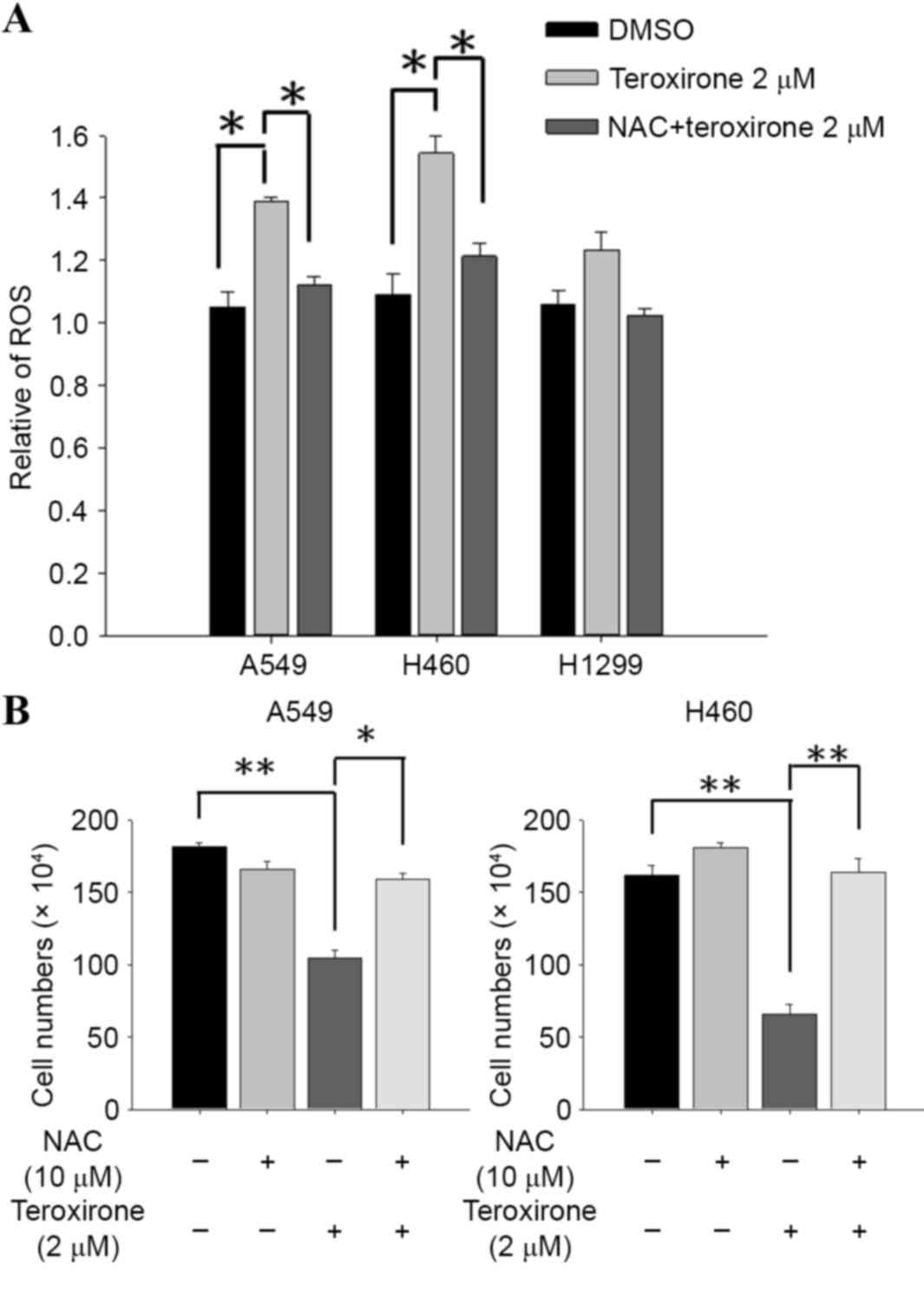 | Figure 2.(A) NAC suppressed teroxirone-induced
ROS. Cells were seeded onto 12-well plates (3×105
cells/well). Following 24 h for allowing complete adherence, the
cells were pretreated with either NAC (10 µM) for 1 h (+) or the
vehicle control (DMSO) (−), followed by treatment with teroxirone
(2 µM) (+) or 0.2% DMSO as the vehicle control (−) for 18 h.
