Introduction
Chronic myelocytic leukemia (CML) is a type of
hematopoietic stem-cell disease, which is characterized by the
presence of the Philadelphia chromosome, t(9;22)(q34;q11) and
generation of the breakpoint cluster region protein/Abelson murine
leukemia viral oncogene homolog 1 (BCR/ABL) fusion gene. Treatment
with tyrosine kinase inhibitors (TKIs) has significantly improved
the prognosis of patients with CML, although certain patients are
resistant to TKIs, leading to treatment failure. CML resistance
involves multifaceted and complex mechanisms, including
BCR/ABL-dependent mechanisms, such as ABL kinase domain mutations,
BCR/ABL overexpression and various BCR/ABL-independent mechanisms
(1,2).
The Wilms Tumor 1 (WT1) gene, which encodes a
regulatory molecule that is important in the process of cell growth
and development, is located on chromosome 11p13, and is a
bispecific gene with antioncogenic and oncogenic properties
(3). Radich et al (4) compared the gene expression profiles of
patients with CML in different phases of the disease state
(chronic, accelerated and blast crisis). The results of the
aforementioned study revealed changes to gene expression in the
early accelerated phase, in which the WT1 gene ranked fifth among
the top 10 differentially expressed genes exhibiting
upregulation/downregulation during disease progression.
Furthermore, certain studies have demonstrated that WT1
overexpression in the K562 cell line (BCR/ABL-positive) results in
resistance to the TKI imatinib (5–7). These
observations suggest that the WT1 gene serves an important role in
CML resistance and progression.
The WT1 gene serves primarily as an oncogene in
hematological malignancies and regulates the expression of
downstream genes. WT1 target genes may be classified according to
their functions, among which the most notable are those associated
with the mitogen-activated protein kinase (MAPK) and Wnt signaling
pathways (8–10). Conversely, WT1 gene expression may be
regulated by upstream genes. Previous in vivo and in
vitro studies revealed that the high-temperature requirement
family (Htr)A family member, HtrA2 serves as an upstream regulator
of WT1 by binding specifically to the WT1 inhibition domain
(11,12). HtrA2 possesses serine protease
activity and degrades WT1 at multiple loci on the N- and C-termini
(11,12).
The present study aimed to investigate the
regulatory role of HtrA2 on WT1 and the effects of imatinib in K562
cells. In addition, the effects of its regulation on cell function
and changes in the downstream signaling pathway were explored.
Materials and methods
Cells and experimental drugs
The K562 cell line (American Type Culture
Collection, Manassas, VA, USA) used in the present study was
derived from a patient in the acute transformation phase of CML and
was preserved in the State Key Laboratory of Experimental
Hematology, Institute of Hematology and Blood Diseases Hospital,
Chinese Academy of Medical Sciences and Peking Union Medical
College, Tianjin, China. The primary drugs used were: Imatinib and
the HtrA2 inhibitor 5-[5-(2-nitrophenyl) furfuryl iodine]-1,
3-diphenyl-2-thiobarbituric acid (UCF-101; both from Calbiochem;
Merck KGaA, Darmstadt, Germany).
Design and synthesis of primers
All primers were synthesized and purified by
Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
upstream and downstream primers were designed using Primer Premier
5.0 (Premier Biosoft International, Palo Alto, CA, USA), and their
amplification specificities were validated using the Basic Local
Alignment Search Tool (https://blast.ncbi.nlm.nih.gov). All primers were
dissolved in deionized water to a concentration of 10 µM and stored
at −20°C for subsequent experiments. The sequences of the primers
were as follows: WT1 forward, 5′-CACGAGGAGCAGTGCCTGAG-3′ and
reverse, 5′-AACCCTGATTGCGAATAGCG-3′; HtrA2 forward,
5′-AGACATCGCAACGCTGAGGATT-3′ and reverse,
5′-GGACGCTGAGCAGAGCTAACAA-3′; BCR/ABL-p210 forward,
5′-GGGCTCTATGGGTTTCTGAATG-3′ and reverse,
5′-CGCTGAAGGGCTTTTGAACT-3′; Internal reference gene GAPDH forward,
5′-GAAGGTGAAGGTCGGAGTC-3′ and reverse,
5′-GAAGATGGTGATGGGATTTC-3′.
Analysis of protein expression
K562 cells (1×106 cells/system) were
treated with half maximal inhibitory concentrations
(IC50) of imatinib (1 µM), and cells were collected and
counted at 0, 3, 6, 12, 24 and 48 h following drug application.
Cells in the drugs+UCF-101 group were pretreated with UCF-101
(final concentration, 2 µM) for 2 h. Protein levels, changes in the
location of WT1 and HtrA2 expression, and changes in
phosphorylation of components of MAPK-associated signaling pathways
following drug treatment were determined by western blot analysis
using the following antibodies: Anti-WT1 rabbit mAb (cat. no.
ab89901; 1:1,000; Abcam, Cambridge, UK), anti-HtrA2 rabbit mAb
(cat. no. ab75982; 1:2,000; Abcam), horseradish peroxidase
(HRP)-labeled goat anti-mouse IgG (cat. no. ab6721; 1:5,000;
Abcam), HRP-labeled goat anti-rabbit IgG (cat. no. ab6789; 1:5,000;
Abcam), anti-poly ADP-ribose polymerase (PARP) rabbit mAb (cat. no.
