Introduction
Hereditary nonpolyposis colorectal cancer (HNPCC) is
one of the most common inherited cancer susceptibility syndromes,
and is characterized by the early development of a number of types
of cancer, including colorectal cancer (1,2). HNPCC is
associated with germline mutations in one of the six DNA mismatch
repair genes (MMR). These are human MutL homolog 1 (hMLH1),
hMSH2, human PMS1 homolog 2 (hPMS2), human MutS
homolog 6 (hMSH6), hPMS1 and hMLH3. The
majority of germline mutations in the HNPCC have been identified in
hMSH2 and hMLH1 (3).
Genetic screening for HNPCC has primarily focused on these two
genes and it was identified that ~half of the germline mutations
identified in patients with HNPCC were detected in hMSH2.
One-third of the germline mutations are missense mutations
(4). The etiology of missense
mutations is difficult to be evaluated with respect to nonsense
mutations, frameshift mutations and large genomic aberrations.
Whether a missense mutation is associated with a disease can be
examined by carrier family data analysis or a rigorous case-control
study based on a molecular epidemiological survey (5). However, there may be incomplete family
data and a difficulty in obtaining epidemiological samples.
Evaluation of missense mutations may lack direct experimental
functional analysis, thus posing a challenge for the genetic
diagnosis of hereditary colorectal cancer. Therefore, assessment of
the pathogenicity of missense mutations is required if using the
functional assays.
The DNA mismatch repair system aids maintenance of
the stability of the genome. The human MSH2 protein is comprised of
934 amino acid residues, and the hMSH6 protein is comprised of 1360
amino acid residues. In vivo, hMSH2 and hMSH6 proteins
constitute a heterologous heterodimer (hMutSα), which binds to the
mismatch site and recognizes single base pair mismatches and small
insertion or deletion ring structure (6,7). hMLH1
dimerizes with hPMS2 to form a hMutL heterodimer. Next, the hMutSα
heterodimer and the hMutL heterodimer constitute a polymer, which
recruits other proteins required for mismatch repair (8). The protein-protein interaction of hMSH2
with hMSH6 is the premise of the hMutSα function. Inhibition of the
hMSH2-hMSH6 interaction, which is the potential consequence of a
missense mutation, may be a major cause of MMR defects in HNPCC
(9).
A previous study established a protein interaction
system for hMLH1 and hPMS2 using the yeast two-hybrid
system, which was applied for the functional evaluation of missense
mutations in hMLH1 genes (10). In the present study, ten missense
mutations of hMSH2 that were frequently detected in patients
with HNPCC in East Asia were selected. The hMSH2 missense
mutations were analyzed for their ability to affect the protein
interaction of hMSH2 with its partner hMSH6 in vivo.
Additionally, Sorting Intolerant from Tolerant tool (SIFT) tool was
applied to predict the effects of amino acid substitution.
Materials and methods
Strains and plasmids
The wild-type hMSH2 and hMSH6 cDNA
were kindly provided by Professor Josef Jiricny (Institute of
Molecular Cancer Research, University of Zurich, Zurich,
Switzerland). Restriction enzymes and ligase were purchased from
Takara Biotechnology Co., Ltd. (Dalian, China). DNA ladders were
purchased from GenScript (Piscataway, NJ, USA). The vectors for the
yeast two-hybrid system pGADT7 and pGBKT7, the reporter strain of
Saccharomyces cerevisiae AH109 and the Escherichia
coli strain Top10 were purchased from Clontech Laboratories,
Inc. (Mountainview, CA, USA). E. coli strain Top10 was used
for the genetic engineering of all plasmids.
Characteristics of the variants
A total of 10 missense mutations localized to the
entire coding region of hMSH2 gene were analyzed. A
frame-shift mutation, c.1664delA, was used as a positive control
(11–18). Mutations were identified in patients
with HNPCC from East Asia (Table
I).
 | Table I.Clinical and functional
characteristics of the 10 hMSH2 missense variants in the
present study. |
Table I.
Clinical and functional
characteristics of the 10 hMSH2 missense variants in the
present study.
| Exon | hMSH2
variation | Nucleotide
substitution | Source (Ref.) | Occurrence in
healthy individuals | SIFT score |
|---|
| 1 | T8M | c.23C>T | Chinese, Japanese
(11,12) | 0/100 | 0.10 |
| 3 | I169V | c.505A>G | Chinese (13) | 0/100 | 0.30 |
| 3 | L173R | c.518T>G | Chinese (13) | 0/100 | 0.00 |
| 3 | C199R | c.595T>C | Chinese (14) | 0/100 | 0.00 |
| 7 | A370T | c.1108G>A | Chinese (15) | 0/100 | 1.00 |
| 7 | Y408C | c.1223A>G | Chinese (13,16) | 0/100 | 0.00 |
| 7 | Q419K | c.1255C>A | Chinese (13,16) | 0/100 | 0.72 |
| 11 | Frameshift | c.1664delA | Chinese (11) |
|
|
| 12 | D603Y | c.1807G>T | Chinese (17) | 0/100 | 0.02 |
| 13 | P696L | c.2087C>T | Chinese (15) | 0/100 | 0.00 |
| 13 | S703Y | c.2108C>A | Chinese (18) | 0/100 | 0.01 |
Mutation scanning of normal
individuals
A total of 100 healthy Chinese controls, 55 males
and 45 females (age range, 35–75 years old), were randomly
recruited in The Jiangsu Cancer Hospital (Nanjing, China) from
October 2015 to December 2015. Peripheral blood was obtained from
normal individuals. Genomic DNA was isolated from 200 µl peripheral
blood using the QIAamp DNA Blood Mini kit (Qiagen GmbH, Hilden,
Germany), according to the manufacturer's protocol. DNA was
subjected to polymerase chain reaction (PCR) amplification of 16
exons of hMSH2 gene using specific primers as listed in
Table II, respectively. PCR
amplification was performed in a volume of 25 µl that contained 50
ng genomic DNA, 0.5 pmol each primer, 2.5 µl 10X Taq buffer, 2.5 µl
25 mM MgCl2, 2 µl 2.5 mM dNTPs and 1 U Taq DNA
Polymerase (Fermentas; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). A thermal cycler (Applied Biosystems Veriti Thermal Cycler;
Thermo Fisher Scientific, Inc.) was used for PCR with the following
cycling conditions: An initial denaturation step at 95°C for 2 min
followed by 35 cycles of denaturation at 95°C for 0.5 min;
annealing at 55°C for 0.5 min; and extension at 72°C for 1 min,
followed by a final extension step at 72°C for 5 min. The PCR
product was detected on denaturing high-performance liquid
chromatography (WAVE System; Transgenomic, Inc., Omaha, NE, USA) to
screen the germline mutations of normal individuals of the specific
exons. Written informed consent was obtained from all individuals.