Following treatment, the trypsinized cells were evaluated using
flow cytometry, with DCFH-DA as a fluorescent oxidation-sensitive
probe. (B) Cell and cell viability determination by teroxirone in
A549 and H460 cells. Cells were seeded onto 6-well plates
(3×105 cells/well). Following 24 h to allow for complete
adherence, the cells were pretreated with either NAC (10 µM) for 1
h (+) or the vehicle control (DMSO) (−), followed by treatment with
teroxirone (2 µM) (+) or 0.2% DMSO vehicle control (−) for 24 h.
The trypsinized cells were counted using a trypsin-exclusion assay.
*P<0.05 and **P<0.01 vs. DMSO control. DCFH-DA,
dichlorodihydrofluorescein diacetate; DMSO, dimethyl sulfoxide;
ROS, reactive oxygen species; NAC, N-acetylcysteine. |
NAC suppressed ROS generation and
disrupted cell cycle distribution by teroxirone
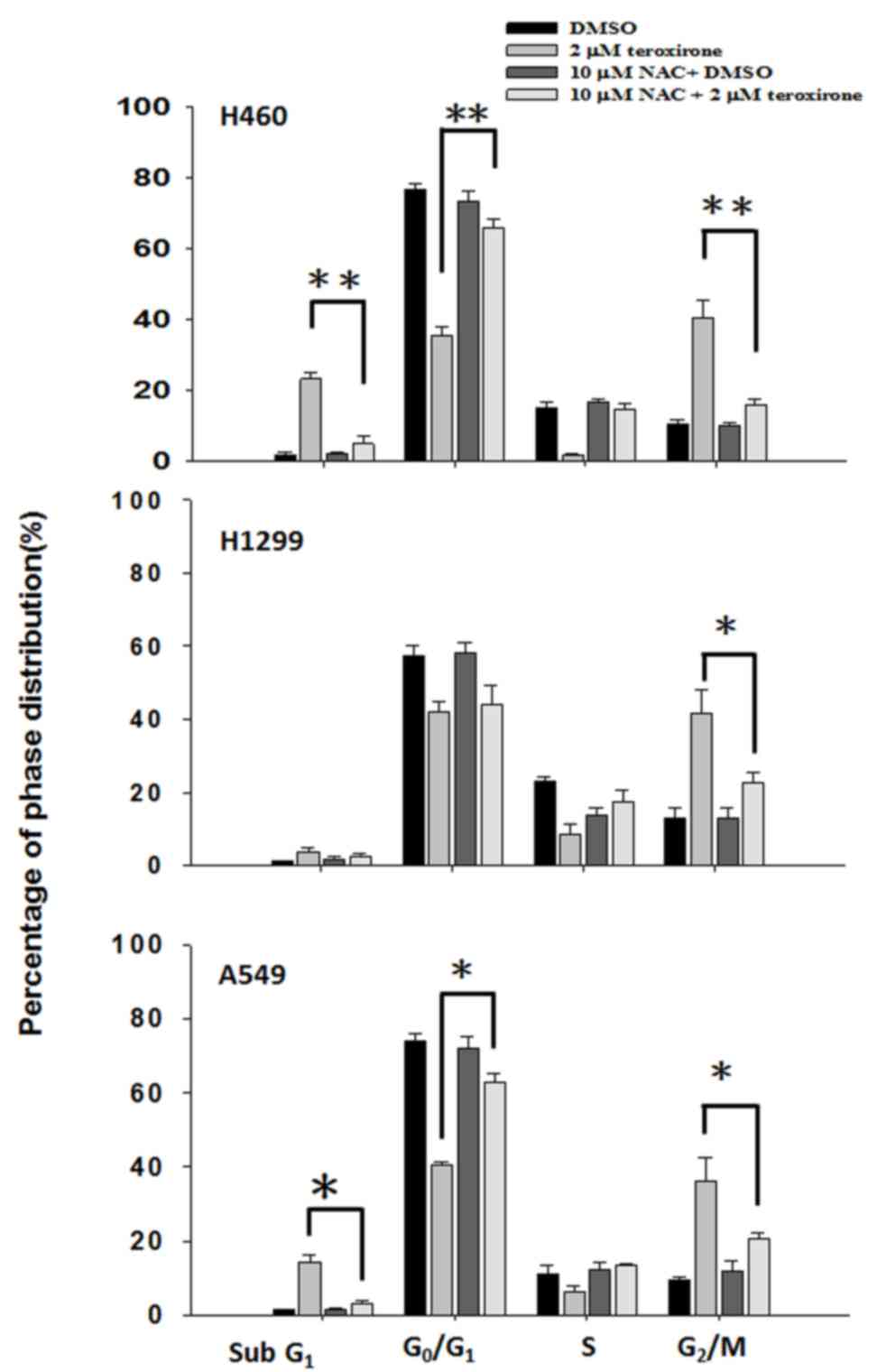
Serving a key role in cell death, ROS produced in
cells resulted in major changes. The ROS products altered cell
growth by influencing cell cycle populations; this was evaluated by
PI-stained flow cytometry analysis. The results revealed that
treatment with teroxirone for 24 h induced cell accumulation at the
sub-G1 phase, at the expense of those at
G0/G1 and S phases, in H460 and A549 cells
but not in H1299 cells. Pretreatment with NAC blocked the
sub-G1 phase cell increase that suggested recovery of
viable cells as suppressed by ROS (Fig.