9532; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA),
anti-Histone H3 rabbit mAb (cat. no. 4499; 1:2,000; Cell Signaling
Technology, Inc.), anti-phospho-p38 MAPK (Thr180/Tyr182) rabbit mAb
(cat. no. 4511; 1:1,000; Cell Signaling Technology, Inc.), anti-p38
MAPK rabbit mAb (cat. no. 8690; 1:1,000; Cell Signaling Technology,
Inc.), anti-phospho-p44/42 extracellular signal-related kinase
(ERK; Thr202/Tyr204) rabbit mAb (cat. no. 8544; 1:1,000; Cell
Signaling Technology, Inc.), anti-p44/42 ERK rabbit mAb (cat. no.
4695; 1:1,000; Cell Signaling Technology, Inc.), and anti-β-actin
mouse mAb (cat. no. SC8432; 1:500; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA). In the western blot analysis, the present study
used RIPA as the protein extraction buffer (Beyotime Institute of
Biotechnology, Haimen, China). The protein determination method was
the BCA method and 20 ug protein were loaded per lane. The present
study used a 10% gel to perform SDS-PAGE for the protein. The
blocking step was performed in 5% BSA buffer at room temperature
for 2 h. In the antibody incubation step, the primary antibodies
were incubated at 4°C overnight and the secondary antibodies were
incubated at room temperature for 1 h. The HRP-goat
anti-mouse/rabbit immunoglobulin G antibodies were supplied by
Abcam (1:5,000). The type of membrane used was nitrocellulose. The
HRP-enhanced chemiluminescence method was used for visualization.
Image J version 2 software was used for result analysis (National
Institutes of Health, Bethesda, MD, USA). β-actin was used as the
control.
Cell proliferation
Cells in the control group, imatinib group, UCF-10
group and imatinib+UCF-101 group were seeded in a 96-well plate
(2×104 cells/well) in 100 µl RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc.) containing 10% fetal bovine serum
(HyClone Company; GE Healthcare, Chicago, IL, USA); three wells
were set up for each group. Then, 10 µl MTT (5 mg/ml) was added at
0, 24, 48 and 72 h following seeding. Subsequent to incubation for
an additional 4 h, 100 µl 10% SDS/0.01 M HCL was added to each
well. Plates were incubated at 37°C overnight and agitated using an
oscillator for 10 min. The optical density (OD) at 546 nm was
measured using a micro-plate reader. The OD value at 0 h in each
group was set arbitrarily as 1, and the relative OD values at the
other time points were calculated to construct the proliferation
curves for comparison of the proliferation rates among different
groups.
Cell apoptosis
K562 cells were collected and analyzed for apoptosis
with an Annexin V-FITC kit (BD Biosciences, Franklin Lakes, NJ,
USA) according to the manufacturer's protocol, and membrane
integrity was simultaneously assessed with propidium iodide (PI)
exclusion (BD Biosciences). The concentration of cells was
1×106 cells/ml, and in each system there were
1×105 cells. The cells were collected using
centrifugation at 140 × g for 5 min at room temperature.
Statistical analysis
Statistical analyses were performed using SPSS19.0
software IBM Corp., Armonk, NY, USA). Measurement data were first
subjected to normality testing using the single-sample
Kolmogorow-Smirnov test. Normally distributed data were analyzed
using a paired t-test, or one-way analysis of variance followed by
the Student-Newman-Keuls method. Non-normally distributed data were
analyzed using the rank sum test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Detection of WT1 and HtrA2 mRNA in
K562 cells treated with imatinib and UCF-101 using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Firstly, the IC50 of imatinib for the
K562 cell line was determined. Following treatment of K562 cells
for 48 h, the IC50 value for imatinib was 1.18±0.2 µM.
