Introduction
Endometrial cancer (EC) is the fourth most common
cancer in women worldwide and is the most common type of
gynecological cancer (1). Patients
with high estrogen levels are at increased risk of developing EC
since estrogen exhibits growth-promoting properties in EC cells.
Thus, estrogen serves as a tumor initiator, since it directly
induces DNA mutations in tumor-suppressor genes and oncogenes
(2). Upon binding to its receptor,
estrogen triggers the transcription of a number of genes. There are
two classes of estrogen receptor (ER), ERα and ERβ, which are
encoded by the estrogen receptor 1 (ESR1) and ESR2 genes, bind to
the same estrogen response elements (EREs) and regulate similar
sets of genes (3). However, during
the early stages of EC, the expression of ERα is increased compared
with that of ERβ (4,5), which activates ERα upon estradiol (E2)
binding. This stimulates the expression of estrogen target genes
and leads to enhanced proliferation of the previously transformed
cells, while causing additional errors in replication and
potentially further DNA mutations (2).
Previous studies have demonstrated that EC is
associated with a shift in the ratio of the two ER subtypes
(4,6,7). In the
classical model, estrogen regulates the downstream expression of
genes by binding to ER and stimulating subsequent receptor
dimerization and nuclear translocation (8–10). At the
transcriptional level, estrogen regulates the expression of
estrogen-responsive genes by binding to ER in the nucleus. In a
number of target cells, ER regulates cell growth and
differentiation by stimulating the transcription of proto-oncogene
c-fos (c-fos), myc proto-oncogene protein (c-myc) and other
proto-oncogenes (11).
Metformin, a biguanide compound, has been
demonstrated to be effective in the treatment of polycystic ovarian
syndrome, diabetes mellitus (DM) and insulin resistance (12). Metformin exhibits chemopreventive and
anti-proliferative effects in a number of cancer types, including
ovarian and breast cancer (13,14). In
addition, application of metformin has been demonstrated to be
significantly associated with a decrease in the incidence of cancer
(15). Previous studies have reported
that metformin inhibits cell proliferation and induces apoptosis in
EC cell lines (16), in addition to
inhibiting cell growth by non-insulin- and insulin-dependent
mechanisms. Metformin regulates systemic insulin levels by
increasing insulin receptor sensitivity and uptake (17). Metformin inhibits cell proliferation
via liver kinase B1-mediated activation of 5′ AMP-activated protein
kinase (AMPK) and reduction in mammalian target of rapamycin (mTOR)
signaling (18). Activation of AMPK
inhibits the mTOR signaling pathway, thereby regulating multiple
signaling pathways involved in cell proliferation (19).
As previously reported, hypertension, DM and obesity
are high risk factors for developing EC, with insulin resistance as
a common pathophysiological basis (12). Estrogen and insulin have been
demonstrated to be important risk factors leading to the
development of EC (20). In
non-diabetic patients with breast cancer, metformin has been
demonstrated to decrease circulating estrogen levels (21). Markowska et al (22) demonstrated that the expression of ER
was decreased in patients with EC and type 2 DM (DM2) receiving
metformin compared with that in patients treated with insulin.
However, the molecular mechanism underlying the effect of metformin
on ER expression remains unclear. Metformin may inhibit the
proliferation of human EC cells by regulating the expression of ER
and estrogen-responsive genes, thereby altering the sensitivity of
cells towards estrogen. To test this hypothesis, in the present
study, the anti-neoplastic activity of metformin in EC, and the
role of metformin in the expression of ER and estrogen-responsive
genes were investigated in vitro. In addition, the
underlying molecular mechanisms of the effects of metformin on EC
cells were identified.
Materials and methods
Cell culture and reagents
The EC cell lines Ishikawa (differentiated) and
HEC-1-A (poorly differentiated) were purchased from The Cell Bank
of Type Culture Collection of Chinese Academy of Sciences
(Shanghai, China). The cell lines were maintained in RPMI-1640
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) and McCoy's 5A
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) medium containing
10% fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.) in a
humidified incubator with 5% CO2 at 37°C. The cells were
passaged every 3–5 days. Metformin and estradiol (E2) were
purchased from Sigma-Aldrich; Merck KGaA. Compound C (AMPK
inhibitor) was purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Primers were purchased from Sangon Biotech Co.,
Ltd. (Shanghai, China). The 5′-bromo-2′-deoxyuridine (BrdU) cell
proliferation ELISA assay was obtained from Roche Molecular
Diagnostics (Branchburg, NJ, USA). A bicinchoninic acid protein
assay kit was obtained from Pierce (Thermo Fisher Scientific,
Inc.). The anti-ERα (cat. no. BS6424), anti-ERβ (cat. no. BS8465),
anti-phosphorylated (p)-AMPK (cat. no. BS4457P), anti-AMPK (cat.
no. BS4457), anti-phospho-S6K (cat. no. BS4440P) and anti-S6K (cat.
no. BS4440) antibodies were purchased from Bioworld Technology,
Inc. (St. Louis Park, MN, USA).
BrdU assays for E2 and metformin
A BrdU ELISA kit was used to measure the effects of
exposure of cells to estrogen (10−6 M) and/or metformin
(5 mM) in the presence or absence of compound C (5 µM). The
Ishikawa and HEC-1-A cell lines were plated on 96-well plates at a
concentration of 8×103 cells/well. After 24 h, the cells
were serum-starved for an additional 24 h and subsequently treated
with E2 (10−6 M) in the presence or absence of metformin
(5 mM) for 24 h at 37°C. To analyze the role of AMPK, cells were
pre-treated with compound C (5 µM) for 24 h at 37°C prior to
treatment with metformin and/or E2. The effect of metformin and
estradiol was calculated as a percentage of the viability of
control cells (cultured with medium without FBS) seeded in 96-well
plates. Serum-free conditions were used for all the assays and
tests. An immunoassay was performed to monitor the synthesis of DNA
based on the incorporation of BrdU into the DNA as follows: Upon
treatment with the aforementioned compounds, the cells were
incubated with 10 µl/well BrdU labeling solution at 37°C (10 µM)
for 24 h. Prior to incubation for 30 min at room temperature, the
labeling medium was removed and 200 µl/well FixDenat (BioVision.
