Introduction
The menin 1 (MEN1) inherited mutation is
associated with multiple endocrine neoplasia type 1 (MEN1), an
autosomal dominant disorder characterized by the occurrence of
multiple endocrine tumors in the parathyroid, anterior pituitary,
pancreatic islets and duodenal endocrine cells (1). Pancreatic neuroendocrine tumors (PNETs)
occur in ~60% of patients with MEN1, and are the leading cause of
mortality from this disease. Numerous MEN1 tumors lead to clinical
manifestations due to abnormalities in hormone secretion, including
insulinoma or gastrinoma (2).
The MEN1 gene locates in chromosomal 11q13.
The MEN1-encoded protein, menin, is a nuclear scaffold
protein, which coordinates chromatin remodeling and functions in
the maintenance of genomic integrity (3). It acts as a tumor repressor via its
involvement in transcriptional and cell signaling regulation
(4), telomere maintenance (5) and homologous recombination-directed DNA
repair (6). The inherited mutation of
MEN1 is heterozygous. Loss of heterozygosity (LOH) or
somatic mutation in the other allele to completely inactivate menin
is detected in the majority of MEN1 tumors (7) and even microadenoma (8), indicating it as an early event of
tumorigenesis. MEN1 is also the most common somatic mutation
in patients with sporadic PNET, and LOH has been frequently
detected (9,10).
Since the identification of the MEN1 gene in
1997, ~1,544 germline and somatic mutations in this gene have been
identified (11,12). However, somatic mutations in other
genes accumulated in the latent period and during the tumor
progression remain to be fully elucidated. In MEN1
heterozygous mice, it takes ~12 months to develop pancreatic
endocrine tumors (13); whereas, in
the conditional β-cell null mutant, it takes ~6 months for the
development of insulinoma (14). This
indicates that, following the functional inactivation of menin, it
takes time to develop clinically identifiable tumors, and other
somatic alterations may be involved.
Another feature of MEN1-associated PNET is that the
tumors are often multicentric, and secrete different hormones that
lead to different clinical manifestations (15). However, as the molecular pathogenesis
of MEN1 tumors remains to be elucidated, whether these multifocal
tumors are polyclonal lacks genetic evidence; and whether the
tumors with the same hormone expression in one patient share common
genetic variations remains to be fully elucidated.
Whole exome sequencing (WES) has been used to
identify somatic mutations in non-functional PNETs (10), sporadic insulinomas (16–18) and
MEN1-associated hyper-parathyroidism (19), but not in MEN1-associated PNETs,
particularly multiple tumors in the same patient. The present study
aimed to improve current understanding of the genetic progression
of MEN1-associated PNETs by performing WES on two
insulin-expressing tumors and peri-tumoral tissue (PT) in one
patient with MEN1.
Materials and methods
Clinical samples
Following partial pancreatectomy in a patient with
MEN1 (36-year-old male, diagnosed with hypoglycemia and Whipple
triad and multiple tumors in the pancreas) in China-Japan
Friendship Hospital on April 8, 2015 (date of surgery and
tissue/blood collection), paraffin section-based pathological
analysis was regularly performed. In addition, regions of two
pancreatic tumors (T1 and T2), a section of PT of T2, and 1 ml of
peripheral blood were preserved at 80°C for further analysis. For
the present study, informed consent was obtained from the patient.
The investigation described was performed in accordance with The
Code of Ethics of the World Medical Association (20), and all protocols were approved by the
Ethics Committee in China-Japan Friendship Hospital (Beijing,
China).
WES
Genomic DNA was extracted from the T1, T2, PT and
blood samples using Gentra Puregene Tissue or Blood kits (Qiagen,
Inc., Valencia, CA, USA). DNA fragment size selection was performed
to remove smaller DNA fragments using AxyPrep Fragment Select I
beads (Corning Costar, Corning, NY, USA). DNA was quantified by
Qubit (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and examined by agarose gel eletrophoresis for integrity.
Whole exome capture was performed on ≥1.0 µg of genomic DNA per
sample, based on KAPA Hyper Prep kits (KapaBiosystems, Inc.,
Wilmington, MA, UA) using the IDT xGen Exome Research Panel v1.0+
IDT xGen® Lockdown® (Integrated DNA
Technologies, Inc., Skokie, IL, USA) according to the
manufacturer's protocol. Paired-end multiplex sequencing of the
samples was performed on the Illumina HiSeq X10 sequencing platform
with a 350-bp insert size. WES was performed at a mean target
region depth of 150X for the blood sample, and 400X for the T1, T2
and PT samples (Geneseeq Technology, Inc., Nanjing, China).
Variant calling and pathway
analysis
The raw reads were subsequently mapped to the human
reference genome [University of California, Santa Cruz (UCSC)
Genome Browserhg19 (ftp://hgdownload.soe.ucsc.edu/goldenPath/hg19)
using Burrows-Wheeler Aligner (version:0.7.15-r1140, http://bio-bwa.sourceforge.net/) software. The
aligned reads were further processed following the GATK Best
Practices of duplicate removal, indel realignment and base
recalibration. Somatic single-nucleotide substitutions were
detected using MuTect in High Confidence mode (21). Small indels were identified using
Strelka (https://sites.google.com/site/strelkasomaticvariantcaller/home)
(22). The mutational consequences
were annotated by ANNOVAR (http://annovar.openbioinformatics.org/) (23) based on UCSC RefGene. Two calling
results were combined, and filtering was performed using an
in-house pipeline implemented in Perl. The following filtering
criteria were applied: i) Total read count in tumor DNA ≥20; ii)
variant allele frequency (VAF)=0 in normal; iii) variants in
positions listed in 1,000 G with MAF>0.01 were removed. Tumor
mutational burden was calculated as the number of only somatic
nonsynonymous missense, nonsense and frame shift indel mutations
with VAF ≥5% divided by the coding region of a tumor genome (45
Mb). Pathway analysis was performed for mutated genes via Ingenuity
Pathway Analysis web software (https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/).
Signal pathway enrichment analysis was used to derive the related
pathways, using novel missense mutations, pathogenic mutations and
previously reported changes to derive regulated genes and P<0.01
to define significantly enriched pathways. Copy number data were
derived from the WES reads using the probabilistic model of
Sequenza (version:2.1.2, https://cran.r-project.org/web/packages/sequenza/index.html)
(24).
Variant validation by Sanger
sequencing
The candidate mutations were validated and verified
by amplification of the targeted genomic region by polymerase chain
reaction (PCR) followed by Sanger sequencing (Sangon Biotech Co.,
Ltd., Shanghai, China).