The study was approved by the Ethics Committee of the Jiangsu
Institute of Cancer Research (Nanjing, China).
| Table II.List of primer sequences used for
polymerase chain reaction amplification of the exons of
hMSH2 gene. |
Table II.
List of primer sequences used for
polymerase chain reaction amplification of the exons of
hMSH2 gene.
| hMSH2 gene | Forward primer
sequence (5′-3′) | Reverse primer
sequence (5′-3′) |
|---|
| Exon 1 |
GCATTTTCTTCAACCAGGAG |
GTCCCTCCCCAGCACGC |
| Exon 2 |
GAAGTCCAGCTAATACAGTGC |
CTTCACATTTTTATTTTTCTACTC |
| Exon 3 |
TTTTAAAGTATGTTCAAGAGTTTG |
ATCTCCTCTATCACTAGACTC |
| Exon 4 |
TTTCATTTTTGCTTTTCTTATTCC |
TGTAATTCACATTTATAATCCATG |
| Exon 5 |
CCAGTGGTATAGAAATCTTCG |
CCATTCAACATTTTTAACCC |
| Exon 6 |
GTTTTCACTAATGAGCTTGCC |
GTGGTATAATCATGTGGG |
| Exon 7 |
GACTTACGTGCTTAGTTG |
TATGAGGACAGCACATTGCC |
| Exon 8 |
GATTTGTATTCTGTAAAATGAGATC |
CTACAAACTTTCTTAAAGTGGC |
| Exon 9 |
TGTCTTTACCCATTATTTATAGG |
CAACCTCCAATGACCCATTC |
| Exon 10 |
TGGTAGTAGGTATTTATGGAATAC |
CATCATGTTAGAGCATTTAGGG |
| Exon 11 |
GTACACATTGCTTCTAGTACAC |
AGCCAGGTGACATTCAGAAC |
| Exon 12 |
ATTCAGTATTCCTGTGTAC |
CGTTACCCCCACAAAGC |
| Exon 13 |
CGCGATTAATCATCAGTG |
GGACAGAGACATACATTTCTATC |
| Exon 14 |
GTTACCACATTTTATGTGATGG |
TTCAAGGGTAGTAAGTTTCCC |
| Exon 15 |
CTCTTCTCATGCTGTCCC |
ATAGAGAAGCTAAGTTAAAC |
| Exon 16 |
TAATTACTAATGGGACATTC |
TACCTTCATTCCATTACTGG |
In silico analysis of hMSH2
variants
The SIFT tool was used to predict whether an amino
acid substitution is deleterious (19,20). SIFT
prediction is based on a degree of conservation of amino acid
residues in sequence alignments derived from closely associated
sequences, obtained from PSI-BLAST (http://sift.jcvi.org/). SIFT calculates normalized
probabilities for all possible substitutions from the alignment.
Positions with normalized probabilities <0.05 are predicted to
be deleterious, and those ≥0.05 are predicted to be tolerated. In
the preset study, 10 missense mutations in the hMSH2 gene
were analyzed to identify deleterious variants.
Plasmid construction
Wild-type hMSH2 cDNA was cloned into pGADT7
using the restriction enzymes SfiI and XhoI to
produce transcriptional AD-fused hMSH2 construct
pGADT7-hMSH2. Wild-type hMSH6 cDNA was ligated into pGBKT7
using the restriction enzymes EcoRI and SalI, to
produce the transcriptional BD-fused hMSH6 construct
pGBKT7-hMSH6. Using pGBKT7-hMSH6 as the template, various domains
of hMSH6 were amplified with primers containing EcoRI and
SalI restriction sites (Table
III). PCR amplification was performed in a volume of 25 µl that
contained 50 ng pGBKT7-hMSH6, 0.5 pmol each primer, 2.5 µl 10X Taq
buffer, 2.5 µl 25 mM MgCl2, 2 µl 2.5 mM dNTPs and 1 U
TaKaRa LA Taq Polymerase (Takara Biotechnology Co., Ltd.). The
Applied Biosystems Veriti Thermal Cycler was used for PCR with the
following cycling conditions: An initial denaturation step at 95°C
for 2 min, followed by 35 cycles of denaturation at 95°C for 0.5
min; annealing at 55°C for 0.5 min; and extension at 72°C for 5
min, followed by a final extension step at 72°C for 10 min. The
PCR-amplified fragments were then cloned into pGBKT7 using
restriction enzymes (EcoRI and SalI), to produce
recombinant pGBKT7 plasmids with various hMSH6 domains. Genetic
sequencing was then performed using the dideoxynucleotide chain
termination method with an automatic sequencer (ABI 3100; Thermo
Fisher Scientific, Inc.) (21).
| Table III.List of primer sequences used for
polymerase chain reaction amplification of various hMSH6
domains. |
Table III.