Inc., Milpitas, CA, USA) was added. Subsequently, the cells were
incubated for 30 min at room temperature and FixDenat solution was
removed, and the cells were incubated with 100 µl/well
anti-BrdU-peroxidase solution for 90 min at 37°C. Following the
incubation, the antibody conjugate was removed and the cells were
rinsed three times with 200 µl/well washing solution. The washing
solution was then removed, 100 µl/well substrate solution was added
and the cells were incubated at room temperature for 30 min. The
absorbance of samples was measured at 490 nm. Each experiment was
performed in triplicate and repeated three times to ensure the
consistency of the results.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Ishikawa and HEC-1-A cells were plated at a
concentration of 105 cells/well in 6-well plates for 24
h at 37°C and subsequently treated with metformin (0, 1, 5 and 15
mM) in McCoy's 5tA or RPMI-1640 medium containing 5% FBS,
respectively, for 24 h at 37°C. In addition, to assess the role of
AMPK on ER expression, cells were treated with metformin (5 mM)
with or without pre-treatment with compound C (5 µM) for 24 h.
Total RNA was extracted from the cells using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The RNA samples were subjected to DNase I
(TIANGEN Biotech Co., Ltd., Beijing, China) digestion to avoid
possible genomic DNA contamination and were reverse transcribed
with oligo-dT primers (Qiagen GmbH, Hilden, Germany) and Moloney
murine leukemia virus reverse transcriptase (Promega Corporation,
Madison, WI, USA). Reactions were performed using LightCycler 480
SYBR-Green I PCR Mastermix (Roche Diagnostics, Indianapolis, IN,
USA) in a 20-µl reaction, containing 1 µl cDNA, 8.2 µl
DNase/RNase-free deionized water, 10 µl SYBR-Green I Mastermix and
0.4 µl each primer (10 µM). Primer sequences are as follows: ERα
forward, 5′-AGTGCCTTGTTGGATGCTG-3′ and reverse,
5′-TGCCAGGTTGGTCAGTAAGC-3′; ERβ forward, 5′-AGTCCCTGGTGTGAAGCAAG-3′
and reverse, 5′-TGAGCATCCCTCTTTGAACC-3′; c-myc forward,
5′-CCTCCACTCGGAAGGACTATC-3′ and reverse,
5′-TTCGCCTCTTGACATTCTCC-3′; c-fos forward,
5′-ACTACCACTCACCCGCAGAC-3′ and reverse, 5′-GGAATGAAGTTGGCACTGGA-3′;
and GAPDH forward, 5′-CAGTCAGCCGCATCTTCTTTT-3′ and reverse,
5′-GTGACCAGGCGCCCAATAC-3′. The cycling conditions for PCR were as
follows: 95°C for 30 sec, followed by 40 cycles (two steps) of 95°C
for 5 sec and 60°C for 31 sec. PCR was performed in triplicate for
each sample on an ABI 7500 Real Time PCR Instrument (Applied
Biosystems; Thermo Fisher Scientific, Inc.) to detect the
fluorescent signals. The accumulation of PCR product was determined
as the increase in SYBR-Green fluorescence. The mRNA levels of ERα,
ERβ, c-myc and c-fos were normalized to GAPDH. The relative mRNA
levels were compared and expressed as the ratio to the control
groups (23).
Western blot analysis
Cells were plated at a density of 2×105
cells/well in 6-well plates for 24 h. To determine the change in
the expression of ERα and ERβ, the plated cell lines were treated
with metformin (0, 5 and 15 mM) with or without compound C (5 µM)
in RPMI-1640 or McCoy's 5A medium containing 5% FBS for 24 h. The
p-AMPK/AMPK and p-p70S6K/p70S6K protein levels were detected to
investigate the relevant signaling targets.
Radioimmunoprecipitation assay buffer, containing 1% NP-40, 0.5%
sodium deoxycholate and 0.1% SDS, was used for the preparation of
cell lysates. Protein extracts (20 µg) were subjected to 10%
SDS-PAGE, subsequently transferred to polyvinylidene fluoride
membranes and blocked in 5% non-fat milk in 10 mM Tris, pH 7.5, 100
mM NaCl and 0.1% Tween-20 for 1 h at room temperature. The
membranes were subsequently incubated with the aforementioned
primary antibodies (dilution, 1:1,000; Bioworld Technology, Inc.)