In subsequent experiments, 1.0 µM imatinib was used. Cells were
collected following treatment with imatinib (± UCF-101) for 0, 3,
6, 12, 24 and 48 h. Total RNA was extracted and reverse transcribed
to obtain cDNA, and the changes of WT1 and HtrA2 mRNA in K562 cells
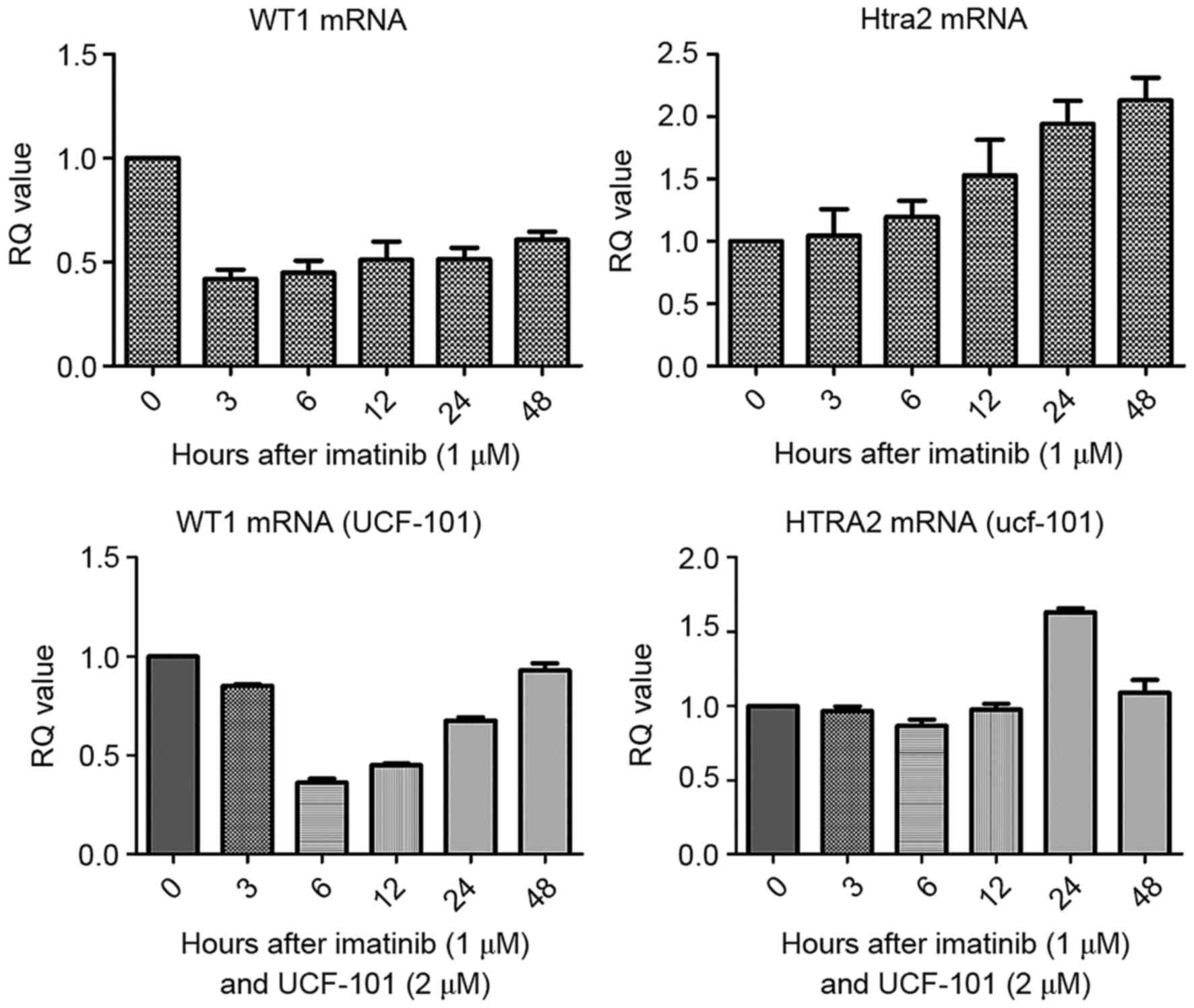
were analyzed by qPCR. As presented in Fig. 1, compared with the control group, WT1
mRNA levels were downregulated in cells treated with imatinib. In
contrast, HtrA2 mRNA levels were gradually upregulated, reaching a
2-fold increase at 48 h compared with the levels in the control
group. Following pretreatment with UCF-101, the downregulation of
WT1 mRNA induced by imatinib was delayed, measuring at its lowest
level at 6 h, and was restored to the levels of the control at 48
h. However, no significant changes in HtrA2 expression were
observed.
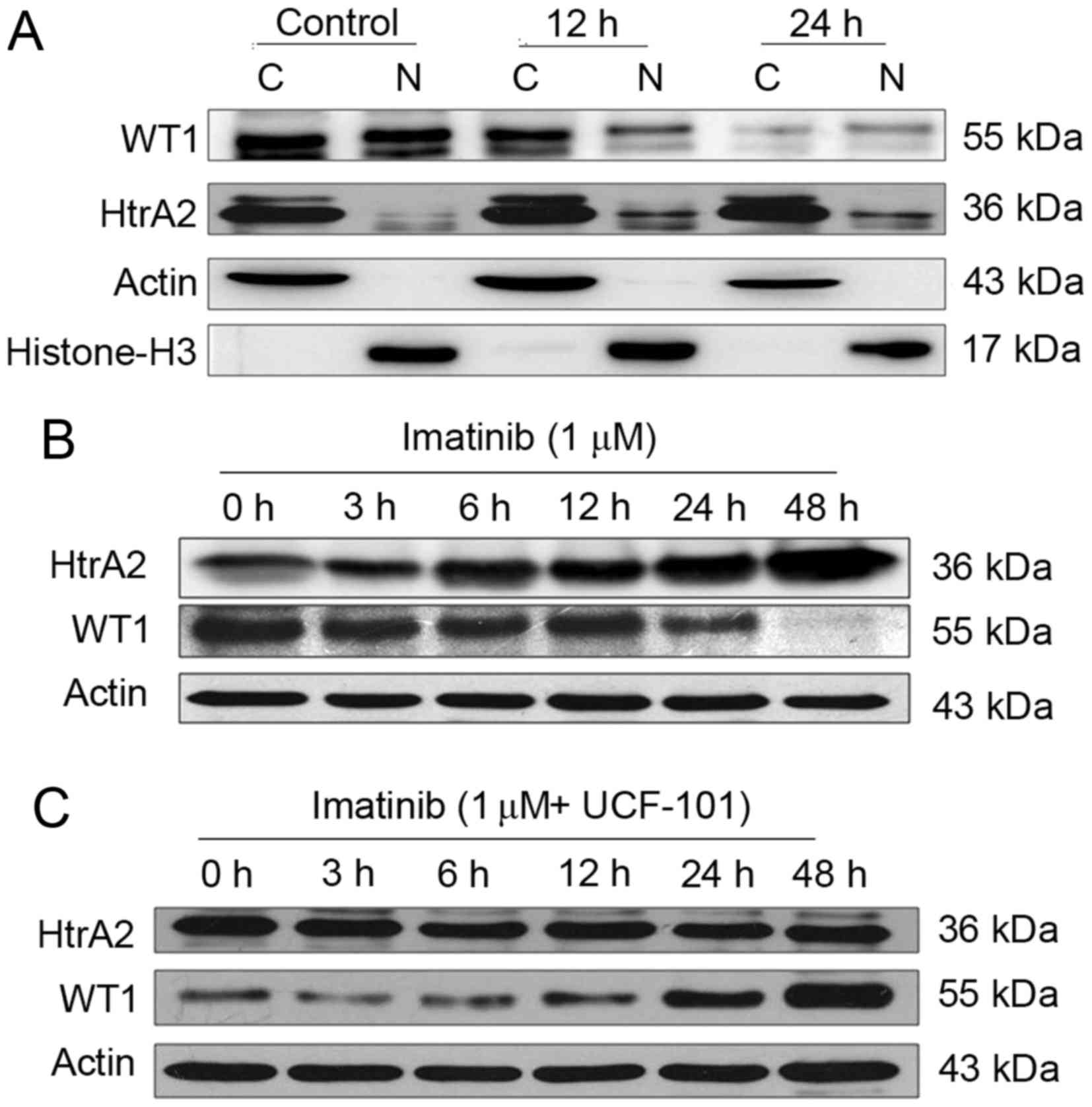
Effects of imatinib
Following treatment of the K562 cells with imatinib
for 12 and 24 h, cells were collected and counted. Cytoplasmic and
nuclear proteins were analyzed by western blotting using β-actin,
and histone H3 as internal controls for the cytoplasmic and nuclear
proteins, respectively. WT1 and HtrA2 were expressed in the
cytoplasm and nuclei, with HtrA2 located primarily in the cytoplasm
(Fig. 2A). Subsequent to treatment
with imatinib, WT1 protein levels were downregulated markedly in
the cytoplasm and nuclei. WT1 protein levels were markedly reduced