Immunofluorescence
The samples were embedded in OCT and cut into 5-µm
frozen sections. The slides were fixed with 4% PFA/PBS and then
permeablized with 0.2% Triton X-100. The staining performed was
according to a standard procedure. The primary antibodies used were
as follows: mouse anti-insulin (Sigma; Merck Millipore) and rabbit
anti-glucagon (Santa Cruz Biotechnology, Inc., Dallas, TX, USA).
Fluorescence images were captured using a Zeiss LSM 800 confocal
microscope (Carl Zeiss AG, Oberkochen, Germany).
Reverse transcription-PCR (RT-PCR)
analysis
Total RNA was extracted using TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Following extraction, 1 µg of total RNA was used for RT
using a FastQuant RT kit with gDNase (Tiangen Biotech Co., Ltd.,
Beijing, China) in a 20 µl system. The PCR was performed on an
Applied Biosystems instrument, (ABI 7500 system; Thermo Fisher
Scientific, Inc.), using 1 µl template cDNA, 0.2 µM primers,
SYBR® Green Realtime PCR master mix (Toyobo Co., Ltd.,
Osaka, Japan) for 40 cycles. The thermocycling steps were as
follows: 95°C for 10 min, and 40 cycles at 95°C for 30 sec, 60°C
for 30 sec and 72°C for 30 sec. The primers used were as follows:
GAPDH, forward 3′-CTGCACCACCAACTGCTTAG-5′ and reverse
3′-GAGCTTCCCGTTCAGCTCAG-5′; Insulin, forward
3′-CTCACACCTGGTGGAAGCTC-5′ and reverse
3′-AGAGGGAGCAGATGCTGGTA-5′.
Results
Characterization of two pancreatic
tumors and PT in a patient with MEN1
As reported in our previous study (25), a 36-year-old male patient with
multiple pancreatic tumors presented with seizures associated with
hypoglycemia (blood glucose, 1.6–1.8 mmol/l; serum insulin levels,
18.6–27.6 µ IU/ml; Whipple triad) for 3 months. The patient had a
history of pituitary tumor, parathyroid tumor and verrucous nodules
on the skin. Together with the multiple pancreatic tumors, the
patient was diagnosed as MEN1. As recorded, the patient's father
was also a MEN1 syndrome patient. The patient underwent laparotomy
at the China-Japan Friendship Hospital. The tumor (T1) in the neck
was first enucleated, which resulted in the normalization of blood
glucose. However, the insulin level in the peripheral and portal
venous blood increased, suggesting that further insulin-secreting
tumors may be present. Therefore, the body and tail of the pancreas
were then excised, and seven additional tumors, including two
insulin-positive, three somatostatin-positive and two
hormone-negative tumors, were identified (25). One of the insulin-positive tumors,
termed T2, and its PT were collected. Pathological analysis based
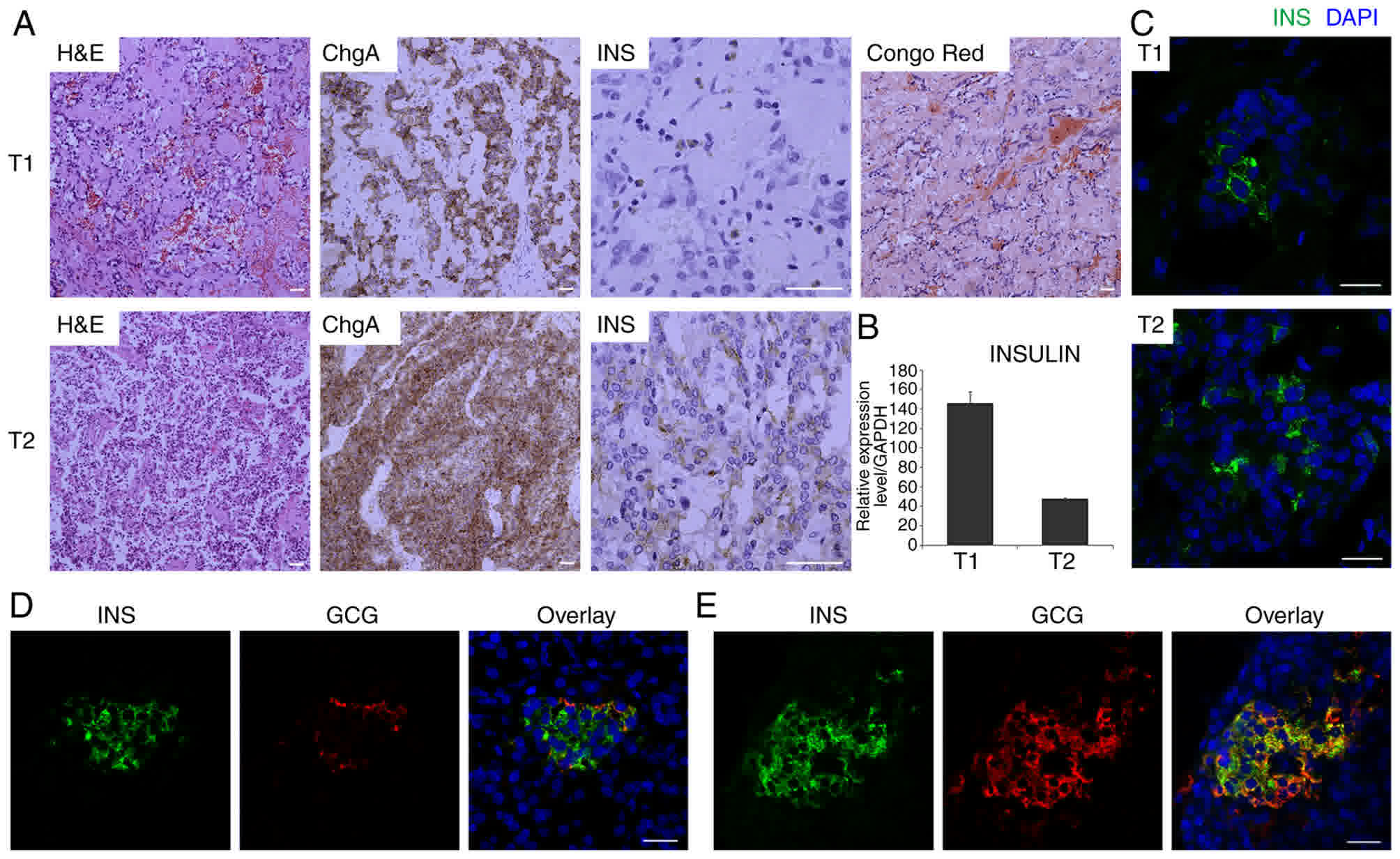
on immunohistochemistry was regularly performed (Fig. 1A and data not shown). Whereas other
tumors were of G1 grade, the T1 and T2 tumors were G2 tumors
according to the Ki67 index, which was generally 3% and focally 18%
in T1, and generally 5% and focally 18% in T2. Chromogranin A was
positive in T1 and T2. Insulin was positive in T2 and was sparsely
positive in T1. Congo Red Staining, which detects IAPP-associated
amyloidosis, showed a positive result in the T1 tumor, which has
been reported to be a common observation in insulinomas (26,27).