3).
Teroxirone induces the p53-dependent
apoptosis of NSCLC cells in an ROS-dependent manner
ROS was revealed to induce cell cycle arrest and
cell death. Western blot analysis was used to investigate if
mitochondrial-mediated apoptosis determinants may be affected by
teroxirone. The results demonstrated that treatment of teroxirone
for 24 h increased expression levels of intrinsic apoptosis-related
markers, including Bax, active caspase-3 and cleaved PARP,
alongside p53 activation in A549 (Fig.
4A) and H460 (Fig. 4B) cells.
Furthermore, teroxirone suppressed the expression levels of the
proliferation marker Akt, and the mitochondrial anti-apoptotic
signal Bcl-2. NAC pretreatment for 1 h prior to teroxirone exposure
suppressed the induced apoptosis characteristics, while survival
signals in A549 and H460 cell lines were recovered. The results
proved that the generated ROS was associated with apoptotic cell
death under the influence of teroxirone.
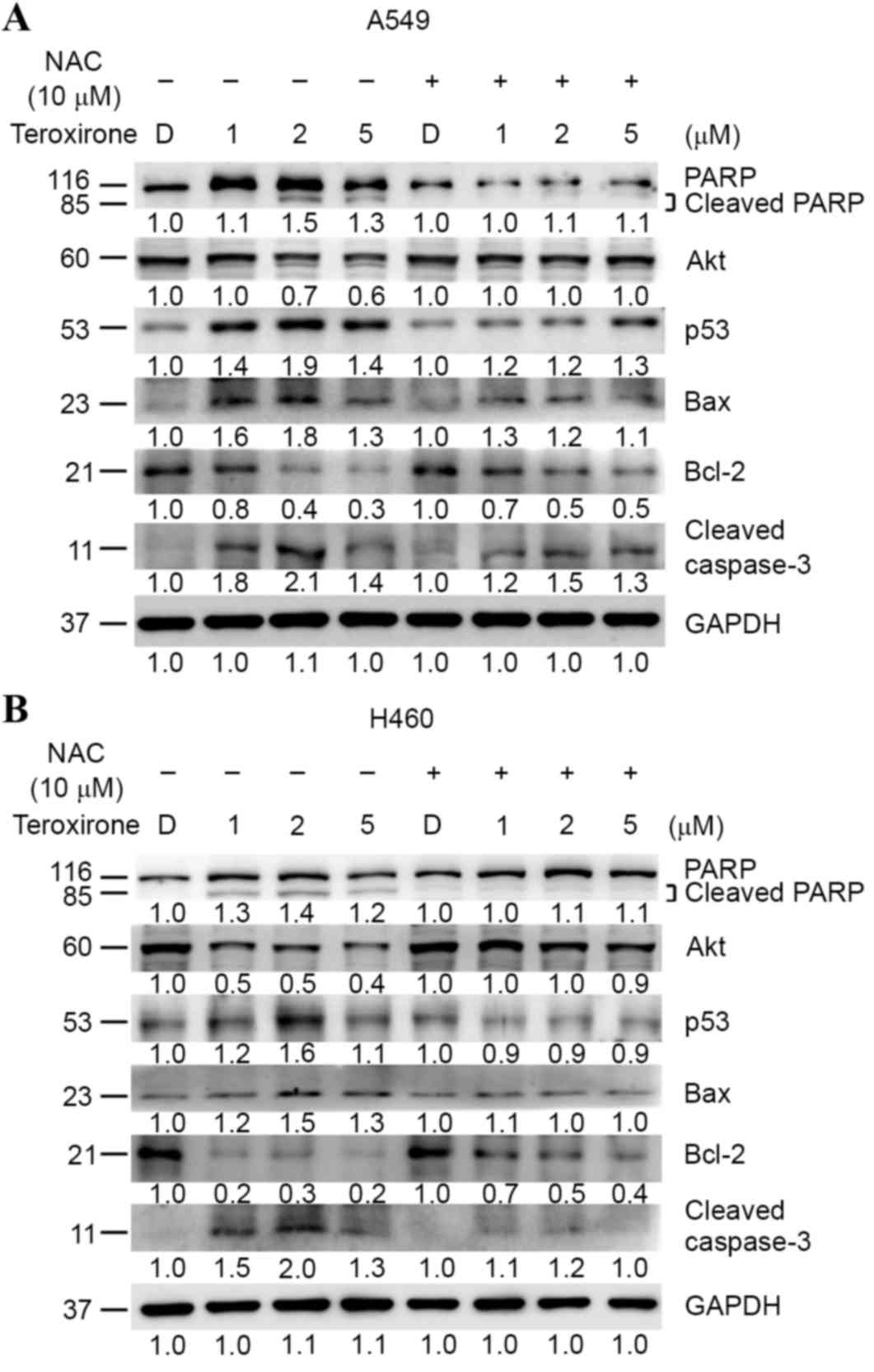 | Figure 4.(A) Antioxidant NAC recovered
ROS-dependent apoptosis in NSCLC A549 cells. Cells were evaluated
using western blot analysis following pre-treatment with NAC (10
µM) (+) or 0.2% DMSO vehicle control (−) for 1 h, followed by
various concentrations of teroxirone treatment for 24 h. The cells
were lysed and the protein concentrations determined. Equal amounts
of cell lysates and protein were separated by SDS-PAGE and
electro-blotted. The blots were subsequently incubated in fresh
blocking solution and probed for 1 h with 1:2,000 dilutions of
PARP, Akt, p53, Bax, Bcl-2, caspase-3 and GAPDH antibodies,
followed by incubation with a 1:3,000 dilution of a horseradish