overnight at 4°C. The membrane was subsequently incubated with a
secondary horseradish peroxidase-linked antibody (cat. no. 7074;
dilution, 1:2,000; Cell Signaling Technology, Inc., Danvers, MA,
USA) for 2 h following washing three times with PBS-Tween-20 for 5
min. Bands were visualized using enhanced chemiluminescence
reagents (SuperSignal™ West Pico PLUS Chemiluminescent Substrate;
cat. no. 34580), according to the manufacturer's protocol (Pierce;
Thermo Fisher Scientific, Inc.). Membranes were subsequently
stripped and re-probed using a primary antibody against β-actin
(cat. no. AP0060; dilution, 1:1,000; Cell Signaling Technology,
Inc.), pan-S6K (cat. no. BS4440; dilution, 1:1,000; Bioworld
Technology, Inc.) or pan-AMPK (cat. no. BS4457; dilution, 1:1,000;
Bioworld Technology, Inc.) to establish equal loading. The relative
protein levels were normalized to β-actin and were expressed as the
ratio to the non-treatment control groups. Densitometry with
Quantity One® 1-D Analysis Software (version 4.6.9 for
PC; Bio-Rad Laboratories, Inc., Hercules, CA, USA) was used for the
quantification of protein bands, including β-actin.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. One-way analysis of variance was used for statistical
analyses and was performed using SPSS software (version 13.0; SPSS,
Inc., Chicago, IL, USA). The data between the two groups were
compared using the least significant difference test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Compound C rescues the decrease in
cell proliferation induced by metformin
A previous study from our group demonstrated that
metformin significantly decreases the viability of Ishikawa and
HEC-1-A cells in a time- and dose-dependent manner (24). Compound C has been identified as an
adenosine triphosphate-competitive AMPK inhibitor (25). In the present study, treatment with
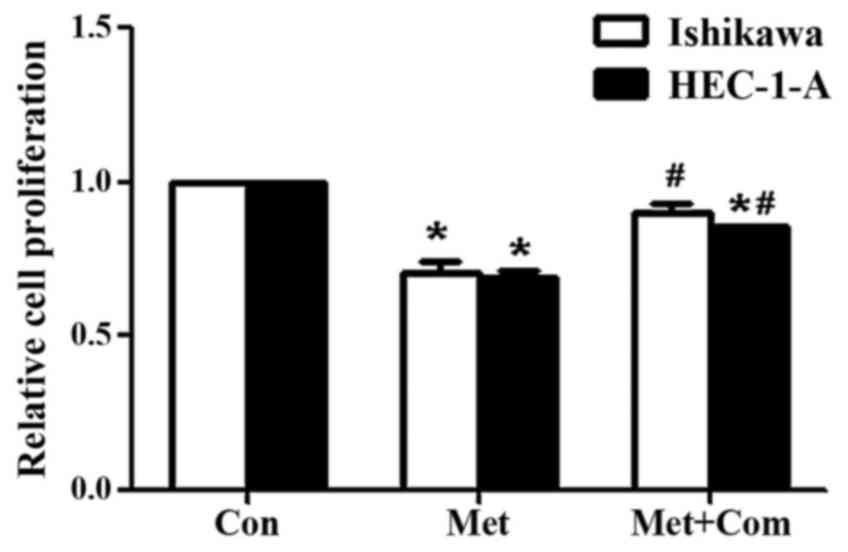
metformin significantly decreased cell proliferation in HEC-1-A and
Ishikawa cells compared with that in the control groups (P<0.05;
Fig. 1), as measured using a BrdU
assay. However, pre-treatment of Ishikawa and HEC-1-A cells with
compoundC significantly rescued the decrease in cell proliferation
induced by metformin (P<0.05; Fig.
1).
Metformin inhibits cell proliferation
through the activation of AMPK
The effects of metformin on the mTOR signaling
pathway were characterized in order to investigate the underlying
molecular mechanisms of the anti-proliferative effects of
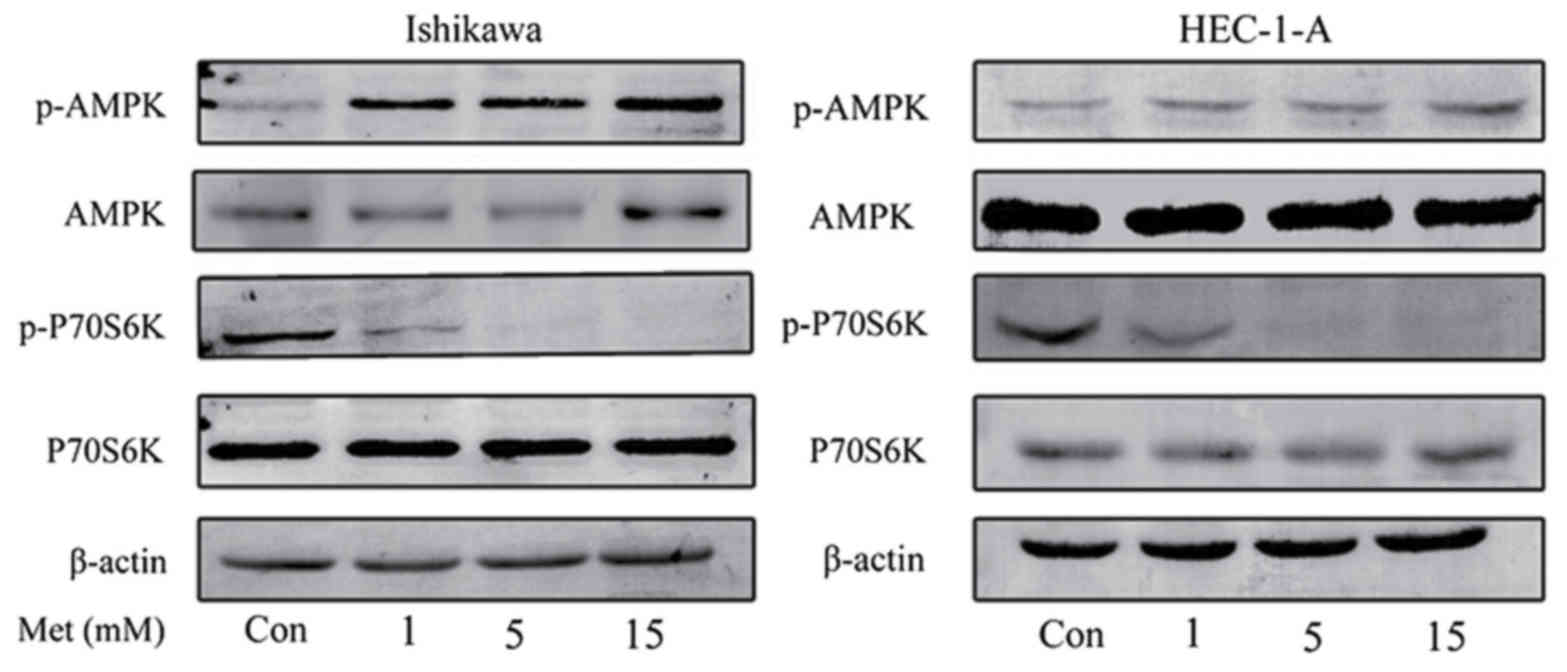
metformin. Western blot analysis demonstrated that metformin
induced the phosphorylation of AMPK in HEC-1-A and Ishikawa cells
in a dose-dependent manner (Fig. 2).
Previous studies have demonstrated that p70S6K is a downstream
target of the mTOR signaling pathway (18,26).