RT-PCR analysis using frozen samples confirmed that insulin was
expressed in the T1 and T2 tumors (Fig.
1B). As frozen samples were used for sequencing in the present
study, insulin immunostaining (Fig.
1C) was also re-examined in the T1 and T2 frozen samples. For
the PT sample, coimmunofluorescence of insulin and glucagon was
performed. In addition to normal islets (Fig. 1D), islets with glucagon and insulin
coexpressing cells were observed (Fig.
1E), which has also been reported as a common occurrence in
insulinomas (28).
MEN1 mutation analysis in T1, T2, PT
and blood samples
Although the patient presented with typical MEN1
syndrome (25), and the patient's
father was also an MEN1 patient, the MEN1 germline mutation
has not been analyzed previously. In the present study, WES was
performed on T1, T2 and PT, controlled by the blood sample of the
patient. The MEN1 gene status was first characterized in the
four samples. The results revealed a G-A mutation, leading to an
R420X germline mutation (Table I;
Fig. 2A), which was further verified
by Sanger sequencing (Fig. 2B). This
mutation is a rare event, which specifically occurs in patients
with MEN1 (11), suggesting that it
was the driver mutation in this patient. Although the reads of the
reference allele and altered allele were almost equal in the blood
and PT samples, indicating heterogenicity of the mutation, the VAF
was increased to 78% in T1, and 84% in T2 (Table I), suggesting a loss of the
chromosomal region harboring the wild-type allele in the two
tumors. This was consistent with the MEN1 LOH detected in
the majority of MEN1 tumors (7), and
was further verified by the chromosomal CNV analysis.
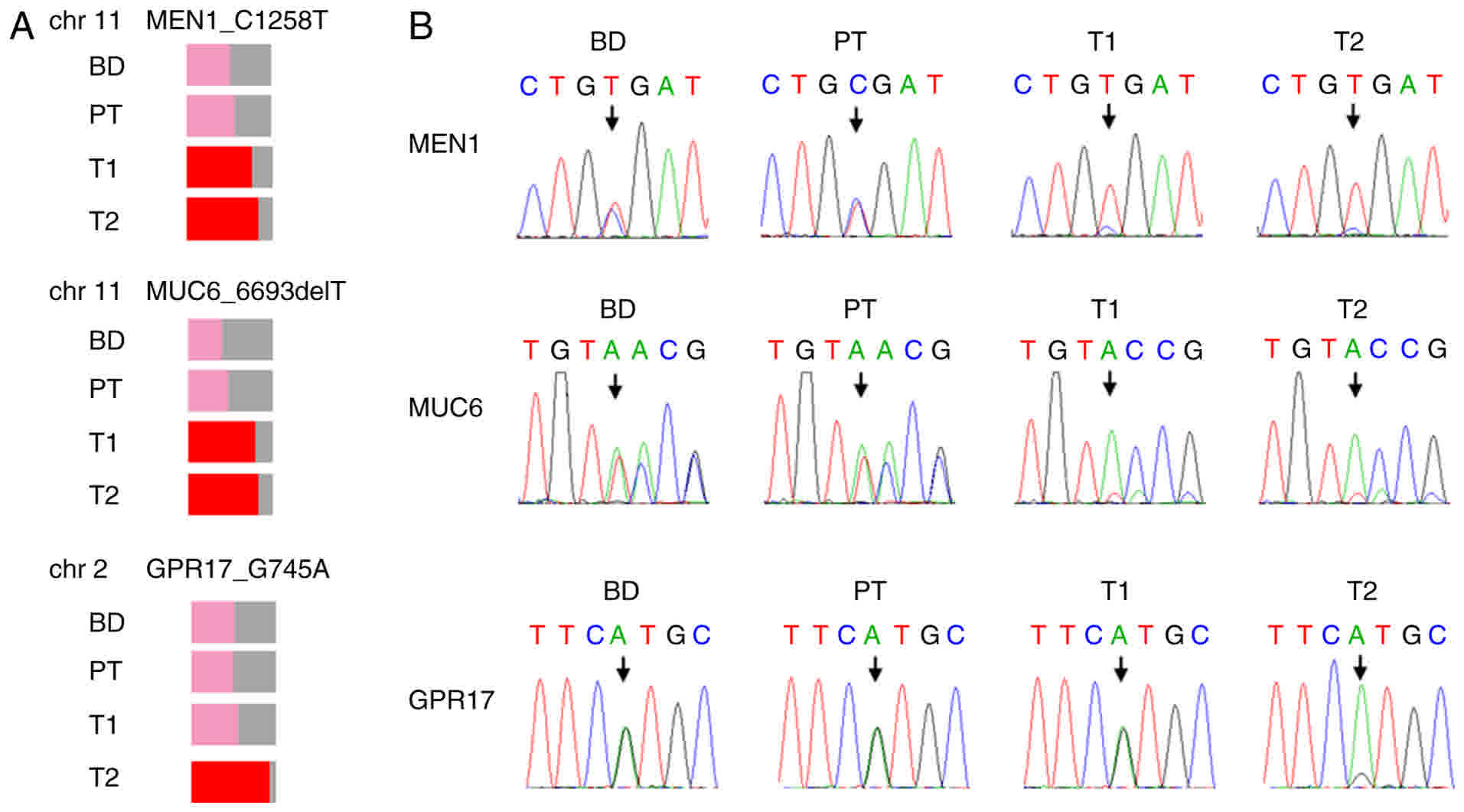 | Figure 2.Germline mutations in BD, T1 and T2,
and PT samples. (A) MEN1 C1258T, MUC6 6693delT and
GPR17 G745A were identified in the BD, PT, T1 and T2 tumor
samples. Variant allele frequency is shown as the percentage of the
colored region. Mutations without LOH are shown in pink, and
mutations with LOH are shown in red. (B) Verification of the
mutations in the BD, PT, T1 and T2 tumor samples by Sanger
sequencing are shown. MEN1, menin 1; MUC6, mucin 6,
oligomeric mucus/gel-forming; GPR17, G protein-coupled
receptor 17; BD, blood; PT, peri-tumoral tissue; LOH, loss of
heterozygosity; chr, chromosome. |
 | Table I.Germline mutations in T1, T2,
peri-tumoral tissue and blood samples. |
Table I.