in the nuclei at 12 h following imatinib treatment, while HtrA2
protein levels were upregulated in the cytoplasm and, more
evidently, in the nuclei.
Effects of imatinib and UCF-101
Subsequent to treatment with imatinib (with and
without UCF-101 pretreatment) for up to 48 h, K562 cells were
collected and counted. Cells were lysed with
radioimmunoprecipitation assay buffer, and total proteins were
collected for western blot analysis. The results are presented in
Fig. 2B and C. Following prolonged
treatment of K562 cells with imatinib in the absence of UCF-101
pretreatment, HtrA2 expression was upregulated, and the WT1 level
was decreased. However, no significant HtrA2 variation was observed
in cells with UCF-101 pretreatment, while WT1 was slightly
downregulated and rapidly restored to a level higher compared with
the baseline. These data suggest that imatinib induces the
upregulation of HtrA2 protein and downregulation of the WT1
protein, that HtrA2 is an upstream regulatory factor of WT1 and
that it is activated by imatinib.
Effect of HtrA2 regulation of WT1 on
the biological function of K562 cells
Cells were treated with imatinib, and divided into
control, UCF-101, imatinib and imatinib+UCF-101 groups. Cells were
then collected at 24 and 48 h. Apoptosis was detected by flow
cytometry following staining with Annexin V and PI and cell
proliferation was measured using the MTT method. Digestion of PARP
was detected by western blotting.