List of primer sequences used for
polymerase chain reaction amplification of various hMSH6
domains.
| Domain | Forward primer
sequence (5′-3′) | Reverse primer
sequence (5′-3′) |
|---|
| MutS I–IV |
CGGAATTCTCTGCCCCTCAAAATTCTG |
GCGTCGACTAAGGACCATCACCCCCTCGAC |
| MutS I–V |
CGGAATTCTCTGCCCCTCAAAATTCTG |
GCGTCGACTATAATTCCTTAATCAAAGTCAG |
| MutS II–IV |
CGGAATTCACCAAGGGTACACAGACTTAC |
GCGTCGACTAAGGACCATCACCCCCTCGAC |
| MutS II–V |
CGGAATTCACCAAGGGTACACAGACTTAC |
GCGTCGACTATAATTCCTTAATCAAAGTCAG |
| MutS III–IV |
CGGAATTCAGCACTACAAGATCTGGTGC |
GCGTCGACTAAGGACCATCACCCCCTCGAC |
| MutS III–V |
CGGAATTCAGCACTACAAGATCTGGTGC |
GCGTCGACTATAATTCCTTAATCAAAGTCAG |
Site-directed mutagenesis and hMSH2
variant construction
The nucleotide substitutions were introduced into
the hMSH2 cDNA using overlapping PCR site-directed
mutagenesis, as described previously (22). Upstream primers Fa, Ra and downstream
primers Fb and Rb were synthesized according to the mutation site
and PCR reactions were applied. In the first PCR reaction, the
upstream fragment was amplified with the upstream primer Fa and the
mutant primer Ra, using pGADT7-hMSH2 as template. In the second PCR
reaction, the downstream fragment was amplified with the downstream
primer Rb and the mutant primer Fb using pGADT7-hMSH2 as template.
In the third PCR reaction, the final fragment with the target
mutant was amplified with primers Fa and Rb using the previous two
overlapping PCR-amplified products as the template. Specific
primers were listed in Table IV. PCR
amplification was performed in a volume of 25 µl that contained 50
ng template DNA, 0.5 pmol of each primer, 2.5 µl 10X Taq buffer,
2.5 µl 25 mM MgCl2, 2 µl 2.5 mM dNTPs and 1 U Taq DNA
Polymerase (Fermentas; Thermo Fisher Scientific, Inc.). The Applied
Biosystems Veriti Thermal Cycler was used for PCR with the
following cycling conditions: An initial denaturation step at 95°C
for 2 min, followed by 35 cycles of denaturation at 95°C for 0.5
min; annealing at 55°C for 0.5 min; and extension at 72°C for 2
min, followed by a final extension step at 72°C for 5 min. The
final PCR product was cut with appropriate restriction enzymes
(Table IV) and cloned into
pGADT7-hMSH2 to replace the wild-type fragment. The recombinant
plasmid pGADT7-hMSH2 with the target mutation was verified by
sequencing using an automatic sequencer (ABI 3100; Thermo Fisher
Scientific, Inc.).
| Table IV.List of primer sequences used for
polymerase chain reaction amplification of hMSH2 variants
and appropriate restriction enzymes for cloning. |
Table IV.
List of primer sequences used for
polymerase chain reaction amplification of hMSH2 variants
and appropriate restriction enzymes for cloning.
| Mutation site | Primer sequence
(5′-3′) | Restriction
enzymes |
|---|
| c.23C>T,