Germline mutations in T1, T2,
peri-tumoral tissue and blood samples.
|
|
|
|
Mutationa (ref. allele, non-ref. allele) VAF
% |
|
|
|
|
|---|
|
|
|
|
|
|
|
|
|
|---|
| Chr. | Position | Base change | T1 | T2 | PT | Blood | Gene | AA change | refGene | 1000g 2015
aug_all |
|---|
| 11 | 64572613 | C1258T | 1 (36,126) 78 | 1 (39,211) 84 | 1 (46,61) 57 | 1 (50,50) 50 | MEN1 | R420X | NM_000244 | NAN |
| 11 | 1016108 | 6693delT | 1 (22,84) 79 | 1 (34,174) 84 | 1 (65,61) 48 | 1 (38,26) 41 | MUC6 | fs | NM_005961 | NAN |
| 2 | 128409054 | G745A | 1 (110,140) 56 | 1 (17,233) 93 | 1 (88,87) 50 | 1 (82,88) 52 | GPR17 | V249M | NM_001161417 | NAN |
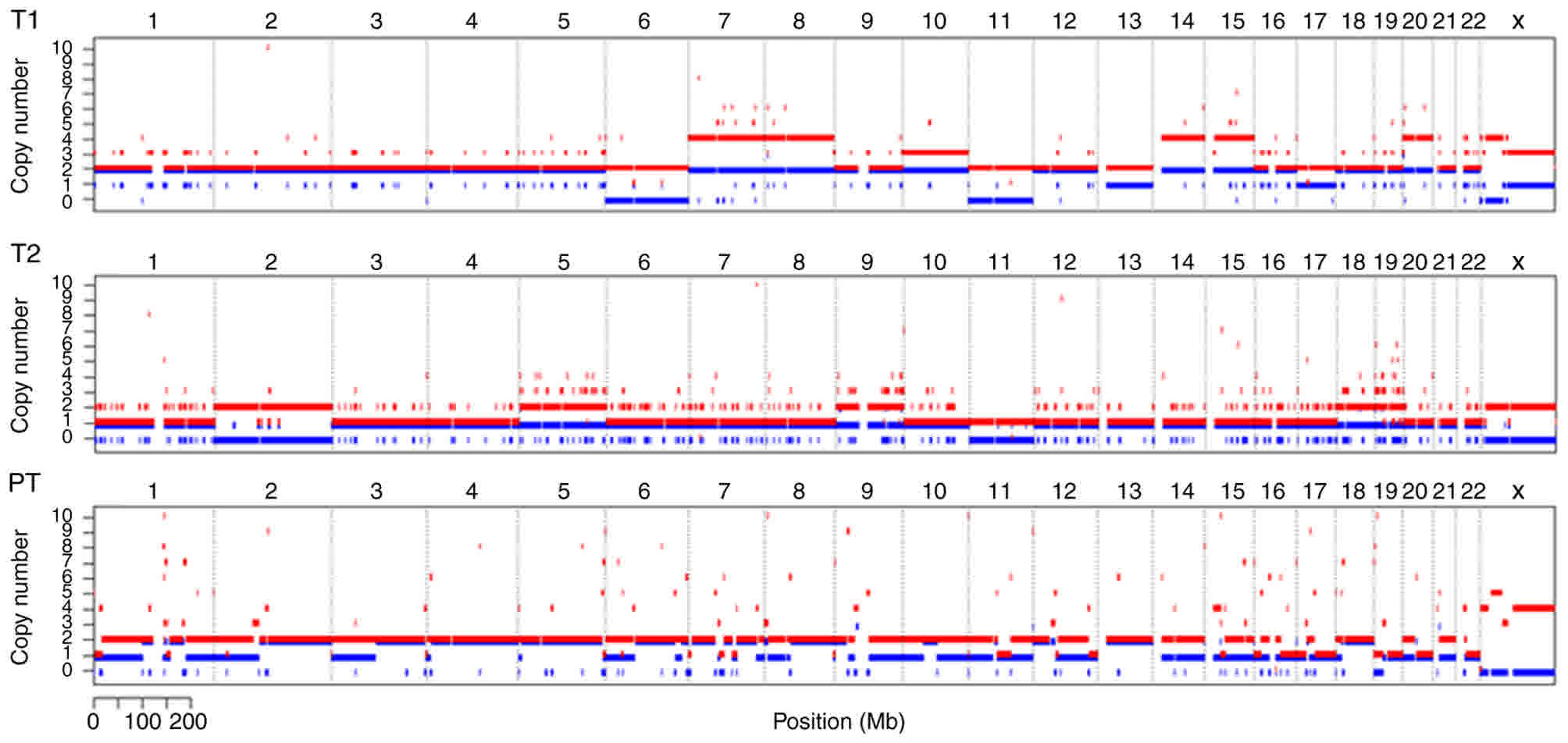
Chromosomal CNV analysis in T1, T2 and
PT samples
The results of the chromosomal CNV analysis showed
that, whereas T2 was a diploid, T1 was generally a tetraploid. LOH
of chromosome 11 occurred in the T1 and T2 tumors. No MEN1
LOH was found in the PT samples (Fig.
3), indicating that LOH may be an important event for MEN1
tumorigenesis. LOH of chromosome 6 in T1 was also present. The B
allele frequency of chromosomes 7, 8, 10, 13, 14, 15, 17 and 20
were also altered in T1 (Fig. 3). In
addition to the LOH of chromosome 11, LOH of chromosome 2 was
detected in T2, presenting as loss of one allele and duplication of
the other allele. CNV with a gain in one allele was detected in
chromosomes 5, 9, 18 and 19 in T2 (Fig.
3). Of note, CNV was also detected in ~12% of the PT cells, and
the estimated ploidy of PT was 2.2 (Fig.
3), indicating that alterations at the chromosome level caused
by a heterozygous MEN1 mutation had already occurred in the
tissues without tumorigenesis.