Metformin markedly inhibited the phosphorylation of p70S6K
following 24 h of treatment; however, the effect of metformin on
AMPK and p70S6K expression were not statistically significant. This
suggests that metformin exhibits its anti-proliferative effect by
activating AMPK and inhibiting the phosphorylation of p70S6K, which
in turn results in the inhibition of the mTOR signaling
pathway.
Inhibition of metformin attenuates the
estrogen-mediated proliferation of EC cells
To establish the role of metformin in attenuating
the estrogen-mediated proliferation of EC cells, Ishikawa and
HEC-1-A cells were pre-treated with compound C for 24 h, and
incubated with metformin and E2 for 24 h prior to performing a BrdU
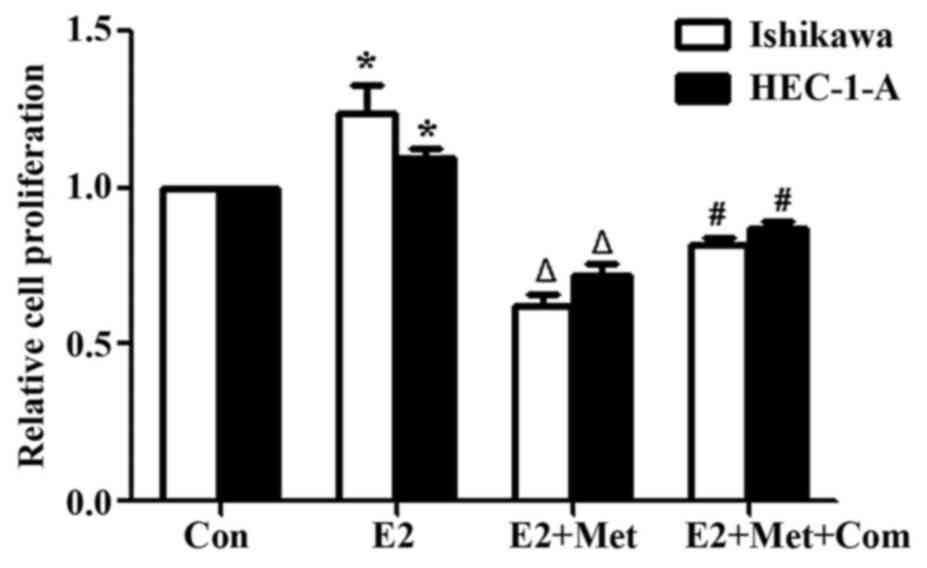
assay to assess cell proliferation. E2 significantly increased the
proliferation of Ishikawa and HEC-1-A cells in comparison with that
of the control group (P<0.05; Fig.
3). Co-treatment of cells with E2 and metformin significantly
decreased cell proliferation compared with that of the E2 alone
group (P<0.05). However, cell proliferation was significantly
rescued when cells were pre-treated with compound C for 24 h
(P<0.05, comparing the E2 + Met + Com group with the E2 + Met
group).
Regulation of ERα and ERβ expression
levels in the Ishikawa and HEC-1-A cell lines
Previous studies from our group have demonstrated
that metformin significantly downregulates ERα and upregulates ERβ
protein levels in Ishikawa and HEC-1-A cells (24). RT-qPCR results from the present study
demonstrated that, following treatment with metformin for 24 h, the
ERα and ERβ mRNA levels in Ishikawa and HEC-1-A cells were
significantly decreased and increased, respectively (P<0.05;
Fig. 4A). Western blot analysis and
RT-qPCR data from the present study suggest that metformin exhibits