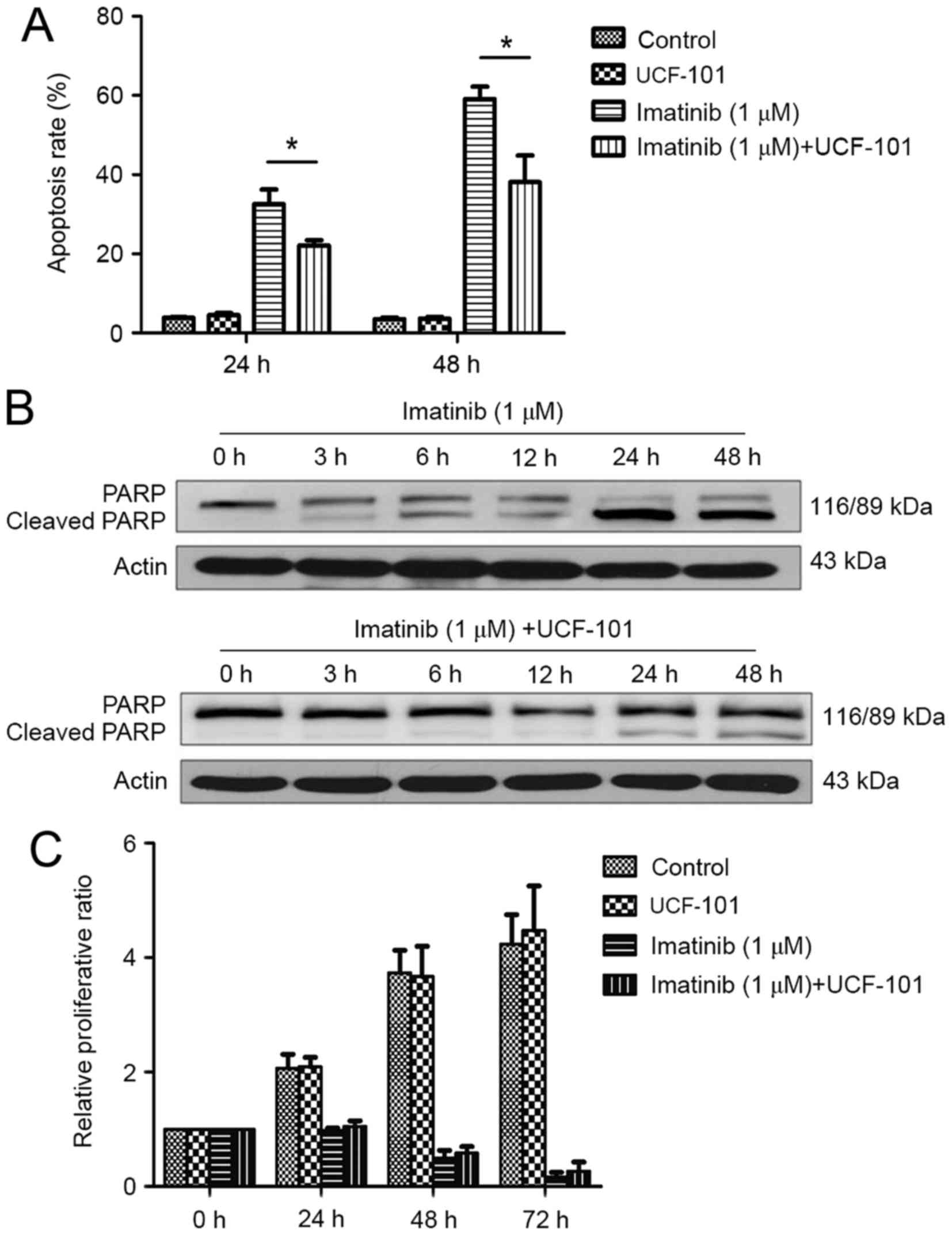
Effect of imatinib and UCF-101 on K562
cell apoptosis
As demonstrated in Fig.
3A, significant increases in apoptosis rates were observed at
24 and 48 h following treatment with imatinib; this effect was
increased with prolonged duration. However, imatinib-induced
apoptosis was significantly reduced by pretreatment with UCF-101,
with significant differences observed between the two groups at 24
and 48 h (both P<0.05). Concomitantly, western blot analysis
demonstrated that PARP digestion was markedly increased following
treatment with imatinib alone, suggesting increased apoptosis
(Fig. 3B). In contrast,
imatinib-induced PARP digestion was remarkably decreased by
pretreatment with UCF-101, which was consistent with the effects
observed on apoptosis (Fig. 3B).
Effect of imatinib and UCF-101 on K562
cell proliferation
Imatinib suppressed K562 cell proliferation
significantly and persistently, while UCF-101 pretreatment
exhibited no significant effect on K562 cell proliferation.
Compared with the imatinib group, the proliferation rate was
slightly increased in the imatinib+UCF-101 group, suggesting that
UCF-101 suppressed the imatinib-induced inhibition of cell
proliferation, although this effect did not reach the level of
statistical significance (Fig.
3C).
Effect of HtrA2 regulation of WT1 on
its downstream signaling pathways
WT1 possesses extensive targets for downstream
signaling, of which the MAPK signaling pathway is particularly
notable. The present study demonstrated that the regulatory effects
of HtrA2 on WT1 affect the apoptosis and proliferation of K562
cells. In this section, to assess the downstream mechanism of the
functional changes induced by the regulatory effects of HtrA2 on
WT1, K562 cells were treated with imatinib, and changes in the
phosphorylation of the MAPK signaling pathway members ERK1/2 and
p38 were investigated by western blot analysis.