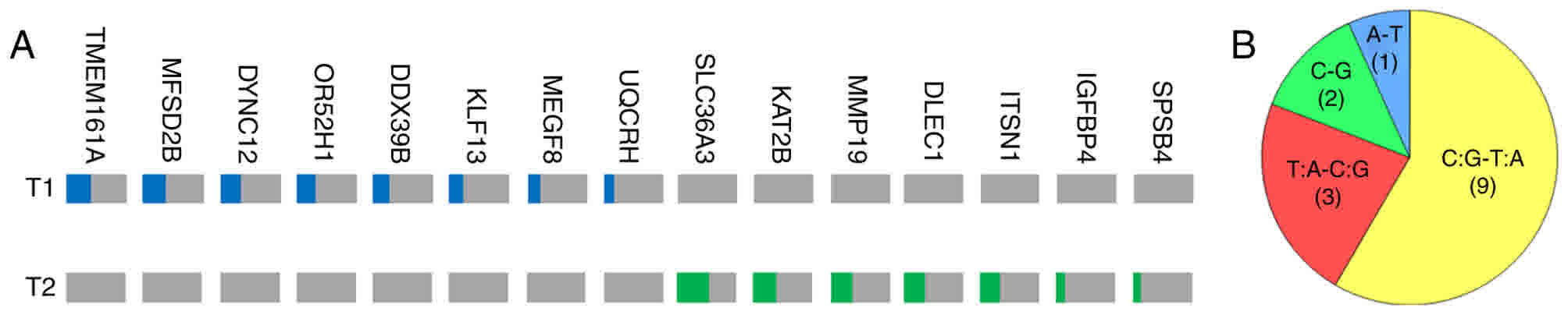
Somatic single nucleotide variants
(SNVs) in T1 and T2 tumors
The present study analyzed somatic SNV status in the
tumor samples. In total, eight and seven missense mutations with
VAF>10% were identified in T1 and T2, respectively (Table II; Fig.
4A). The mutation burden was low in the two tumors (0.16 and
0.18 per megabase). The majority of the mutations in T1 and T2 were
verified by Sanger sequencing, with the exception of KAT2B
and IGFBP4 due to difficulties in amplifying the particular
DNA fragments (data not shown). These mutations were completely
different in T1 and T2, further demonstrating their high level of
heterogeneity and polyclonal origin.
| Table II.Somatic mutations in MEN1-pancreatic
neuroendocrine tumors identified by whole exome sequencing. |
Table II.
Somatic mutations in MEN1-pancreatic
neuroendocrine tumors identified by whole exome sequencing.
|
|
|
|
Mutationa (ref. allele, non-ref. allele) VAF
% |
|
|
|
|
|---|
|
|
|
|
|
|
|
|
|
|---|
| Chr. | Position | Base change | T1 | T2 | PT | Blood | Gene | AA change | refGene | 1000g 2015
aug_all |
|---|
| 19 | 19240997 | G563A | 1 (177,109) 41 | 0 (665,1) 0 | 0 (101,0) 0 | 0 (87,0) 0 | TMEM161A | R188Q | NM_017814 | NAN |
| 2 | 24247120 | C1469T | 1 (162,101) 40 | 0 (365,0) 0 | 0 (110,0) 0 | 0 (78,1) 0 | MFSD2B | S490F | NM_001080473 | NAN |
| 2 | 172583350 | A896T | 1 (136,70) 34 | 0 (328,0) 0 | 0 (128,0) 0 | 0 (70,0) 0 | DYNC1I2 | H299L | NM_001271786 | NAN |
| 11 | 5566506 | C248T | 1 (143,70) 33 | 0 (351,0) 0 | 0 (190,0) 0 | 0 (111,0) 0 | OR52H1 | T83I | NM_001005289 | NAN |
| 6 | 31506952 | T311C | 1 (161,66) 28 | 0 (596,0) 0 | 0 (150,0) 0 | 0 (99,0) 0 | DDX39B | L104P | NM_004640 | NAN |
| 15 | 31664484 | C849G | 1 (410,137) 25 | 0 (717,0) 0 | 0 (151,0) 0 | 0 (99,0) 0 | KLF13 | S283R | NM_015995 | NAN |
| 19 | 42872733 | C6199T | 1 (285,79) 21 | 0 (858,0) 0 | 0 (151,0) 0 | 0 (111,0) 0 | MEGF8 | P2067S | NM_001410 | NAN |
| 1 | 46775912 | G140A | 1 (495,103) 17 | 0 (860,0) 0 | 0 (246,1) 0 | 0 (158,0) 0 | UQCRH | R47H | NM_001297566 | NAN |
| 5 | 150663732 | T847C | 0 (288,0) 0 | 1 (266,301) 54 | 0 (146,1) 0 | 0 (61,0) 0 | SLC36A3 | F283L | NM_181774 | NAN |
| 3 | 20082151 | C182T | 0 (166,0) 0 | 1 (140,90) 39 | 0 (67,0) 0 | 0 (61,0) 0 | KAT2B | A61V | NM_003884 | NAN |
| 12 | 56233474 | A572G | 0 (431,0) 0 | 1 (422,236) 36 | 0 (147,0) 0 | 0 (122,0) 0 | MMP19 | D191G | NM_002429 | NAN |
| 3 | 38104084 | C886T | 0 (278,0) 0 | 1 (271,145) 35 | 0 (137,0) 0 | 0 (74,1) 0 | DLEC1 | P296S | NM_007335 | NAN |
| 21 | 35237565 | G4001A | 0 (332,0) 0 | 1 (269,149) 34 | 0 (126,1) 0 | 0 (94,0) 0 | ITSN1 | S1334N | NM_003024 | NAN |
| 17 | 38600109 | C122G | 0 (208,0) 0 | 1 (282,53) 15 | 0 (86,1) 0 | 0 (58,1) 0 | IGFBP4 | P41R | NM_001552 | NAN |
| 3 | 140785241 | G295A | 0 (632,0) 0 | 1 (758,126) 14 | 0 (282,0) 0 | 0 (232,0) 0 | SPSB4 | A99T | NM_080862 | NAN |
For the mutation spectrum, 9/15 (60%) SNVs in T1 and
T2 were C/G-T/A mutations, which was the absolute dominant form.
This frequency was higher than in previously reported sporadic
PNETs (41.8%) (10) and sporadic
insulinomas (29.2 or 39.18%) (16,17), which
may represent a characteristic for MEN1-associated PNETs. In the
other six mutations, three T/A-C/G, two C-G and one A-T mutations
were present (Fig. 4B).