T8M | Fa:
CTAACGTTCATGATAACTTCATG Ra: CTCCAACTGCAGCaTCTCCTTCGGC Fb:
GCCGAAGGAGAtGCTGCAGTTGGAG Rb: AATTCTGCATCTTCTACAAAAGC | AclI
HindIII |
| c.505A>G,
I169V | Fa:
CGGGCCATGGAGGCCCCTGGCAATCTCTCTCAGTTTG Ra:
GTTTCCTCTGTAcGGAATCCACATAC Fb: GTATGTGGATTCCgTACAGAGGAAAC Rb:
AATTCTGCATCTTCTACAAAAGC | SfiI
HindIII |
| c.518T>G,
L173R | Fa:
CGGGCCATGGAGGCCCCTGGCAATCTCTCTCAGTTTG Ra:
CACACAGTCCTcGTTTCCTCTGTATG Fb: CATACAGAGGAAACgAGGACTGTGTG Rb:
AATTCTGCATCTTCTACAAAAGC | SfiI
HindIII |
| c.595T>C,
C199R | Fa:
CGGGCCATGGAGGCCCCTGGCAATCTCTCTCAGTTTG Ra: CGGGTAAAACACgTTCCTTTGGTCC
Fb: GGACCAAAGGAAcGTGTTTTACCCG Rb: AATTCTGCATCTTCTACAAAAGC | SfiI
HindIII |
| c.1108G>A,
A370T | Fa:
CGGGCCATGGAGGCCCCTGGCAATCTCTCTCAGTTTG Ra:
GCCTCAATTCTGtATCTTCTACAAAAG Fb: CTTTTGTAGAAGATaCAGAATTGAGGC Rb:
GCCTCGAGTCACGTAGTAACTTTTATTCGTG | SfiI
XhoI |
| c.1223A>G,
Y408C | Fa:
GAGAGATTGAATTTAGTGGAAGC Ra: GATTTATACCCTGAcAGAGTCGGTAAC Fb:
GTTACCGACTCTgTCAGGGTATAAATC Rb:
GCCTCGAGTCACGTAGTAACTTTTATTCGTG | HindIII
XhoI |
| c.1255C>A,
Q419K | Fa:
GAGAGATTGAATTTAGTGGAAGC Ra: GTTTTTCCAGAGCCTtTATAACATTAGG Fb:
CCTAATGTTATAaAGGCTCTGGAAAAAC Rb:
GCCTCGAGTCACGTAGTAACTTTTATTCGTG | HindIII
XhoI |
| c.1664delA,
frameshift | Fa:
GAGAGATTGAATTTAGTGGAAGC Ra: CATTTAAAGAAGTCAATTGCTGTTGGTAAATTTAAC
Fb: GTTAAATTTACCAACAGCAATTGACTTCTTTAAATG Rb:
GCCTCGAGTCACGTAGTAACTTTTATTCGTG | HindIII
XhoI |
| c.1807G>T,
D603Y | Fa:
GAGAGATTGAATTTAGTGGAAGC Ra: GACAACAGCATaTAGCTGAGCTAAC Fb:
GTTAGCTCAGCTAtATGCTGTTGTC Rb: GCCTCGAGTCACGTAGTAACTTTTATTCGTG | HindIII
XhoI |
| c.2087C>T,
P696L | Fa:
GAGAGATTGAATTTAGTGGAAGC Ra: GCTGACTCACATaGCACAAAACACC Fb:
GGTGTTTTGTGCtATGTGAGTCAGC Rb: GCCTCGAGTCACGTAGTAACTTTTATTCGTG | HindIII
XhoI |
| c.2108C>A,
S703Y | Fa:
GAGAGATTGAATTTAGTGGAAGC Ra: CAGTCCACAATGtACACTTCTGCTG Fb:
CAGCAGAAGTGTaCATTGTGGACTG Rb: GCCTCGAGTCACGTAGTAACTTTTATTCGTG | HindIII
XhoI |
Yeast two-hybrid screening
The Matchmaker GAL4-based two-hybrid system
(Clontech Laboratories, Inc.) was used in the present study. The
recombinant plasmids pGBKT7-hMSH6 and pGADT7-hMSH2 (wild type or
mutant) were co-transformed into S. cerevisiae strain AH109
and plated onto dropout medium SD/-Trp/-Leu (Clontech Laboratories,
Inc.) at 30°C for 2–4 days. A total of 10 clones were then selected
and inoculated on the dropout medium SD/-Trp/-leu/-His (Clontech
Laboratories, Inc.) at 30°C for 2–6 days. The growth status of each
transformant in the defect plate was then observed. The clone had
normal growth when the transformant grew in two days, whereas slow
growth was observed following 2–4 days of culture. No growth was
observed >4 days of culture.
Results
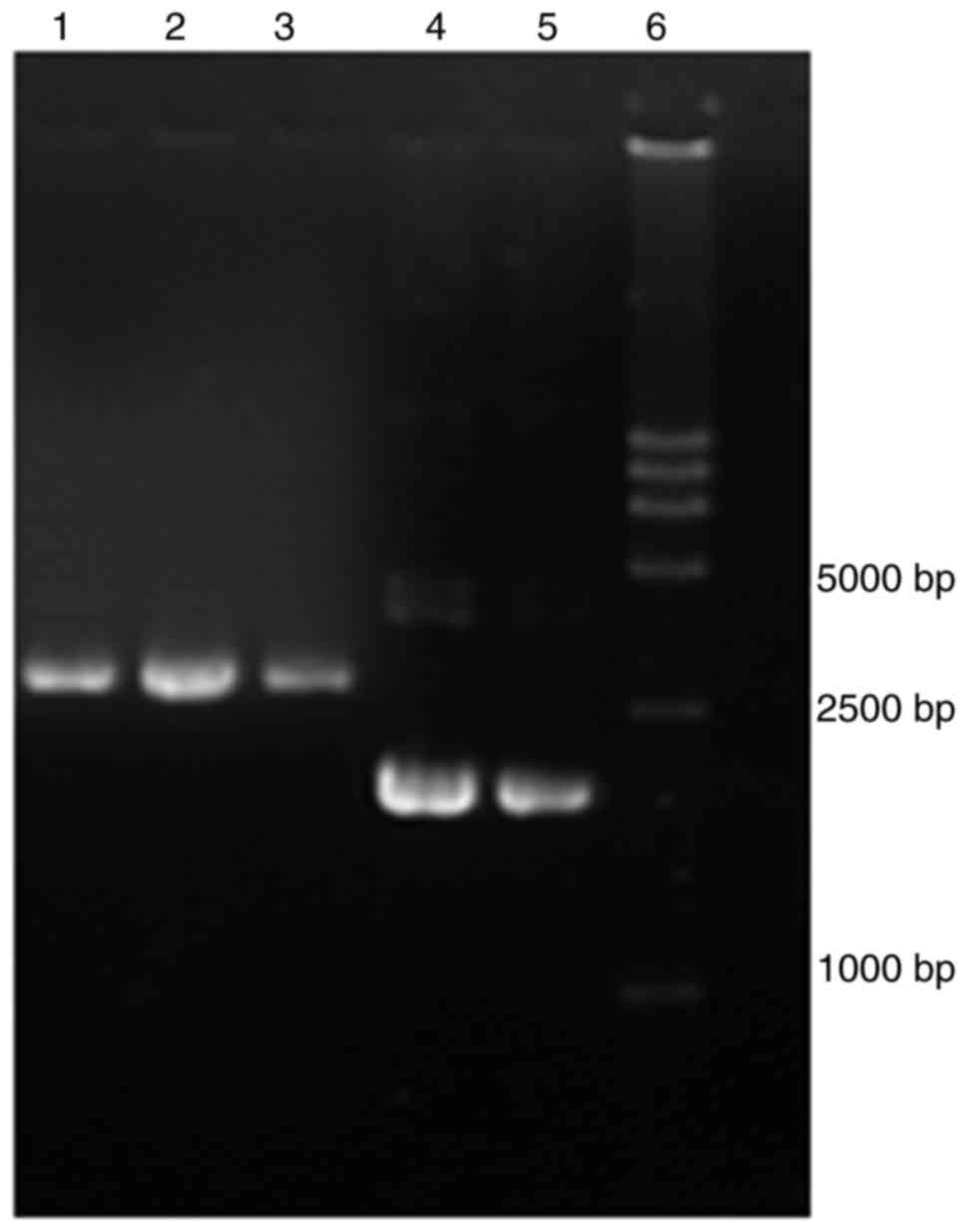
Plasmid construction
Gene engineering was used to construct the
recombinant plasmids pGADT7-hMSH2, pGBKT7-hMSH6 from wild-type
hMSH2 and hMSH6 cDNA (Fig.