peroxidase-conjugated secondary antibody, prior to being evaluated
using an ECL detection system. The numbers below each lane
signified relative intensities of cleaved PARP, Akt, p53, Bax,
Bcl-2 and GAPDH at each concentration compared with the results of
the DMSO vehicle control with or without NAC. (B) NAC recovered
ROS-dependent apoptosis in NSCLC H460 cells. Cells were analyzed by
western blotting. NAC, N-acetyl cysteine; ROS, reactive oxygen
species; NSCLC, non-small cell lung cancer cells; DMSO, dimethyl
sulfoxide; PARP, poly ADP-ribose polymerase; Akt, protein kinase B;
Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; ECL,
enhanced chemiluminescence; p53, tumor protein 53. |
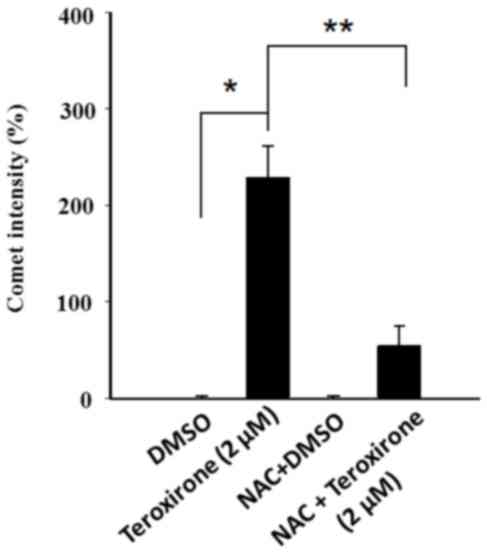
ROS enhanced cytochrome c release from
mitochondria and DNA damage by teroxirone, in NSCLC cells
In order to investigate mitochondrial injury,
immunofluorescence image analysis of cytochrome c release
was used to reveal the extent of damage leading to apoptosis. The
results demonstrated that teroxirone treatment enhanced the
intensities of cytosolic cytochrome c and the effects were
concentration-dependent in H460 (Fig. 5A
and B) and A549 (Fig. 5C and D)
cells following treatment for 24 h. The intensity of accumulated
cytochrome c in the cytoplasm was reduced following 1 h of
NAC pretreatment. The diminished DNA lesions by teroxirone
treatment, subsequent to a 24-h pretreatment with NAC in A549
cells, provided further evidence that DNA damage was resulted from
generated ROS (Fig. 6). Together,
these results demonstrated that ROS induced mitochondrial
disturbance and DNA damage in NSCLC cells, which contributed to the
effectiveness of teroxirone.