The present study then analyzed whether these
mutations were tumor-associated mutations. No mutations in
frequently mutated genes identified in sporadic insulinoma or
non-functional PNET were present, for example YY1, DAXX, ATRX,
MUTYH or genes in the mammalian target of rapamycin pathway
(9,10,16–18). Only
one mutation in TMEM161A was a recurrent mutation in the
Catalogue of Somatic Mutations in Cancer (COSMIC) database, but
this was predicted to be benign by several software programs used
for protein function prediction, including SIFT, Polyphen2 and LRT
(29). As shown by pathway analysis,
only two mutated genes in T1 and one mutated gene in T2 were
associated with other diseases or dysfunctions: DYNC1I2 in
Huntington's disease signaling, UQCRH in mitochondrial
dysfunction, and IGFBP4 in hepatic fibrosis and hepatic
stellate cell activation. However, no therapeutic implications were
drawn based on these SNVs.
Tumor suppressor genes mucin 6,
oligomeric mucus/gel-forming (MUC6) and G protein-coupled receptor
17 (GPR17) show germline mutations and tumor-specific LOH
As no other suspicious driver mutations were
identified in the somatic SNVs, it was hypothesized that the
consequence of chromosome alterations, including LOH of other tumor
suppressor genes, gene amplification and gene fusion, may
contribute to the frequent tumorigenesis in this patient. The
germline mutations in chromosome 11 were examined first, which
showed LOH in T1 and T2, and chromosome 6 and 2, which showed LOH
in T1 and T2, respectively. In addition to MEN1, a frame
shift deletion was identified in the C-terminus of MUC6
located in chromosome 11, which showed evidence of LOH in T1 and
T2; and a recurrent missense mutation in GPR17 located in
chromosome 2, predicted to be deleterious by several software
programs, (29) showed evidence of
LOH in T2 only (Table I; Fig. 2A and B). Taking the ploidy change into
consideration, while MUC6 and GPR17 showed one normal
allele and one altered allele in the blood and PT samples,
MUC6 had one and two copies of the altered allele in T2 and
T1, respectively, with no normal alleles; and GPR17 had two
normal and two altered alleles in T1, and only two altered alleles
in T2. MUC6 is a tumor suppressor gene, which may inhibit
tumor invasion. The C-terminal domain is critical for its function
(30,31). There is experimental evidence that
GPR17 has an anti-proliferative role in glioma cells
(32). Based on these data, the
present study demonstrated that tumor suppressor genes other than
MEN1 may undergo LOH upon chromosome alterations and
contribute to tumorigenesis.
As WES is not suitable for the detection of specific
gene amplifications, it is only possible to estimate the gene copy
number changes based on the information of chromosome CNVs. For
example, the present study found that chromosomes harboring
pro-tumor genes, including EGFR, MYC, MET and YY1,
amplified to six copies, whereas chromosomes harboring TP53
had three copies in T1, demonstrating that chromosome instability
may have an effect on the copy number and expression levels of
tumor-associated genes, thus promoting tumorigenesis in
MEN1-associated PNETs.
Discussion
MEN1-associated PNETs are always multiple, with an
average of three or four per patient (15). Although the inherited mutation in the
MEN1 gene and the secondary LOH of MEN1 is known to
be associated with tumorigenesis, the molecular pathogenesis
remains to be fully elucidated. In the present study, in a case of
MEN1 with multiple PNETs, WES was performed on two
insulin-expressing tumors and a PT sample, in order to provide an
initial understanding of the molecular tumorigenesis in
MEN1-associatedpancreatic tumors (Fig.
5).
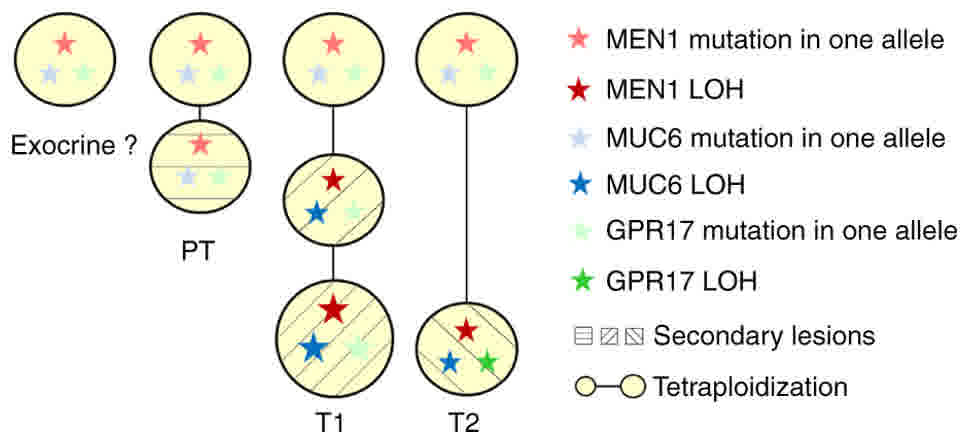 | Figure 5.Schematic diagram showing the deduced
molecular evolutionary processes of T1 and T2 tumors and other
pancreatic cells. Mutations in MEN1, MUC6 and GPR17
were the common genetic background of the pancreatic cells. In the
endocrine cells without tumorigenesis, mild CNV without loss of
chromosome 11 was observed. MEN1 LOH and MUC6 LOH
accompanying the loss of chromosome 11 occurred in T1 and T2
tumorigenesis. T1 underwent further tetraploidization. GPR17
LOH accompanying loss of chromosome 2 occurred in T2. T1 and T2
underwent distinct molecular evolutionary processes, as suggested
by the different CNV and SNV spectra. PT, peri-tumoral tissue;
MEN1, menin 1; MUC6, mucin 6, oligomeric
mucus/gel-forming; GPR17, G protein-coupled receptor 17;
CNV, copy number variation; SNV, single nucleotide variant; LOH,
loss of heterozygosity. |
The results of the present study demonstrated that
the PT region already had chromosome number variations without LOH
of MEN1, indicating that the genetic alterations may occur
independently of MEN1 LOH. Multiple low frequent SNVs
(VAF<10%) were also present in the PT sample, for which Sanger
sequencing-based verification was difficult. However, considering
that the pancreatic islets accounts for <5% of the pancreatic
volume, the frequency of CNVs and SNVs detected in the PT may not
be low if the variations are concentrated in the endocrine
compartments. Therefore, the separation of endocrine and exocrine
cells prior to WES is required for the identification of early
events prior to tumorigenesis in the islets; it is also useful to
examine the genetic changes in exocrine cells, which are presumably
considered to be unaffected by MEN1 mutations. In the
present study, frozen samples were used for WES. The technical
limitation of isolating endocrine compartments from the pancreas
made it impossible to achieve the above. In addition, abnormal
insulin and glucagon coexpression were detected in the islet cells
in the PT samples, which are commonly observed in pancreatic
endocrine tumors (28). Whether this
is one of the phenotypic clues for the genetic variations requires
further investigation.