1). Overlapping PCR site-directed mutagenesis was used to
construct 11 mutated pGADT7-hMSH2 plasmids. All wild-type and
variant plasmids were constructed successfully and verified by
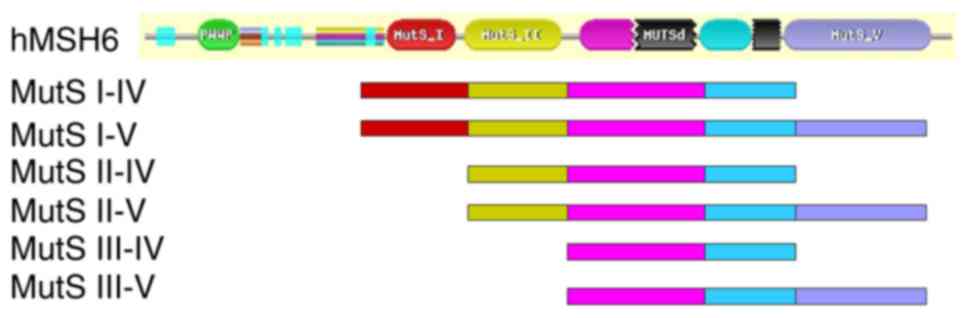
sequencing. The hMSH6 protein is comprised of 1,360 amino-acid
residues, which can be divided into a N-terminal domain (residues,
1–409), and MutS I, II, III, IV and V functional domains (MutS I is
the mismatch-binding domain, MutS II is the connector domain, MutS
III is the core domain, MutS IV is the clamp domain and MutS V is
the ATPase domain) (6,7). These domains all interact with the
corresponding functional domains of hMSH2 protein to complete the
mismatch repair function in vivo (8) hMSH6 has a large molecular weight which
may prevent proper protein folding in yeast. A total of six
recombinant pGBKT7 plasmids with various hMSH6 domains were
constructed, including pGBKT7-hMSH6-MutS I–IV (residues,
341–1,073), MutS I–V (residues, 341–1,360), MutS II–IV (residues,
518–1,073), MutS II–V (residues, 518–1,360), MutS III–IV (residues,
718–1,073) and MutS III–V (residues, 718–1,360) (Fig. 2) (6,7). All of
the pGBKT7 plasmids with different hMSH6 domains were constructed
successfully and verified by sequencing.
Mutation identification in 100 normal
individuals
A total of 100 individuals were recruited in The
Jiangsu Cancer Hospital. Denaturing high-performance liquid
chromatography analysis was used to screen the germline mutations
of the normal individuals by PCR of the specific exons. None of the
missense mutations examined in the present study were identified in
the normal controls (Table I).
Prediction of hMSH2 protein activity
and structure
The SIFT score of hMSH2 mutations
demonstrated that six of them were considered to be intolerable
(L173R, C199R, Y408C, D603Y, P696L and S703Y), indicating that
these amino acid substitutions may affect the function of the
protein. The remaining four mutations (T8M, I169V, A370T and Q419K)
were considered to be tolerable (Table
I).
Yeast two-hybrid assay of hMSH2 and
hMSH6 domains
Recombinant plasmids pGADT7-hMSH2 (wild type) and
pGBKT7 with various hMSH6 domains were co-transformed into S.
cerevisiae strain AH109 and selected on nutritional
defect plates. The growth status of each transformant was observed
(Table V). The negative control was
pGADT7/pGBKT7 and the positive control was
pGADT7-hMLH1/pGBKT7-hPMS2 (10). It
was demonstrated that the AH109 two-hybrid transformant
pGADT7-hMSH2/pGBKT7-hMSH6-MutS II–V and
pGADT7-hMSH2/pGBKT7-hMSH6-MutS III–V grew on the
histidine-deficient medium (SD/-Trp/-leu/-His). The results
demonstrated that hMSH6-MutS II–V and MutS III–V may interact with
hMSH2 in the yeast strain AH109. Additionally, the two-hybrid
transformants pGADT7/pGBKT7-hMSH6-MutS II–V and
pGADT7/pGBKT7-hMSH6-MutS III–V did not grow on the
histidine-deficient medium. These results precluded the
self-activation of pGADT7/pGBKT7-hMSH6-MutS II–V and
pGADT7/pGBKT7-hMSH6-MutS III–V for His reporter gene in yeast
strain AH109.
| Table V.Yeast two-hybrid assay of hMSH2 and
various hMSH6 domains. |
Table V.