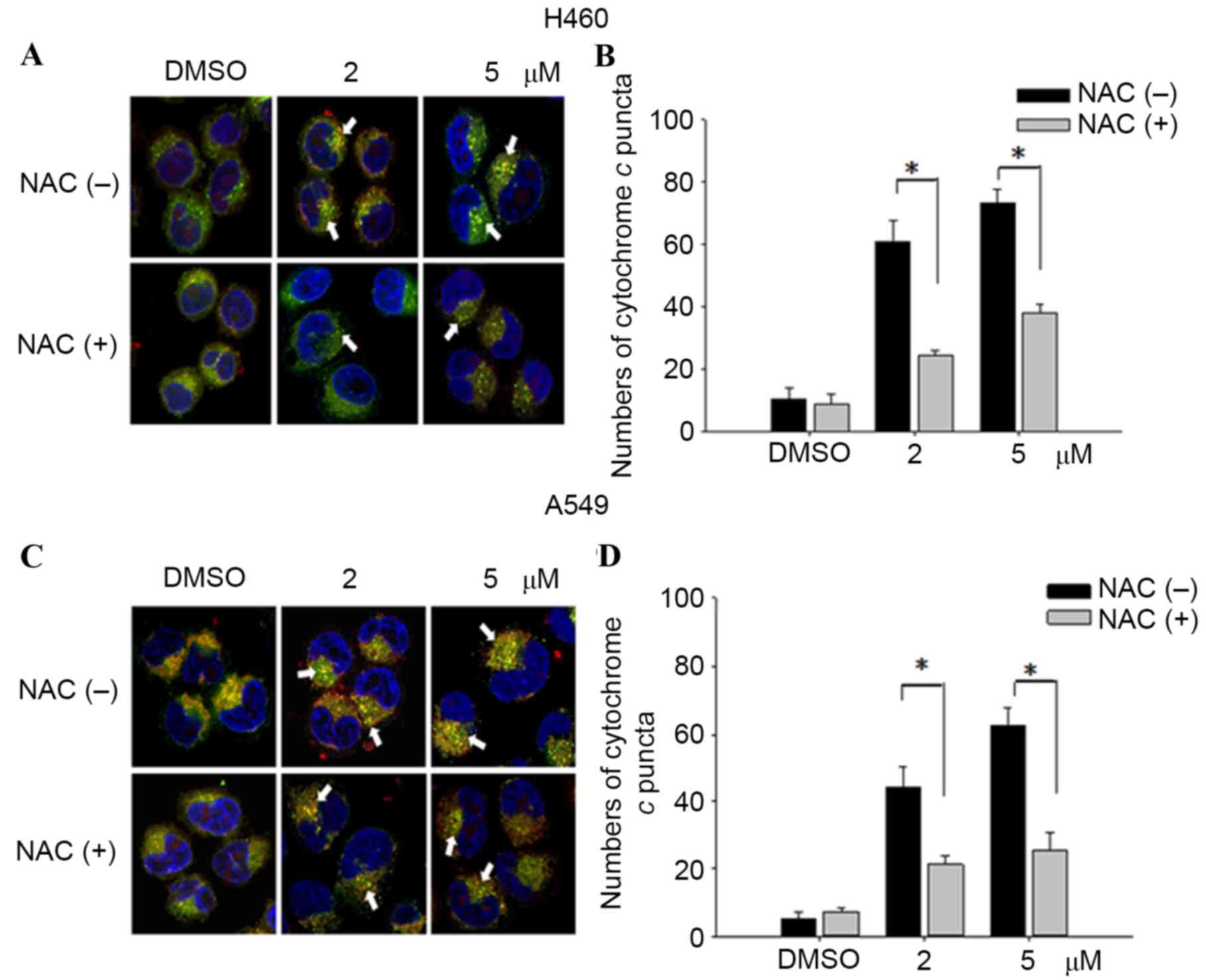 | Figure 5.Cytochrome c release in NSCLC
cells. The release of cytochrome c was reduced from
mitochondria in H460 and A549 cells pretreated with NAC (10 µM) for
1 h, followed by incubation with 2/5 µM teroxirone or the vehicle
control (0.2% DMSO) for 24 h. Cells were fixed with 4% of
formaldehyde, permeabilized and stained with an anti-cytochrome
c antibody (dilution, 1:200) at 4°C for 18 h. Subsequent to
washing, cells were stained with Mitotracker Green (mitochondrial
staining), DAPI (nuclear staining) and secondary antibody
conjugated with TRITC for cytochrome c. The pointed arrow
signified the co-localization of cytochrome c (red) and
mitochondria (green), whereas the nucleus is stained blue (scale
bar, 100 µm). The release of cytochrome c punctae was
quantified in H460 (A and B) and A549 (C and D) cells using ImageJ
software. The results in the presence of NAC (+) and absence NAC
(−) were compared. *P<0.05 vs. DMSO control. ROS, reactive
oxygen species; NSCLC, non-small cell lung cancer cells; TRITC,
tetramethylrhodamine; NAC, N-acetylcysteine; DMSO, dimethyl
sulfoxide. |
Discussion
In response to disruption of electron transport, MMP
loss, ATP level decrease, ROS production and mitochondria signal
transducer activation contributed to p53-dependent apoptosis
(8), suggesting that the
mitochondrion-dependent pathway serves a crucial role in
ROS-mediated apoptosis. The increased levels of ROS may result from
oxidative phosphorylation uncoupling, hyperbaric O2
treatment, ischemia and alterations of mitochondrial lipids
(9).
It has been previously revealed that teroxirone
induced apoptosis in human NSCLC cells, and that this development
depended on the status of p53 (6).
Activation of p53 promoted the intrinsic apoptosis pathway by
triggering permeabilization of the outer mitochondrial membrane and
coordinating pro-apoptotic Bax and anti-apoptotic Bcl-2 (10). This process involved cytochrome
c, mitochondrial lipids, proteins regulating bio-energetic
metabolic flux and the components within the permeability
transition pore (9,10). Once the outer mitochondrial membrane
was disrupted, a set of proteins between the inner and outer
mitochondrial membranes became active and promoted cytochrome
c release (8). The activated
intrinsic pathway was crucial in initiating apoptotic cell death
under the influence of ROS (11). The