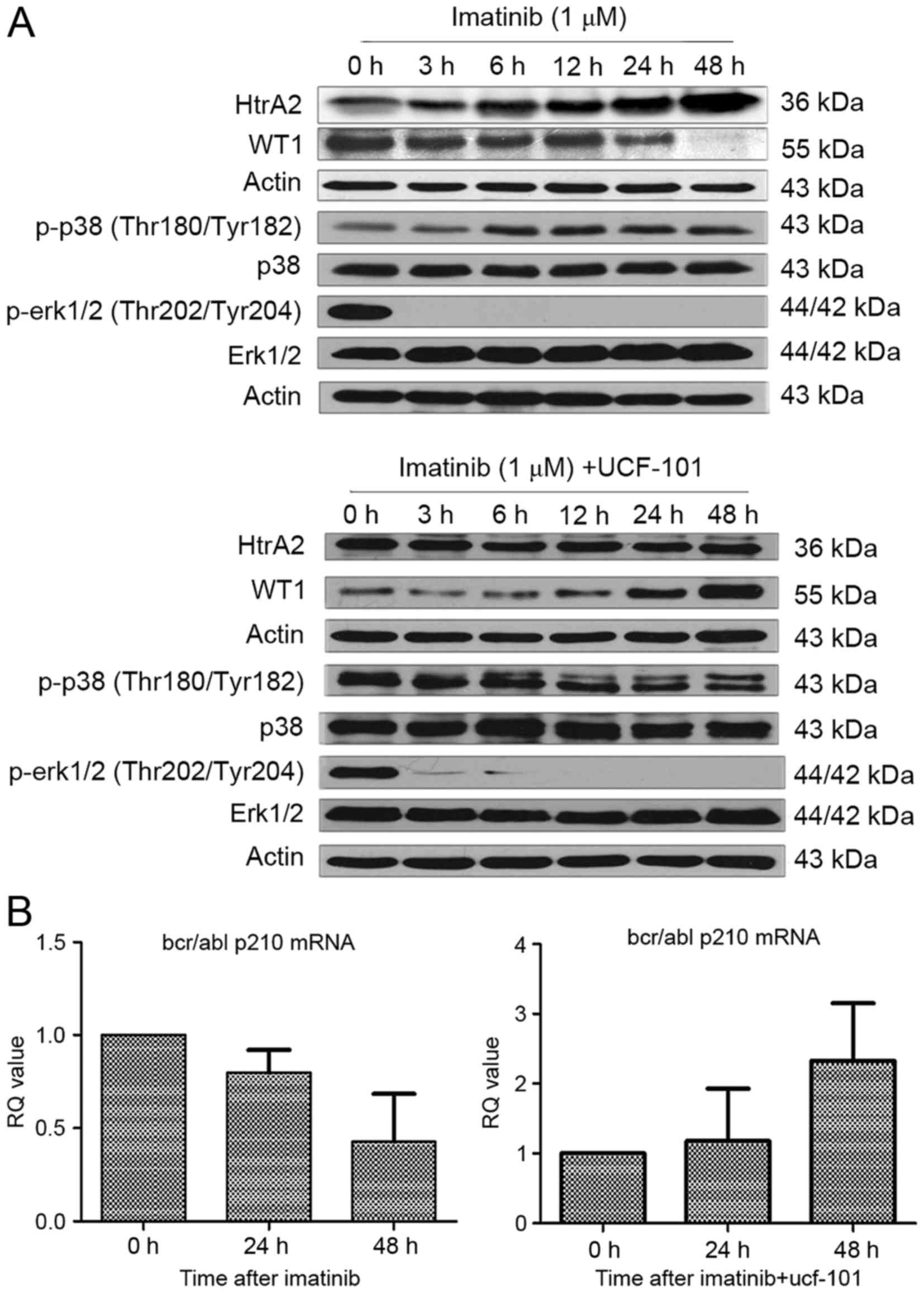
Effect of imatinib and UCF-101 on
signaling pathways
As aforementioned, HtrA2 expression was upregulated
and WT1 expression was downregulated following treatment with
imatinib in K562 cells. However, the downregulation of WT1 protein
expression was reversed subsequent to pretreatment with UCF-101 to
suppress HtrA2 function. Under similar conditions and time points,
it was identified that p38-MAPK phosphorylation began to increase
following treatment with imatinib for 6 h, and was sustained to 48
h (Fig. 4A). Concurrently, the ERK1/2
phosphorylation started to decrease significantly from 3 h
following imatinib treatment and was sustained to 48 h without
restoration. In cells pretreated with UCF-101, p38-MAPK
phosphorylation was not upregulated and ERK1/2 phosphorylation
remained at a low level (Fig.
4A).
Association between HtrA2 regulation
of WT1 and BCR/ABL expression
Expression of the BCR/ABL-p210 fusion gene is a
feature of K562 cell lines. To investigate the association of HtrA2
regulation on WT1 with the expression of BCR/ABL-p210 fusion gene,
RNA was extracted from cells treated with imatinib ±UCF-101. cDNA
was reverse transcribed, and alterations to BCR/ABL-p210 fusion
gene expression were determined by qPCR. Imatinib downregulated
expression of the BCR/ABL-p210 fusion gene significantly, while
UCF-101 pretreatment resulted in a gradual upregulation of
BCR/ABL-p210 fusion gene expression, which was consistent with the
variation observed in the expression of the WT1 gene under similar
conditions (Fig. 4B).