a regulatory effect on ER protein and mRNA levels. However, the
regulatory effect of metformin on ER expression was rescued by
pre-treatment with compound C at the mRNA and protein level
(P<0.05; Fig. 4B and C).
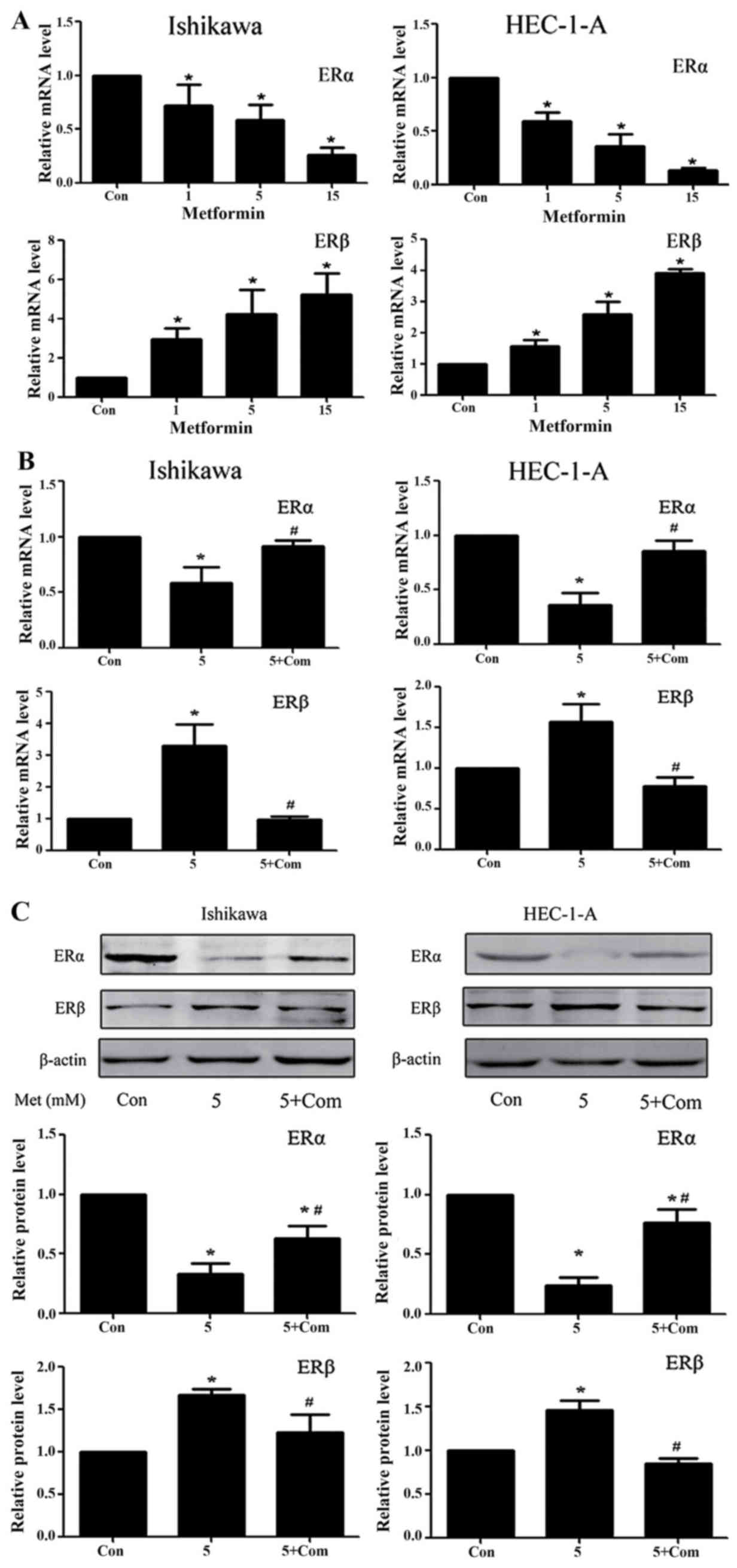 | Figure 4.Metformin downregulates ERα and
upregulates ERβ expression in Ishikawa and HEC-1-A cells in an
AMPK-dependent manner. Reverse transcription-quantitative
polymerase chain reaction analysis of ERα and ERβ expression in
Ishikawa and HEC-1-A cells following treatment with (A) metformin
(0, 1, 5 or 15 mM) or (B) 5 mM metformin pre-treatment with or
without 5 µM compound C for 24 h. ERα and ERβ mRNA levels in each
sample were calculated from a standard curve and normalized using
GAPDH mRNA levels. Results represent the mean ± standard error of
the mean of three independent experiments, each performed in
triplicate (C) Western blot analysis of ERα and ERβ expression in
Ishikawa and HEC-1-A cells following treatment with metformin (5
mM) and/or pre-treatment with compound C (5 µM) for 24 h. β-actin
was used as a loading control. Results represent the mean ±
standard error of the mean of three independent experiments.
*P<0.05 compared with control. #P<0.05, 5+ Com
group compared with the 5 mM metformin group. Con, control; 5, 5 mM
metformin; 5+ Com, 5 mM metformin combined with pre-treatment with
5 µM compound C; Met, metformin; ER, estrogen receptor; AMPK, 5′
AMP-activated protein kinase. |
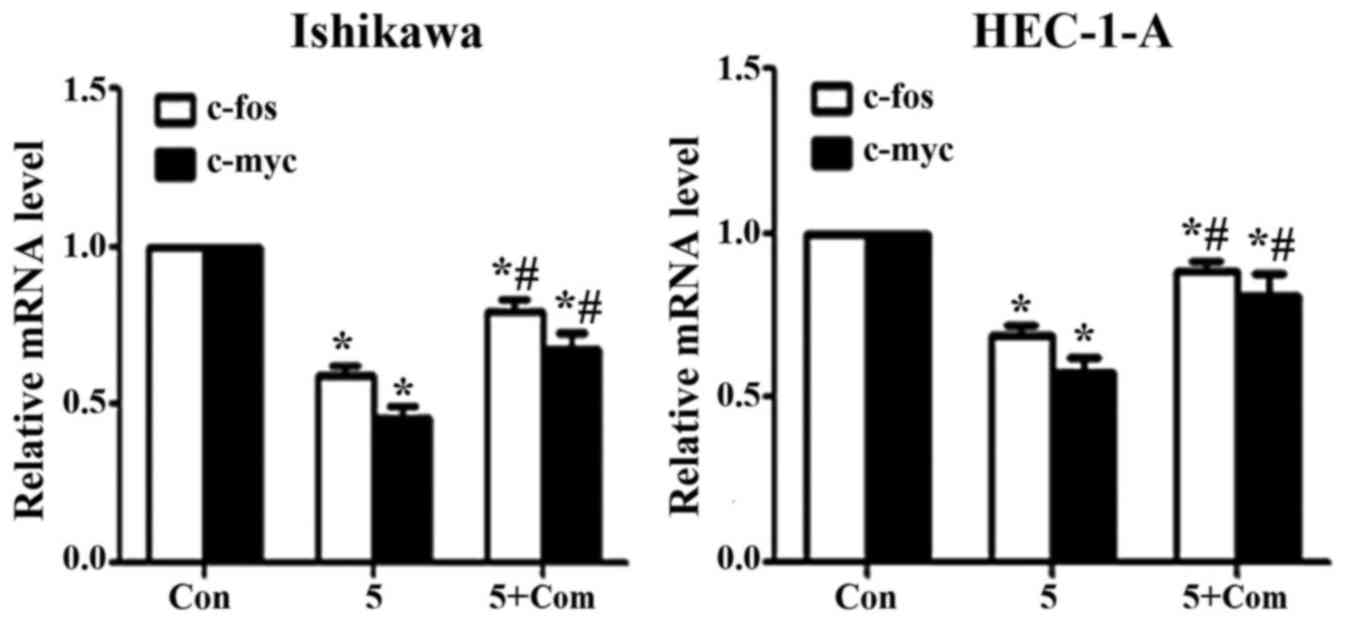
Effect of metformin on c-fos and c-myc
mRNA levels
Treatment of Ishikawa and HEC-1-A cells with
metformin significantly decreased the expression of c-fos and c-myc
at the mRNA level (P<0.05; Fig.
5); however, the effect of metformin was reversed when cells
were pre-treated with compound C prior to metformin addition
(P<0.05).