released cytochrome c forms a cytochrome c/apoptotic
protease activating factor 1/caspase-9 apoptosome complex, as
recruited during apoptosis progression and included Bax and the
initiator caspase-9 (12).
Subsequently, caspase-3 and −7 was activated, causing the
activation of caspase-3 downstream substrates which are critical to
apoptosis (12).
The results of the present study demonstrated that
teroxirone exerted oxidative stress on human NSCLC cells by
disrupting the MMP, generating ROS and promoting ultimate apoptotic
cell death. The decrease of the MMP was identified 12 h following
teroxirone treatment, the increase of ROS 18 h following treatment
and DNA damage subsequent to this. Furthermore, pretreatment with
NAC significantly restored MMP (Fig.
1) and reduced ROS accumulation (Fig.
2). As demonstrated by the cell viability determination,
teroxirone inhibited the proliferation of H460 and A549 cells
following 24 h of teroxirone treatment. Pretreatment with NAC
recovered cell growth rates and suppressed the effects of
teroxirone. The decreased cell viability and the emergent
sub-G1 and G2/M cell phase populations by
teroxirone was blocked by NAC pretreatment (Fig. 3). Western blot analysis demonstrated
that teroxirone induced apoptosis by increasing the expression
levels of p53 and Bax, activating procaspase-3 and PARP
fragmentation in addition to reducing the expression levels of
Bcl-2 following 24 h of treatment (Fig.
4). Teroxirone treatment resulted in a significant increase in
Bax expression and a decrease in Bcl-2 expression. A direct
association exists between ROS and PI3K/Akt signal inactivation, in
which ROS functions as an upstream modulator of Akt (13). The reduced Akt expression level due to
teroxirone and reverted by NAC suggested the role of ROS in
attenuating the PI3k/Akt signaling pathway that contributed to drug
potency (14). Furthermore, the
presence of NAC abolished the intrinsic pathway by suppressing p53
and Bax, in addition to decreasing caspase-3 and PARP
fragmentation. In immunofluorescence analysis, NAC was revealed to
block the release of cytoplasmic cytochrome c accumulation
by teroxirone (Fig. 5). These
combined results suggested the existence of teroxirone-induced ROS
mediated cytotoxicity in human NSCLC cells associated with ROS.
Antioxidant NAC pretreatment blocked the inhibitory effects of
teroxirone by attenuating ROS (Fig.