Discussion
In previous years, extensive studies of CML
resistance have demonstrated that this phenomenon involves
multi-faceted and complex mechanisms, including BCR/ABL-dependent
mechanisms and a variety of non-BCR/ABL-dependent mechanisms, such
as ABCB1 and OCT1-mediated intake and efflux of drugs, clonal cell
evolution, and bone marrow stroma-mediated resistance in addition
to resistance mechanisms associated with CML stem cells (1,2). Otahalova
et al (6) identified that the
sensitivity to imatinib was predictable based on the WT1 expression
level in peripheral blood lymphocytes in patients with CML
following in vitro culture and treatment with imatinib. In
addition, specific studies revealed that the overexpression of WT1
protein following gene transfection of K562 cell lines induced
imatinibresistance (7). These studies
indicate that the WT1 gene serves an important role in the
progression of CML and TKI-resistance.
As a transcription factor, WT1 possesses extensive
downstream targets, thereby regulating the biological behavior of
cells (3). Certain studies
functionally classified the target genes of WT1 in the Wilms cell
line CCG99-11 using the CHIP-CHIP method; the most important genes
were identified to be associated with the MAPK and Wnt pathways
(8). Our previous studies on the K562
cell line also indicated that WT1 target genes involved a variety
of MAKP and Wnt/b-catenin signaling pathway genes, including MAPK6,
MAPK7, Wnt2b and Wnt11 (9,10). However, the WT1 upstream regulatory
factors have rarely been studied. It is currently unknown whether
HtrA2 regulates WT1 in BCR/ABL-positive cells, including K562
cells. In the present study, the classical CML-targeted therapy
drug imatinib caused upregulation of HtrA2 protein expression and
downregulation of the WT1. UCF-101, which is a specific inhibitor
of HtrA2 and competitively inhibits the activity of the HtrA2
protease, was used to determine their regulatory association in
K562 cell lines (13). When cells
were pretreated with UCF-101 to suppress HtrA2 activity, the
drug-induced downregulation of WT1 protein expression was reversed,
and WT1 expression was maintained at a high level. These results
indicate that drug stimulation induced the HtrA2 protein
upregulation and increased WT1 protein degradation in K562 cell
lines.
Furthermore, it was identified that imatinib
upregulated HtrA2 expression and downregulated WT1 expression at
the transcriptional level, while UCF-101 pretreatment reversed this
effect. These results suggest that HtrA2 exerted a regulatory
effect on WT1 at the protein level (protein degradation) and at the
transcriptional level, although the mechanism remains to be
elucidated. Regulation of HtrA2 may ultimately lead to a
downregulation of WT1 protein expression, which may affect the
binding of WT1 with the promoters, and may lead to changes in gene
regulation. Therefore, HtrA2 functions as a regulatory factor of
WT1 under the effects of drug stimulation.
Our previous studies revealed that the WT1 protein
was expressed in the cytoplasm and nuclei of K562 cells, with
higher expression in the cytoplasm (9). However, HtrA2 protein is expressed as a
45-kDa precursor protein, which is translated and translocated to
the mitochondria, where it is lysed to form a 36-kDa mature protein
located in the inner mitochondrial membrane region, and partly
located in the nuclei. In the present study, following treatment
with imatinib, the WT1 protein level was decreased in the cytoplasm
and nuclei of K562 cells. This effect was enhanced with prolonged
imatinib exposure, and the reduction was more rapid in the nuclei
compared with that observed in the cytoplasm. Conversely, under
similar conditions, HtrA2 protein expression was identified to
increase in the cytoplasm and nuclei. These results indicate that
HtrA2 protein activation is increased in the cytoplasm under drug
stimulation, leading to WT1 protein degradation in the cytoplasm,
and effects on WT1 localization and transcriptional regulation.
Alternatively, it is possible that HtrA2 protein migrates to the
nuclei under the effect of external stimulation, leading to WT1
protein degradation in the nuclei, therefore affecting its
transcriptional regulation.
WT1 is an important transcription regulation factor
involved in maintaining cell growth and self-renewal (9). It is unknown whether the regulation of
HtrA2 on WT1 affects the biological behavior of cells. In the
present study, K562 cell apoptosis and proliferation as
investigated under HtrA2 regulation, and it was identified that the
proportion of apoptotic cells was decreased significantly
(P<0.05) by pretreatment with UCF-101 to suppress the
imatinib-induced HtrA2 activation. This was also verified by the
results of PARP digestion analysis, suggesting that the regulation
of HtrA2 on WT1 affects K562 cell apoptosis. Under the effects of
external apoptotic stimulation (imatinib), HtrA2 was activated and
upregulated, while WT1 protein expression was downregulated,
affecting its transcriptional regulation of downstream genes, and
promoting the occurrence of apoptosis. Conversely, inhibition of
HtrA2 activation may affect its regulatory effect on WT1, leading
to sustained WT1 expression and an anti-apoptotic effect on cells.
Therefore, HtrA2 regulation of WT1 affects cell apoptosis, where a
loss of this regulatory ability prevents apoptosis. It may be
hypothesized that this effect represents one of the
non-BCR/ABL-dependent mechanisms for the treatment of CML.