Discussion
A previous study from our group demonstrated the
roles of metformin and estrogen in inhibiting and promoting the
proliferation of EC cells (24). The
results from the present study demonstrate the role of metformin in
regulating ER expression; metformin significantly increased ERα
while significantly decreasing ERβ expression at the mRNA and
protein level in Ishikawa and HEC-1-A cells. In addition, metformin
significantly inhibited the expression of c-fos and c-myc at the
mRNA level. However, the molecular mechanisms underlying this
regulation remain unclear.
Although its etiology remains unclear, previous
studies have demonstrated the role of endocrine and genetic factors
in its initiation and progression (27). According to a number of
epidemiological studies, EC is associated with chronic exposure to
high levels of estrogen (28). In
addition, abnormalities in glucose and insulin levels are
associated with EC (29).
A previous study demonstrated that metformin
decreases the neoplastic proliferation of cells by modulating
glucose metabolism, insulin sensitivity and intracellular signaling
pathways (12). Data from the present
study demonstrated that metformin inhibits the proliferation of EC
cell lines; an effect that is reversible with pre-treatment with
compound C, an AMPK inhibitor. Metformin treatment resulted in the
activation of AMPK and its immediate downstream target, p70S6K.
These results suggest that metformin exerts its effects through the
mTOR signaling pathway and reveal a potential molecular mechanism
for its antitumor effects on EC. These results are consistent with
those from previous studies on ovarian, colon and prostate cancer
cell lines (30,31).
It has previously been demonstrated that estrogen
serves a critical role in the progression and development of EC
(32,33). Previous studies have reported that
women treated with metformin (1,500 mg/day) exhibit a significant
reduction in E2 levels (−38%; P=0.02) and a borderline significant
reduction in estrone (−10%; P=0.06) (21). Similar results were obtained in a
randomized study in obese postmenopausal female patients with a
history of polycystic ovary syndrome and/or insulin resistance
(34); treatment with metformin
(2,000 mg/day) resulted in a significant reduction in E2 levels
(−27%). Additionally, metformin may decrease the concentration of
estrogen in neoplastic tissue by locally inhibiting aromatase
activity and suppressing the synthesis of the enzyme by interacting
with its promoter, PII (35).
Estrogen has been demonstrated to promote the
proliferation of EC cell lines. Results from the present study
revealed that metformin attenuates the effect of E2 on the
proliferation of EC cells. In addition, compound C rescued the
anti-proliferative effect of metformin. Erdemoglu et al
(36) demonstrated that metformin
reduces estrogen-induced endometrial hyperplasia by inhibiting
mTOR-mediated S6K1 activation, which acts as a potent regulator of
protein synthesis and growth. Therefore, the metformin-induced
attenuation of the effect of E2 on cell proliferation may be
attributed to the activation of AMPK followed by the inhibition of
the mTOR signaling pathway.
Estrogen-induced regulation of cell proliferation
occurs via the ERα and ERβ isoforms. Estrogen and ERs serve
critical roles in the initiation and development of EC (37). ERα is associated with estrogen-induced
mitogenic signaling, whereas the function of ERβ is opposite to
that of ERα (38). In the human
mammary gland, E2 binds to ER isoforms, thereby regulating the
proliferation and differentiation of cells (39). The E2-ERα complex functions as a
regulator of the transcription of genes involved in the
proliferation, differentiation and survival of cells. In various
types of cancer, ERβ serves a critical role in inhibiting the
ERα-mediated transcription and E2-induced proliferation of cells
(38,40). Furthermore, in normal mammary
epithelial cells, ERα and ERβ differentially regulate proliferation
and apoptosis (41). The ERα/ERβ
ratio has been reported to be a key element in modulating the
activity of E2 in breast cancer cells (42). Markowska et al (22) reported a significant decrease in the
expression of ER in female patients with DM2 and EC following
administration of metformin compared with that in patients treated
with insulin (P=0.004). However, the study did not demonstrate the
effects of metformin on ER subtypes.
The data from the present study demonstrate that
metformin reduces the expression of ERα while promoting the
expression of ERβ, which may be associated with the activation of
AMPK. Therefore, the regulation of ER subtype expression following
metformin treatment may affect cell proliferation and influence the
prognosis of patients with EC. However, further studies are
required to validate this hypothesis.
Studies on breast cancer cells (43) and rat uterus (44) have demonstrated estrogen-induced
induction of c-fos, c-myc and N-myc proto-oncogene protein. In
mammalian uterus, estrogen induces the expression of c-fos
(45), c-myc (46) and c-jun (47). In addition, metformin acts as a
chemopreventive agent by downregulating the expression of c-myc to
restrict the initiation and transformation of prostatic neoplasia
(48). According to Zhang et
al (17), metformin treatment
attenuates the estrogen-dependent proliferation of c-myc and c-fos
expression in the endometrium of obese rats compared with that in
untreated controls. The findings from the present study indicate
that metformin treatment results in reduced expression of c-myc and
c-fos in vitro. Metformin treatment activated the AMPK
signaling pathway and concomitantly downregulated the expression of
c-myc and c-fos. It has previously been demonstrated that metformin
reduces the expression of c-myc in breast tumors (49) and prostatic neoplasia (48) by activating AMPK. According to Dang
(50), c-myc and c-fos oncogenes are
critical in the tumorigenesis of a number of human cancers.
In conclusion, the results from the present study
indicate that metformin inhibits the proliferation of EC cells
through the activation of AMPK and subsequent inhibition of the
mTOR signaling pathway. Further studies are required to identify
the role of metformin as chemopreventive agent in populations with
a high risk of cancer.
Glossary
Abbreviations
Abbreviations:
|
Met
|
metformin
|
|
EC
|
endometrial cancer
|
|
ER
|
estrogen receptor
|
|
E2
|
estradiol
|
|
AMPK
|
5′ AMP-activated protein kinase
|
|
Com
|
compound C
|
|
Con
|
control
|
References
|
1
|
Fader AN, Arriba LN, Frasure HE and von
Gruenigen VE: Endometrial cancer and obesity: Epidemiology,
biomarkers, prevention and survivorship. Gynecol Oncol.