4). Pretreatment with NAC reversed the expression of Bax, Bcl-2
and cytochrome c, and also inhibited teroxirone-induced MMP
collapse, suggesting that ROS was capable of functioning as an
initial mediator in the p53-dependent mitochondrial apoptotic
pathway. These results also support the notion that ROS serves a
primary role in triggering apoptosis by activating the intrinsic
pathway. Thus, as a promising potential therapy to treat NSCLC lung
cancer, teroxirone affected cellular oxidative stress prior to
final apoptosis.
Certain triepoxide compounds demonstrated antitumor
activities against lung cancer and previous studies have identified
that the triepoxide diterpenoid triptolide, as a major bioactive
component of the Chinese herb, and its water-soluble analog,
minnelide, promoted apoptosis and decreased proliferation in human
NSCLC cells (15,16). Triptolide is an inhibitor of RNA
polymerase activity and affects the transcriptional machinery. A
previous study identified that the compound promoted ROS, decreased
MMP and activated caspase-3 by disturbing mitochondrial functions
(17). The epoxide analog
benzo(a)pyrene-7,8-diol-9,10-epoxide mediated apoptosis by
activating the caspase-9-dependent mitochondria pathway and the
formation of ROS, loss of MMP and upregulation of p53, in human
bronchiolar epithelial cells (18).
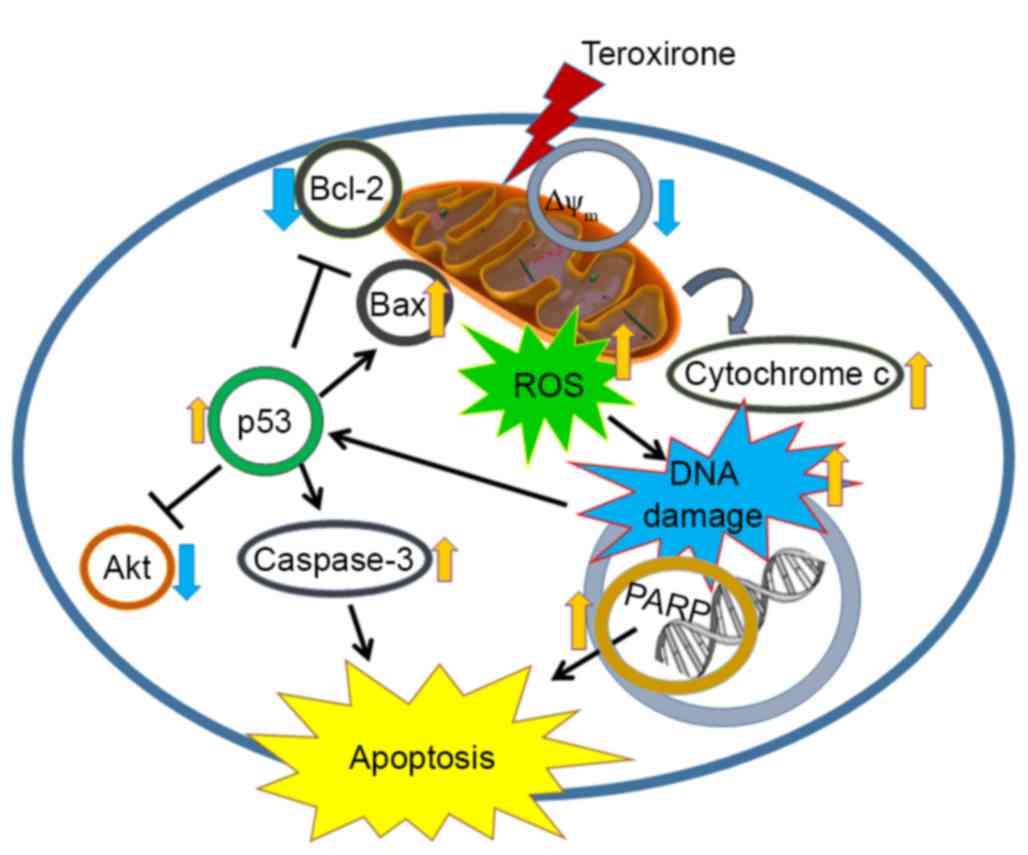
The present study revealed that the triepoxide teroxirone induced
apoptosis via the p53-associated intrinsic apoptosis pathway, which
involves the production of ROS, the decline of MMP with implication
of mitochondrial function injury and DNA damage (Fig. 7). The study provides a novel
perspective of teroxirone in providing drug development and therapy
for the treatment of lung cancer.