Yeast two-hybrid assay of hMSH2 and
various hMSH6 domains.
| Transformant | Growth status |
|---|
| pGADT7/pGBKT7
(negative control) | − |
|
pGADT7-hMSH2/pGBKT7-hMSH6 | − |
|
pGADT7-hMSH2/pGBKT7-hMSH6-MutS I–IV | − |
|
pGADT7-hMSH2/pGBKT7-hMSH6-MutS I–V | − |
|
pGADT7-hMSH2/pGBKT7-hMSH6-MutS II–IV | − |
|
pGADT7-hMSH2/pGBKT7-hMSH6-MutS II–V | + |
|
pGADT7-hMSH2/pGBKT7-hMSH6-MutS III–IV | − |
|
pGADT7-hMSH2/pGBKT7-hMSH6-MutS III–V | + |
|
pGADT7-hMLH1/pGBKT7-hPMS2 (positive
control) | + |
|
pGADT7/pGBKT7-hMSH6-MutS II–V | − |
|
pGADT7/pGBKT7-hMSH6-MutS III–V | − |
Yeast two-hybrid assay of hMSH2
variants and hMSH6-MutS II–V
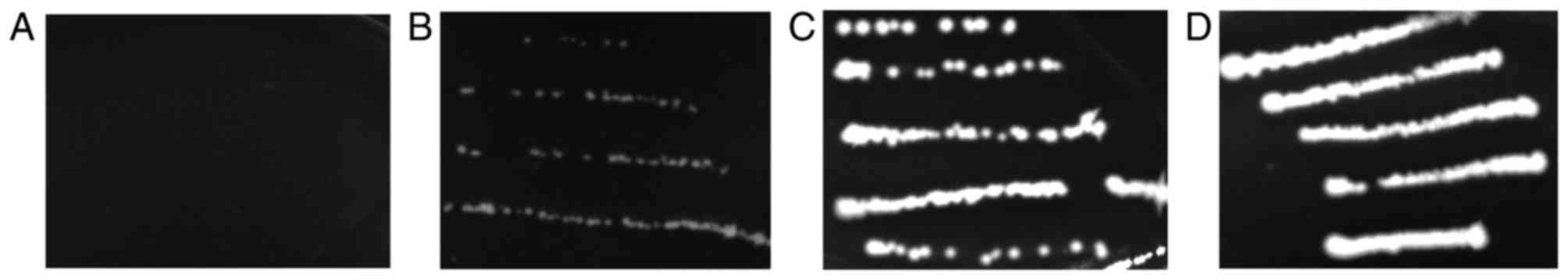
Mutant pGADT7-hMSH2 and pGBKT7-hMSH6-MutS II–V
plasmids were co-transformed into S. cerevisiae strain AH109
and selected on a SD/-Leu-Trp plate. The selected clones were then
inoculated on a SD/-Trp/-leu/-His plate. The growth status of each
transformant was observed (Table
VI). The negative control was pGADT7/pGBKT7 and positive
control was pGDAT7-hMSH2/pGBKT7-hMSH6-MutS II–V. It was
demonstrated that the yeast two-hybrid transformants T8M, I169V,
A370T and Q419K grew normally, the transformants Y408C, D603Y,
P696L and S703Y grew slowly, whereas the transformants L173R, C199R
and c.1664delA did not grow on the histidine-deficient medium in
yeast strain AH109 compared with wild-type hMSH2
(Fig. 3). The Y408C, D603Y, P696L and
S703Y mutations may partly affect the function of hMSH2 in yeast
strain AH109. c.1664delA is a frameshift mutation, thus
hMSH2 c.1664delA can not bind hMSH6-MutS II–V. The yeast
two-hybrid transformant pGDAT7-hMSH2-c.1664delA/pGBKT7-hMSH6-MutS
II–V did not grow in the histidine-deficient medium in yeast strain
AH109. The L173R and C199R mutants gave similar results to those of
c.1664delA, indicating that the mutations prevented the interaction
between hMSH2 and hMSH6.
| Table VI.Yeast two-hybrid Assay of hMSH2
variants and hMSH6-MutS II–V. |
Table VI.
Yeast two-hybrid Assay of hMSH2
variants and hMSH6-MutS II–V.
| hMSH2
variation | Growth status |
|---|
| Wild-type
hMSH2 | + |
| T8M | + |
| I169V | + |
| L173R | − |
| C199R | − |
| A370T | + |
| Y408C | +/− |
| Q419K | + |
| c.1664delA | − |
| D603Y | +/− |
| P696L | +/− |
| S703Y | +/− |
Discussion
In the present study, 10 missense mutations to
hMSH2 were examined in patients with HNPCC from East Asia.
The functional consequences of the variants were analyzed based on
their genetic, epidemiological and functional characteristics.
In vitro measurement of MMR function has been
used as a direct method to evaluate the variants in the
hMSH2 gene (23). It was
difficult to perform this complex experiment since it required
expression and purification of proteins. The yeast two-hybrid
system has been used as a method to study the interaction between
proteins (24). A previous study
established a protein interaction system for hMLH1 and hPMS2 using
the yeast two-hybrid system, which was applied for the functional
evaluation of missense mutations in hMLH1 genes (10). The yeast two-hybrid system has
advantages for detecting protein binding: Proteins may maintain a
natural conformation to mimic the physiological state in
vivo, and this system eliminates the time-consuming steps
necessary for protein purification in vitro. A previous
study used a similar system to evaluate the partial hMSH2
missense mutations I169V, L173R, Y408C, Q419K and S703Y (25). In the present study, the yeast
two-hybrid experimental method has been improved and the results
have been evaluated using SIFT to evaluate the enlarged
hMSH2 missense mutations. The yeast two-hybrid system was
used to analyze the ability of hMSH2 missense mutations to
affect the interaction of hMSH2 with its partner hMSH6 in
vivo. The growth status of each transformant provided an
indication of the protein interaction strength, as the enzyme
activity of reporter genes was associated with the degree of
protein binding.