For the two tumors showing insulin expression, they
were considered to be from different origins based on the high
genetic heterogeneity. With the exception of the loss of chromosome
11, the ploidy and CNVs in T1 and T2 were entirely different. As
loss of chromosome 11 has been reported in several cases of MEN1
(19,33), it was considered to be a non-small
probability event. Therefore, the loss of chromosome 11 may occur
independently in these two tumors and may not be considered as
co-evolutionary evidence. The SNVs with VAF>10% were also
completely different in T1 and T2, further confirming their
polyclonal origins.
With the exception of one recurrent but possibly
benign mutation, no other suspicious tumor-associated mutations
were identified in the somatic mutations; the relevance of these
somatic mutations with tumor formation was unclear. By contrast,
accompanying the chromosome alterations, germline mutations in
tumor suppressor genes MUC6 and GPR17 showed LOH in
the two tumors, or in T2, respectively; chromosomes harboring
pro-tumor genes, including EGFR, MYC, MET and YY1,
amplified to six copies, whereas chromosome harboring TP53
had three copies in T1. These data demonstrated that chromosome
instability may aggravate inherited mutations other than
MEN1, affecting the copy number and expression levels of
tumor-associated genes, and thus contributing to the tumorigenesis
in MEN1-associated PNETs.
From the therapeutic viewpoint, the low tumor
mutation burden in these two MEN1 tumors suggested that they may
not be suitable for immune checkpoint therapy (34). Although menin is involved in DNA
repair (6), the low mutation burden
suggests a different genomic signature from BRCA1/2 mutant
tumors (35). This is also consistent
with a previous finding that MEN1-mutant spontaneous PNETs
exhibited a longer telomere and reduced gene fusion event,
suggesting that MEN1-mutant tumors do not show significant
DNA repair deficiency (9). The high
level of heterogeneity of the pancreatic tumors in the same MEN1
patient, but with the same hormone-expression, suggested that
personalized treatment for patients with MEN1 should be based on
the alterations in the specific tumors of clinical concern. Rather
than genetic mutations, which may not provide sufficient
therapeutic clues in certain cases, alterations at the epigenetic,
transcriptional and translational level, in addition to the
activation status of key signaling pathways, requires systematic
investigation in the future.
Acknowledgements
The authors would like to thank Dr. Gang Niu and Dr.
Qiangzu Zhang (LemonData Biotech Co., Ltd) for their assistance
with data analysis and manuscript revisions.
Funding
This study was supported by China-Japan Friendship
Hospital Youth Science and Technology Excellence Project (grant no.
2015-QNYC-B-06 to ZW) and the National Nature Science Foundation of
China (grant no. 81302334 to ZW and 81370873 to WZ).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZW and LL were the major contributors in the
experimental design, sequencing data analysis and manuscript
writing; JL interpreted the histological data; JG, MZ and WZ
contributed to the real time PCR and immunofluorescence, ZY was
responsible for the clinical samples and the whole project.
Ethics approval and consent to
participate
Informed consent was obtained from the patient. The
investigation was performed in accordance with The Code of Ethics
of the World Medical Association, and all protocols were approved
by the Ethics Committee in China-Japan Friendship Hospital.
Consent for publication
Informed consent was obtained from the patient.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chandrasekharappa SC, Guru SC, Manickam P,
Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z,
Lubensky IA, Liotta LA, et al: Positional cloning of the gene for
multiple endocrine neoplasia-type 1. Science. 276:404–407. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tonelli F, Giudici F, Fratini G and Brandi
ML: Pancreatic endocrine tumors in multiple endocrine neoplasia
type 1 syndrome: Review of literature. Endocr Pract. 17 Suppl
3:S33–S40. 2011. View Article : Google Scholar
|
|
3
|
Busygina V, Suphapeetiporn K, Marek LR,
Stowers RS, Xu T and Bale AE: Hypermutability in a Drosophila model
for multiple endocrine neoplasia type 1. Hum Mol Genet.
13:2399–2408. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Agarwal SK, Guru SC, Heppner C, Erdos MR,
Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS,
Spiegel AM, et al: Menin interacts with the AP1 transcription
factor JunD and represses JunD-activated transcription. Cell.
96:143–152. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin SY and Elledge SJ: Multiple tumor
suppressor pathways negatively regulate telomerase. Cell.
113:881–889. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fang M, Xia F, Mahalingam M, Virbasius CM,
Wajapeyee N and Green MR: MEN1 is a melanoma tumor suppressor that
preserves genomic integrity by stimulating transcription of genes
that promote homologous recombination-directed DNA repair. Mol Cell
Biol. 33:2635–2647. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lubensky IA, Debelenko LV, Zhuang Z,
Emmert-Buck MR, Dong Q, Chandrasekharappa S, Guru SC, Manickam P,
Olufemi SE, Marx SJ, et al: Allelic deletions on chromosome 11q13
in multiple tumors from individual MEN1 patients. Cancer Res.
56:5272–5278. 1996.PubMed/NCBI
|
|
8
|
Perren A, Anlauf M, Henopp T, Rudolph T,
Schmitt A, Raffel A, Gimm O, Weihe E, Knoefel WT, Dralle H, et al:
Multiple endocrine neoplasia type 1 (MEN1): Loss of one MEN1 allele
in tumors and monohormonal endocrine cell clusters but not in islet
hyperplasia of the pancreas. J Clin Endocrinol Metab. 92:1118–1128.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Scarpa A, Chang DK, Nones K, Corbo V,
Patch AM, Bailey P, Lawlor RT, Johns AL, Miller DK, Mafficini A, et
al: Whole-genome landscape of pancreatic neuroendocrine tumours.
Nature. 543:65–71. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiao Y, Shi C, Edil BH, de Wilde RF,
Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA,
et al: DAXX/ATRX, MEN1, and mTOR pathway genes are frequently
altered in pancreatic neuroendocrine tumors. Science.