114:121–127. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rižner T: Estrogen biosynthesis, phase I
and phase II metabolism, and action in endometrial cancer. Mol Cell
Endocrinol. 381:124–139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Klinge CM: Estrogen receptor interaction
with estrogen response elements. Nucleic Acids Res. 29:2905–2919.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sakaguchi H, Fujimoto J, Aoki I, Toyoki H,
Khatun S and Tamaya T: Expression of oestrogen receptor alpha and
beta in uterine endometrial and ovarian cancers. Eur J Cancer. 38
Suppl 6:S74–S75. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Utsunomiya H, Suzuki T, Harada N, Ito K,
Matsuzaki S, Konno R, Sato S, Yajima A and Sasano H: Analysis of
estrogen receptor alpha and beta in endometrial carcinomas:
Correlation with ER beta and clinicopathologic findings in 45
cases. Int J Gynecol Pathol. 19:335–341. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hu K, Zhong G and He F: Expression of
estrogen receptors ERalpha and ERbeta in endometrial hyperplasia
and adenocarcinoma. Int J Gynecol Cancer. 15:537–541. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saegusa M and Okayasu I: Changes in
expression of estrogen receptors alpha and beta in relation to
progesterone receptor and pS2 status in normal and malignant
endometrium. Jpn J Cancer Res. 91:510–518. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bolton JL and Thatcher GR: Potential
mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol.
21:93–101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hammes SR and Levin ER: Minireview: Recent
advances in extranuclear steroid receptor actions. Endocrinology.
152:4489–4495. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Prossnitz ER and Barton M: The
G-protein-coupled estrogen receptor GPER in health and disease. Nat
Rev Endocrinol. 7:715–726. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamashita S, Takayanagi A and Shimizu N:
Temporal and cell-type specific expression of c-fos and c-jun
protooncogenes in the mouse uterus after estrogen stimulation.
Endocrinology. 137:5468–5475. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bjørge T, Lukanova A, Jonsson H, Tretli S,
Ulmer H, Manjer J, Stocks T, Selmer R, Nagel G, Almquist M, et al:
Metabolic syndrome and breast cancer in the me-can (metabolic
syndrome and cancer) project. Cancer Epidemiol Biomarkers Prev.
19:1737–1745. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Del Barco S, Vazquez-Martin A, Cufi S,
Oliveras-Ferraros C, Bosch-Barrera J, Joven J, Martin-Castillo B
and Menendez JA: Metformin: Multi-faceted protection against
cancer. Oncotarget. 2:896–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dowling RJ, Goodwin PJ and Stambolic V:
Understanding the benefit of metformin use in cancer treatment. BMC
Med. 9:332011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rocha GZ, Dias MM, Ropelle ER,
Osório-Costa F, Rossato FA, Vercesi AE, Saad MJ and Carvalheira JB:
Metformin amplifies chemotherapy-induced AMPK activation and
antitumoral growth. Clin Cancer Res. 17:3993–4005. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cantrell LA, Zhou C, Mendivil A, Malloy
KM, Gehrig PA and Bae-Jump VL: Metformin is a potent inhibitor of
endometrial cancer cell proliferation-implications for a novel
treatment strategy. Gynecol Oncol. 116:92–98. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Q, Celestino J, Schmandt R,
McCampbell AS, Urbauer DL, Meyer LA, Burzawa JK, Huang M, Yates MS,
Iglesias D, et al: Chemopreventive effects of metformin on
obesity-associated endometrial proliferation. Am J Obstet Gynecol.
209:24.e1–24.e12. 2013. View Article : Google Scholar
|
|
18
|
Dowling RJ, Zakikhani M, Fantus IG, Pollak
M and Sonenberg N: Metformin inhibits mammalian target of
rapamycin-dependent translation initiation in breast cancer cells.
Cancer Res. 67:10804–10812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Viollet B, Guigas B, Garcia Sanz N,
Leclerc J, Foretz M and Andreelli F: Cellular and molecular
mechanisms of metformin: An overview. Clin Sci (Lond). 122:253–270.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Esteva FJ, Moulder SL, Gonzalez-Angulo AM,
Ensor J, Murray JL, Green MC, Koenig KB, Lee MH, Hortobagyi GN and
Yeung SC: Phase I trial of exemestane in combination with metformin
and rosiglitazone in nondiabetic obese postmenopausal women with
hormone receptor-positive metastatic breast cancer. Cancer
Chemother Pharmacol. 71:63–72. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Campagnoli C, Berrino F, Venturelli E,
Abbà C, Biglia N, Brucato T, Cogliati P, Danese S, Donadio M, Zito
G and Pasanisi P: Metformin decreases circulating androgen and
estrogen levels in nondiabetic women with breast cancer. Clin
Breast Cancer. 13:433–438. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Markowska A, Pawałowska M, Filas V, Korski
K, Gryboś M, Sajdak S, Olejek A, Bednarek W, Spiewankiewicz B,
Lubin J and Markowska J: Does Metformin affect ER, PR, IGF-1R,
β-catenin and PAX-2 expression in women with diabetes mellitus and
endometrial cancer? Diabetol Metab Syndr. 5:762013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xie Y, Wang YL, Yu L, Hu Q, Ji L, Zhang Y
and Liao QP: Metformin promotes progesterone receptor expression
via inhibition of mammalian target of rapamycin (mTOR) in
endometrial cancer cells. J Steroid Biochem Mol Biol. 126:113–120.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang J, Zhang B, Yin Z, Chen F, Liu T, Xu
H, Liu Y and Zhou X: Effects of metformin on the estrogen-induced
proliferation and the expression of ER in human endometrial cancer
cells. Zhonghua Fu Chan Ke Za Zhi. 49:932–937. 2014.(In Chinese).