Genetic engineering was used to construct
recombinant plasmids pGADT7-hMSH2 and pGBKT7-hMSH6. Next, the
recombinant plasmids pGADT7-hMSH2 and pGBKT7-hMSH6 were
co-transformed into S. cerevisiae strain AH109. The results
demonstrated that the yeast two-hybrid transformant couldn't grow
in histidine-deficient medium, indicating that the interaction
between hMSH2 and hMSH6 may be inhibited in yeast AH109. The
possible reason behind this may be the inappropriate folding of
hMSH6 protein when expressed in yeast strain AH109, owing to its
size. Additionally, six recombinant pGBKT7 plasmids with various
hMSH6 domains were constructed. The results demonstrated that the
transformant pGADT7-hMSH2/pGBKT7-hMSH6-MutS II–V and
pGADT7-hMSH2/pGBKT7-hMSH6-MutS III–V were able to grow on the
histidine-deficient medium (Table V).
These results indicate that hMSH6-MutS II–V and MutS III–V may
interact with hMSH2 in yeast strain AH109. The hMSH6 MutS II–V
domain contains amino acid residues 518–1,360 of the hMSH6 protein,
which comprises the core regions of the full protein. The yeast
two-hybrid transformant pGADT7-hMSH2/pGBKT7-hMSH6-MutS II–V was
used to construct the interaction system of hMSH2-hMSH6 and to
evaluate hMSH2 missense variants (Table VI).
SIFT scores revealed that T8M, I169V, A370T and
Q419K were tolerant substitutions. The results of the present study
indicated that these variants may be nonfunctional polymorphisms or
may affect the protein function through other molecular
mechanisms.
SIFT scores revealed that Y408C, D603Y, P696L and
S703Y were intolerable substitutions. In the Y408C mutant, tyrosine
and cysteine belong to neutral amino acids, but their
stereochemistry differs. For the D603Y mutant, aspartic acid is an
acidic amino acid, whereas tyrosine is an aromatic amino acid,
meaning their reactivities may differ. P696L was associated with a
loss of hMSH2 protein expression and high level of microsatellite
instability in tumors (15). For
S703Y, homology modeling revealed that steric hindrance was
increased because of the serine-to-tyrosine mutation at residue
703. Finally, the hydrogen bonds of Ala700 and Glu706 were not
formed, affecting the whole molecular structure and leading to
inactivation of hMSH2 function (18).
The yeast two-hybrid transformant of the four variants grew slowly
on the histidine-deficient medium. Therefore, these variants may
affect the partial interaction and partly affect the function of
hMSH2.
L173R and C199R are highly conserved from yeast to
humans (6). SIFT scores revealed that
they were intolerable substitutions. For the L173R mutation,
leucine is an uncharged hydrophobic amino acid, whereas arginine is
a positively charged, weak-basic amino acid. Their functions may
differ as a result of differences in solubility and ionization
properties. For the C199R mutation, cysteine is an uncharged
hydrophilic amino acid, whereas arginine is a positively charged,
weak-basic amino acid. Their functions may also differ as a result
of differences in stereochemistry and ionization properties. The
yeast two-hybrid transformant of the two variants, including
c.1664delA, did not grow on histidine-deficient medium. c.1664delA
is a frameshift mutation, thus hMSH2 c.1664delA is not able to bind
hMSH6-MutS II–V. L173R and C199R may have a similar effect as
c.1664delA. Therefore, it was considered that they these mutations
may be pathological.
In the present study, segregation studies were not
conducted because of the unavailability of family samples.
Therefore, the synergistic effect of low-risk alleles of MMR genes,
which was previously described (26),
was not evaluated in the present study.
In conclusion, the present study established the
preliminary hMSH2/hMSH6 protein-interaction system using a yeast
two-hybrid system. Combined with SIFT analysis and amino acid
analysis, an evaluation system for missense mutations in
hMSH2 was established. Using this system, 10 missense
mutations in hMSH2 were examined. The results demonstrated
that the hMSH2 mutations L173R and C199R caused a functional
change in the hMutSα complex and were identified as pathological
mutations. The Y408C, D603Y, P696L and S703Y variants partially
affected interaction, and may partly affect the function of hMSH2.
The remaining four variants, T8M, I169V, A370T and Q419K, may be
non-functional polymorphisms or may affect the protein function
through other molecular mechanisms. The present study evaluated the
functional consequences of several previously unknown missense
mutations in the hMSH2 gene, which could contribute to
clinical diagnosis and mutation screening of HNPCC.
Acknowledgements
The authors thank Professor Josef Jiricny (Institute
of Molecular Cancer Research, University of Zurich, Switzerland)
for providing the hMSH2 and hMSH6 cDNAs.
Funding
The present study was supported by the Project of
Jiangsu Provincial Commission of Health and Family Planning (grant
nos. H201510 and H2017035), Jiangsu Provincial Medical Youth Talent
(grant no. QNRC2016652), the Natural Science Foundation of Jiangsu
Province (grant nos. BK20141490 and BK20161598) and the Research
Project of Jiangsu Cancer Hospital (grant no. ZM201107).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MZ designed the study, performed the research and
wrote the manuscript. XZ and SC constructed all the plasmids. JY
analyzed the data. YZ performed the yeast two-hybrid assay. ML
collected the blood samples and performed mutation scanning of
normal individuals. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This research was approved by the Ethics Committee
of the Jiangsu Institute of Cancer Research (Nanjing, China) and
written informed consent was obtained from all individuals.
Consent for publication
Informed consent for the publication of any
associated data and accompanying images was obtained from all the
normal individuals.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Brosens LA, Offerhaus GJ and Giardiello
FM: Hereditary colorectal cancer: Genetics and screening. Surg Clin
North Am. 95:1067–1080. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lynch HT and de la Chapelle A: Genetic
susceptibility to non-polyposis colorectal cancer. J Med Genet.
36:801–818. 1999.PubMed/NCBI
|
|
3
|
In SiGHT: InSiGHT variant databases.
https://www.insight-group.org/variants/databases/June
29–2017
|
|
4
|
Woods MO, Williams P, Careen A, Edwards L,
Bartlett S, McLaughlin JR and Younghusband HB: A new variant
database for mismatch repair genes associated with Lynch syndrome.