The mechanism investigations of the present study
demonstrated that imatinib activated the p38 MAPK pathway, leading
to upregulation of p38 MAPK phosphorylation. However, activation of
the p38 MAPK phosphorylation was inhibited by pretreatment of cells
with UCF-101. The changes in p38 MAPK phosphorylation caused by
HtrA2 regulation of WT1 were partially consistent with the changes
in K562 cell apoptosis under drug treatment, indicating that the
p38 MAPK signaling pathway is a downstream target pathway of WT1
and its activation is indirectly affected by HtrA2 regulation of
WT1, which in turn affects the biological function of cells.
The ERK-MAPK signaling pathway is the most prevalent
MAPK signaling pathway, and it serves a significant role in cell
proliferation. The present study demonstrated that imatinib
downregulated ERK1/2 phosphorylation significantly and
persistently. However, UCF-101 pretreatment failed to reverse
p-ERK1/2 downregulation. This was identical to the features of cell
proliferation under imatinib treatment with/without UCF-101
pretreatment. Furthermore, no significant association was observed
between the phosphorylation levels of the ERK1/2-MAPK pathway and
WT1 protein level, indicating that the ERK1/2 pathway is not the
primary downstream target of WT1. Therefore, the imatinib-induced
downregulation of ERK1/2 phosphorylation may be regulated primarily
by other upstream factors.
In the present study, it was also observed that
imatinib led to the downregulation of BCR/ABL p210 fusion gene
expression, which was reversed by UCF-101-mediated suppression of
HtrA2, and even exhibited a trend of upregulation. These
observations suggest that the pattern of BCR/ABL p210 fusion gene
expression is consistent with that of WT1 under the effects of
HtrA2. This indicates that HtrA2 and its regulatory effect on WT1
may affect the sensitivity of BCR/ABL-positive cell lines to target
therapy drugs through different mechanisms, whereby
BCR/ABL-dependent and -independent mechanisms may be involved.
The results of the present study indicate that HtrA2
functions as an upstream regulatory factor of WT1, and affects
imatinib-induced K562 cell apoptosis. These data provide an insight
into novel targets for treatment of CML in the future.
Acknowledgements
The present study was supported by the National
Science and Technology Pillar Program (grant no., 2014BAI09B12),
the National Natural Science Foundation of China (grant no.,
30870913) and the Tianjin Research Program of Application
Foundation and Advanced Technology (grant no. 15JCZDJC36400).
References
|
1
|
Apperley JF: Part I: Mechanisms of
resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol.
8:1018–1029. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Deininger M, Buchdunger E and Druker BJ:
The development of imatinib as a therapeutic agent for chronic
myeloid leukemia. Blood. 105:2640–2653. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang L, Han Y, Suarez Saiz F and Minden
MD: A tumor suppressor and oncogene: The WT1 story. Leukemia.
21:868–876. 2007.PubMed/NCBI
|
|
4
|
Radich J, Dai H, Mao M, Oehler V, Schelter
J, Druker B, Sawyers C, Shah N, Stock W, Willman CL, et al: Gene
expression changes associated with progression and response in
chronic myeloid leukemia. Proc Natl Acad Sci USA. 103:2794–2799.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Varma N, Anand MS, Varma S and Juneja SS:
Role of hTERT and WT1 gene expression in disease progression and
imatinib responsiveness of patients with BCR-ABL positive chronic
myeloid leukemia. Leuk Lymphoma. 52:687–693. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Otahalova E, Ullmannova-Benson V, Klamova
H and Haskovec C: WT1 expression in peripheral leukocytes of
patients with chronic myeloid leukemia serves for the prediction of
Imatinib resistance. Neoplasma. 56:393–397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Svensson E, Vidovic K, Lassen C, Richter
J, Olofsson T, Fioretos T and Gullberg U: Deregulation of the
Wilms' tumour gene 1 protein (WT1) by BCR/ABL1 mediates resistance
to imatinib in human leukaemia cells. Leukemia. 21:2485–2494. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim MK, McGarry TJ, Broin OP, Flatow JM,
Golden AA and Licht JD: An integrated genome screen identifies the
Wnt signaling pathway as a major target of WT1. Proc Natl Acad Sci
USA. 106:11154–11159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li Y, Wang J, Li X, Jia Y, Huai L, He K,
Yu P, Wang M, Xing H, Rao Q, et al: Role of the Wilms' tumor 1 gene
in the aberrant biological behavior of leukemic cells and the
related mechanisms. Oncol Rep. 32:2680–2686. 2014.PubMed/NCBI
|
|
10
|
Li X, Li Y, Yuan T, Zhang Q, Jia Y, Li Q,
Huai L, Yu P, Tian Z, Tang K, et al: Exogenous expression of WT1
gene influences U937 cell biological behaviors and activates MAPK
and JAK-STAT signaling pathways. Leuk Res. 38:931–939. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hartkamp J, Carpenter B and Roberts SG:
The Wilms' tumor suppressor protein WT1 is processed by the serine
protease HtrA2/Omi. Mol Cell. 37:159–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hartkamp J and Roberts SG: HtrA2, taming
the oncogenic activities of WT1. Cell Cycle. 9:2508–2514. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Klupsch K and Downward J: The protease
inhibitor Ucf-101 induces cellular responses independently of its
known target, HtrA2/Omi. Cell Death Differ. 13:2157–2159. 2006.
View Article : Google Scholar : PubMed/NCBI
|