Acknowledgements
The present study was supported by the Ministry of
Science and Technology (grant no. MOST-103-2311-B-003-001) and the
National Taiwan Normal University (grant nos. 102T3040B2,
103T3040D2 and 104T3040C2; Taipei, Taiwan).
References
|
1
|
Rathos MJ, Khanwalkar H, Joshi K, Manohar
SM and Joshi KS: Potentiation of in vitro and in vivo antitumor
efficacy of doxorubicin by cyclin-dependent kinase inhibitor
P276-00 in human non-small cell lung cancer cells. BMC Cancer.
13:292013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Erridge SC, Møller H, Price A and Brewster
D: International comparisons of survival from lung cancer: Pitfalls
and warnings. Nat Clin Pract Oncol. 4:570–577. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Charoensinphon N, Qiu P, Dong P, Zheng J,
Ngauv P, Cao Y, Li S, Ho CT and Xiao H: 5-Demethyltangeretin
inhibits human nonsmall cell lung cancer cell growth by inducing
G2/M cell cycle arrest and apoptosis. Mol Nutr Food Res.
57:2103–2111. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Spreafico F, Atassi G, Filippeschi S,
Malfiore C, Noseda S and Boschetti D: A characterization of the
activity of alpha-1,3,5-triglycidyl-s-triazinetrione, a novel
antineoplastic compound. Cancer Chemother Pharmacol. 5:103–108.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dombernowsky P, Lund B and Hansen HH:
Phase-I study of alpha-1,3,5-triglycidyl-s-triazinetrione (NSC
296934). Cancer Chemother Pharmacol. 11:59–61. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang JP, Lin KH, Liu CY, Yu YC, Wu PT,
Chiu CC, Su CL, Chen KM and Fang K: Teroxirone inhibited growth of
human non-small cell lung cancer cells by activating p53. Toxicol
Appl Pharmacol. 273:110–120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Okon IS, Coughlan KA, Zhang M, Wang Q and
Zou MH: Gefitinib-mediated reactive oxygen species (ROS) instigates
mitochondrial dysfunction and drug resistance in lung cancer cells.
J Biol Chem. 290:9101–9110. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brodská B and Holoubek A: Generation of
reactive oxygen species during apoptosis induced by DNA-damaging
agents and/or histone deacetylase inhibitors. Oxid Med Cell Longev.
2011:2535292011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ricci JE, Muñoz-Pinedo C, Fitzgerald P,
Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH
and Green DR: Disruption of mitochondrial function during apoptosis
is mediated by caspase cleavage of the p75 subunit of complex I of
the electron transport chain. Cell. 117:773–786. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Herrera B, Alvarez AM, Sánchez A,
Fernández M, Roncero C, Benito M and Fabregat I: Reactive oxygen
species (ROS) mediates the mitochondrial-dependent apoptosis
induced by transforming growth factor (beta) in fetal hepatocytes.
FASEB J. 15:741–751. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu H, Li X, Ding W, Zeng X, Kong H, Wang H
and Xie W: Deguelin induces the apoptosis of lung cancer cells
through regulating a ROS driven Akt pathway. Cancer Cell Int.
15:252015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rousalova I, Banerjee S, Sangwan V,
Evenson K, McCauley JA, Kratzke R, Vickers SM, Saluja A and D'Cunha
J: Minnelide: A novel therapeutic that promotes apoptosis in
non-small cell lung carcinoma in vivo. PLoS One. 8:e774112013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reno TA, Kim JY and Raz DJ: Triptolide
inhibits lung cancer cell migration, invasion and metastasis. Ann
Thorac Surg. 100:1817–1825. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vispé S, DeVries L, Créancier L, Besse J,
Bréand S, Hobson DJ, Svejstrup JQ, Annereau JP, Cussac D, Dumontet
C, et al: Triptolide is an inhibitor of RNA polymerase I and
II-dependent transcription leading predominantly to down-regulation
of short-lived mRNA. Mol Cancer Ther. 8:2780–2790. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sang H, Zhang L and Li J:
Anti-benzopyrene-7,8-diol-9,10-epoxide induces apoptosis via
mitochondrial pathway in human bronchiolar epithelium cells
independent of the mitochondria permeability transition pore. Food
Chem Toxicol. 50:2417–2423. 2012. View Article : Google Scholar : PubMed/NCBI
|