Hum Mutat. 28:669–673. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peltomaki P and Vasen HF: Mutations
predisposing to hereditary nonpolyposis colorectal cancer: Database
and results of a collaborative study. The International
Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer.
Gastroenterology. 113:1146–1158. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Obmolova G, Ban C, Hsieh P and Yang W:
Crystal structures of mismatch repair protein MutS and its complex
with a substrate DNA. Nature. 407:703–710. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lamers MH, Perrakis A, Enzlin JH,
Winterwerp HH, de Wind N and Sixma TK: The crystal structure of DNA
mismatch repair protein MutS binding to a GxT mismatch. Nature.
407:711–717. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiricny J: The multifaceted
mismatch-repair system. Nat Rev Mol Cell Biol. 7:335–346. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Geng H, Sakato M, DeRocco V, Yamane K, Du
C, Erie DA, Hingorani M and Hsieh P: Biochemical analysis of the
human mismatch repair proteins hMutSα MSH2(G674A)-MSH6 and
MSH2-MSH6(T1219D). J Biol Chem. 287:9777–9791. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fan Y, Wang W, Zhu M, Zhou J, Peng J, Xu
L, Hua Z, Gao X and Wang Y: Analysis of hMLH1 missense mutations in
East Asian patients with suspected hereditary nonpolyposis
colorectal cancer. Clin Cancer Res. 13:7515–7521. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang XL, Yuan Y, Zhang SZ, Cai SR, Huang
YQ, Jiang Q and Zheng S: Clinical and genetic characteristics of
Chinese hereditary nonpolyposis colorectal cancer families. World J
Gastroenterol. 12:4074–4077. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nomura S, Sugano K, Kashiwabara H,
Taniguchi T, Fukayama N, Fujita S, Akasu T, Moriya Y, Ohhigashi S,
Kakizoe T and Sekiya T: Enhanced detection of deleterious and other
germline mutations of hMSH2 and hMLH1 in Japanese hereditary
nonpolyposis colorectal cancer kindreds. Biochem Biophys Res
Commun. 271:120–129. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fan Y, Liu X, Zhang H, Dai J, Zhang X, Zhu
M, Gao X and Wang Y: Variations in exon 7 of the MSH2 gene and
susceptibility to gastrointestinal cancer in a Chinese population.
Cancer Genet Cytogenet. 170:121–128. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Leung SY, Chan TL, Chung LP, Chan AS, Fan
YW, Hung KN, Kwong WK, Ho JW and Yuen ST: Microsatellite
instability and mutation of DNA mismatch repair genes in gliomas.
Am J Pathol. 153:1181–1188. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang R, Hsiung C, Wang JY, Lai CH, Chien
HT, Chiu LL, Liu CT, Chen HH, Wang HM, Chen SX, et al: Germ line
MLH1 and MSH2 mutations in Taiwanese Lynch syndrome families:
Characterization of a founder genomic mutation in the MLH1 gene.
Clin Genet. 75:334–345. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Y, Liu X, Fan Y, Ding J, Xu A, Zhou
X, Hu X, Zhu M, Zhang X, Li S, et al: Germline mutations and
polymorphic variants in MMR, E-cadherin and MYH genes associated
with familial gastric cancer in Jiangsu of China. Int J Cancer.
119:2592–2596. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yuen ST, Chan TL, Ho JW, Chan AS, Chung
LP, Lam PW, Tse CW, Wyllie AH and Leung SY: Germline, somatic and
epigenetic events underlying mismatch repair deficiency in
colorectal and HNPCC-related cancers. Oncogene. 21:7585–7592. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jin HY, Yan HL, Song LH, Cui L, Ding YJ
and Sun SH: Identification and functional analysis of a novel
germ-line mutation in hMSH2 from a Chinese hereditary nonpolyposis
colorectal cancer family. Acad J Sec Mil Med Univ. 26:888–891.
2005.(In Chinese).
|
|
19
|
Ng PC and Henikoff S: Predicting
deleterious amino acid substitutions. Genome Res. 11:863–874. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vander Molen J, Frisse LM, Fullerton SM,
Qian Y, Del Bosque-Plata L, Hudson RR and Di Rienzo A: Population
genetics of CAPN10 and GPR35: Implications for the evolution of
type 2 diabetes variants. Am J Hum Genet. 76:548–560. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nystrom-Lahti M, Perrera C, Raschle M,
Panyushkina-Seiler E, Marra G, Curci A, Quaresima B, Costanzo F,
D'Urso M, Venuta S and Jiricny J: Functional analysis of MLH1
mutations linked to hereditary nonpolyposis colon cancer. Genes
Chromosomes Cancer. 33:160–167. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Clark AB, Cook ME, Tran HT, Gordenin DA,
Resnick MA and Kunkel TA: Functional analysis of human MutSalpha
and MutSbeta complexes in yeast. Nucleic Acids Res. 27:736–742.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rodríguez-Negrete E, Bejarano ER and
Castillo AG: Using the yeast two-hybrid system to identify
protein-protein interactions. Methods Mol Biol. 1072:241–258. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu M, Fan YM, Zhu YB and Wang YP:
Establishment of a hMSH2/hMSH6 protein interaction system and
functional evaluation of hMSH2 gene missense mutations. Zhonghua Yi
Xue Yi Chuan Xue Za Zhi. 30:559–564. 2013.(In Chinese). PubMed/NCBI
|
|
26
|
Duraturo F, Liccardo R, Cavallo A, De Rosa
M, Grosso M and Izzo P: Association of low-risk MSH3 and MSH2
variant alleles with Lynch syndrome: Probability of synergistic
effects. Int J Cancer. 129:1643–1650. 2011. View Article : Google Scholar : PubMed/NCBI
|