PubMed/NCBI
|
|
25
|
Emerling BM, Viollet B, Tormos KV and
Chandel NS: Compound C inhibits hypoxic activation of HIF-1
independent of AMPK. FEBS Lett. 581:5727–5731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jefferies HB, Fumagalli S, Dennis PB,
Reinhard C, Pearson RB and Thomas G: Rapamycin suppresses 5′TOP
mRNA translation through inhibition of p70s6k. EMBO J.
16:3693–3704. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ellenson LH and Wu TC: Focus on
endometrial and cervical cancer. Cancer Cell. 5:533–538. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Farnell YZ and Ing NH: The effects of
estradiol and selective estrogen receptor modulators on gene
expression and messenger RNA stability in immortalized sheep
endometrial stromal cells and human endometrial adenocarcinoma
cells. J Steroid Biochem Mol Biol. 84:453–461. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Soliman PT, Wu D, Tortolero-Luna G,
Schmeler KM, Slomovitz BM, Bray MS, Gershenson DM and Lu KH:
Association between adiponectin, insulin resistance, and
endometrial cancer. Cancer. 106:2376–2381. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zakikhani M, Dowling RJ, Sonenberg N and
Pollak MN: The effects of adiponectin and metformin on prostate and
colon neoplasia involve activation of AMP-activated protein kinase.
Cancer Prev Res (Phila). 1:369–375. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gotlieb WH, Saumet J, Beauchamp MC, Gu J,
Lau S, Pollak MN and Bruchim I: In vitro metformin anti-neoplastic
activity in epithelial ovarian cancer. Gynecol Oncol. 110:246–250.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Terry KL and Missmer SA: Epidemiology of
ovarian and endometrial cancers. Pathol Epidemiol Cancer. 2017.
View Article : Google Scholar
|
|
33
|
Zhao H, Jiang Y, Liu Y, Yun C and Li L:
Endogenous estrogen metabolites as biomarkers for endometrial
cancer via a novel method of liquid chromatography-mass
spectrometry with hollow fiber liquid-phase microextraction. Horm
Metab Res. 47:158–164. 2015.PubMed/NCBI
|
|
34
|
Patel SM, Iqbal N, Kaul S, Ratcliffe SJ,
Rickels MR, Reilly MP, Scattergood T, Basu A, Fuller C and Cappola
AR: The effects of metformin and leuprolide acetate on insulin
resistance and testosterone levels in non-diabetic postmenopausal
women: A randomized, placebo-controlled trial. Fertil Steril.
94:2161–2166. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brown KA, Hunger NI, Docanto M and Simpson
ER: Metformin inhibits aromatase expression in human breast adipose
stromal cells via stimulation of AMP-activated protein kinase.
Breast Cancer Res Treat. 123:591–596. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Erdemoglu E, Güney M, Giray SG, Take G and
Mungan T: Effects of metformin on mammalian target of rapamycin in
a mouse model of endometrial hyperplasia. Eur J Obstet Gynecol
Reprod Biol. 145:195–199. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Burns KA and Korach KS: Estrogen receptors
and human disease: An update. Arch Toxicol. 86:1491–1504. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ström A, Hartman J, Foster JS, Kietz S,
Wimalasena J and Gustafsson JA: Estrogen receptor beta inhibits
17beta-estradiol-stimulated proliferation of the breast cancer cell
line T47D. Proc Natl Acad Sci USA. 101:1566–1571. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Helguero LA, Faulds MH, Gustafsson JA and
Haldosén LA: Estrogen receptors alfa (ERalpha) and beta (ERbeta)
differentially regulate proliferation and apoptosis of the normal
murine mammary epithelial cell line HC11. Oncogene. 24:6605–6616.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Paruthiyil S, Parmar H, Kerekatte V, Cunha
GR, Firestone GL and Leitman DC: Estrogen receptor beta inhibits
human breast cancer cell proliferation and tumor formation by
causing a G2 cell cycle arrest. Cancer Res. 64:423–428. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Grober OM, Mutarelli M, Giurato G, Ravo M,
Cicatiello L, De Filippo MR, Ferraro L, Nassa G, Papa MF, Paris O,
et al: Global analysis of estrogen receptor beta binding to breast
cancer cell genome reveals an extensive interplay with estrogen
receptor alpha for target gene regulation. BMC Genomics. 12:362011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Matthews J and Gustafsson JA: Estrogen
signaling: A subtle balance between ER alpha and ER beta. Mol
Interv. 3:281–292. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ali SH, O'Donnell AL, Balu D, Pohl MB,
Seyler MJ, Mohamed S, Mousa S and Dandona P: Estrogen
receptor-alpha in the inhibition of cancer growth and angiogenesis.
Cancer Res. 60:7094–7098. 2000.PubMed/NCBI
|
|
44
|
Ali SH, O'Donnell AL, Balu D, Pohl MB,
Seyler MJ, Mohamed S, Mousa S and Dandona P: High levels of
oestrogen receptor-alpha in tumorigenesis: Inhibition of cell
growth and angiogenic factors. Cell Prolif. 34:223–231. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Loose-Mitchell DS, Chiappetta C and
Stancel GM: Estrogen regulation of c-fos messenger ribonucleic
acid. Mol Endocrinol. 2:946–951. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Murphy LJ, Murphy LC and Friesen HG:
Estrogen induction of N-myc and c-myc proto-oncogene expression in
the rat uterus. Endocrinology. 120:1882–1888. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Weisz A, Cicatiello L, Persico E, Scalona
M and Bresciani F: Estrogen stimulates transcription of c-jun
protooncogene. Mol Endocrinol. 4:1041–1050. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Akinyeke T, Matsumura S, Wang X, Wu Y,
Schalfer ED, Saxena A, Yan W, Logan SK and Li X: Metformin targets
c-MYC oncogene to prevent prostate cancer. Carcinogenesis.
34:2823–2832. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Blandino G, Valerio M, Cioce M, Mori F,
Casadei L, Pulito C, Sacconi A, Biagioni F, Cortese G, Galanti S,
et al: Metformin elicits anticancer effects through the sequential
modulation of DICER and c-MYC. Nat Commun. 3:8652012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012. View Article : Google Scholar : PubMed/NCBI
|