331:1199–1203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lemos MC and Thakker RV: Multiple
endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations
reported in the first decade following identification of the gene.
Hum Mutat. 29:22–32. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Concolino P, Costella A and Capoluongo E:
Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new
germline variants reported in the last nine years. Cancer Genet.
209:36–41. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fontaniere S, Tost J, Wierinckx A, Lachuer
J, Lu J, Hussein N, Busato F, Gut I, Wang ZQ and Zhang CX: Gene
expression profiling in insulinomas of Men1 beta-cell mutant mice
reveals early genetic and epigenetic events involved in pancreatic
beta-cell tumorigenesis. Endocr Relat Cancer. 13:1223–1236. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Crabtree JS, Scacheri PC, Ward JM, McNally
SR, Swain GP, Montagna C, Hager JH, Hanahan D, Edlund H, Magnuson
MA, et al: Of mice and MEN1: Insulinomas in a conditional mouse
knockout. Mol Cell Biol. 23:6075–6085. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Le Bodic MF, Heymann MF, Lecomte M, Berger
N, Berger F, Louvel A, De Micco C, Patey M, De Mascarel A, Burtin F
and Saint-Andre JP: Immunohistochemical study of 100 pancreatic
tumors in 28 patients with multiple endocrine neoplasia, type I. Am
J Surg Pathol. 20:1378–1384. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cao Y, Gao Z, Li L, Jiang X, Shan A, Cai
J, Peng Y, Li Y, Huang X, Wang J, et al: Whole exome sequencing of
insulinoma reveals recurrent T372R mutations in YY1. Nat Commun.
4:28102013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cromer MK, Choi M, Nelson-Williams C,
Fonseca AL, Kunstman JW, Korah RM, Overton JD, Mane S, Kenney B,
Malchoff CD, et al: Neomorphic effects of recurrent somatic
mutations in Yin Yang 1 in insulin-producing adenomas. Proc Natl
Acad Sci USA. 112:4062–4067. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lichtenauer UD, Di Dalmazi G, Slater EP,
Wieland T, Kuebart A, Schmittfull A, Schwarzmayr T, Diener S, Wiese
D, Thasler WE, et al: Frequency and clinical correlates of somatic
Ying Yang 1 mutations in sporadic insulinomas. J Clin Endocrinol
Metab. 100:E776–E782. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Arenas Romero MA, Fowler RG, Lucas San FA,
Shen J, Rich TA, Grubbs EG, Lee JE, Scheet P, Perrier ND and Zhao
H: Preliminary whole-exome sequencing reveals mutations that imply
common tumorigenicity pathways in multiple endocrine neoplasia type
1 patients. Surgery. 156:1351–1358. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
World Medical Association: World Medical
Association declaration of Helsinki: Ethical principles for medical
research involving human subjects. Jama. 310:2191–2194. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cibulskis K, Lawrence MS, Carter SL,
Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES
and Getz G: Sensitive detection of somatic point mutations in
impure and heterogeneous cancer samples. Nat Biotechnol.
31:213–219. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Saunders CT, Wong WS, Swamy S, Becq J,
Murray LJ and Cheetham RK: Strelka: Accurate somatic small-variant
calling from sequenced tumor-normal sample pairs. Bioinformatics.
28:1811–1817. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Favero F, Joshi T, Marquard AM, Birkbak
NJ, Krzystanek M, Li Q, Szallasi Z and Eklund AC: Sequenza:
Allele-specific copy number and mutation profiles from tumor
sequencing data. Ann Oncol. 26:64–70. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang Z, Tan H, Sun Y, Si S, Xu L, Liu X,
Liu L, Zhou W and Huang J: Intraoperative portal vein insulin assay
combined with occlusion of the pancreas for complex pancreatogenous
hypoglycemia: Two cases report. Medicine (Baltimore). 95:e39282016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cao P, Abedini A and Raleigh DP:
Aggregation of islet amyloid polypeptide: From physical chemistry
to cell biology. Curr Opin Struct Biol. 23:82–89. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
O'Brien TD, Butler AE, Roche PC, Johnson
KH and Butler PC: Islet amyloid polypeptide in human insulinomas.
Evidence for intracellular amyloidogenesis. Diabetes. 43:329–336.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Larsson LI, Grimelius L, Hakanson R,
Rehfeld JF, Stadil F, Holst J, Angervall L and Sundler F: Mixed
endocrine pancreatic tumors producing several peptide hormones. Am
J Pathol. 79:271–284. 1975.PubMed/NCBI
|
|
29
|
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle
E, Wang K and Liu X: Comparison and integration of deleteriousness
prediction methods for nonsynonymous SNVs in whole exome sequencing
studies. Hum Mol Genet. 24:2125–2137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Leir SH and Harris A: MUC6 mucin
expression inhibits tumor cell invasion. Exp Cell Res.
317:2408–2419. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Betge J, Schneider NI, Harbaum L,
Pollheimer MJ, Lindtner RA, Kornprat P, Ebert MP and Langner C:
MUC1, MUC2, MUC5AC, and MUC6 in colorectal cancer: Expression
profiles and clinical significance. Virchows Arch. 469:255–265.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dougherty JD, Fomchenko EI, Akuffo AA,
Schmidt E, Helmy KY, Bazzoli E, Brennan CW, Holland EC and
Milosevic A: Candidate pathways for promoting differentiation or
quiescence of oligodendrocyte progenitor-like cells in glioma.
Cancer Res. 72:4856–4868. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bhatti TR, Ganapathy K, Huppmann AR,
Conlin L, Boodhansingh KE, MacMullen C, Becker S, Ernst LM, Adzick
NS, Ruchelli ED, et al: Histologic and molecular profile of
pediatric insulinomas: Evidence of a paternal parent-of-origin
effect. J Clin Endocrinol Metab. 101:914–922. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Takahashi M: Genetic mutation accumulation
and clinical outcome of immune checkpoint blockade therapy. Gan To
Kagaku Ryoho. 43:678–682. 2016.(In Japanese). PubMed/NCBI
|
|
35
|
Birkbak NJ, Kochupurakkal B, Izarzugaza
JM, Eklund AC, Li Y, Liu J, Szallasi Z, Matulonis UA, Richardson
AL, Iglehart JD and Wang ZC: Tumor mutation burden forecasts
outcome in ovarian cancer with BRCA1 or BRCA2 mutations. PLoS One.
8:e800232013. View Article : Google Scholar : PubMed/NCBI
|