Introduction
Malignant melanoma is the most dangerous form of
skin tumor, accounting for the majority of cancer-associated
mortalities (1). Due to the high rate
of metastasis at an early stage of the disease and resistance to
conventional chemotherapy and radiotherapy, the morbidity and
mortality rates for malignant melanoma increase faster each year
than any other type of cancer (1,2). Previous
studies have demonstrated that melanoma is a heterogeneous disease
(3,4)
and the optimal treatment for patients with melanoma is
personalized based on the presence of specific molecular
abnormalities. Numerous abnormalities have been identified in
melanoma, including mutations in receptor tyrosine kinase (RTK)
genes [including KIT proto-oncogene (KIT), EPH receptor A2, erb-b2
receptor tyrosine kinase 4 and platelet derived growth factor
receptor (PDGFR)], the RAS family of small G protein genes
[including NRAS proto-oncogene GTPase (NRAS) and HRAS
proto-oncogene GTPase] and cytoplasm kinase genes [B-Raf
proto-oncogene serine/threonine kinase (BRAF), RAF family,
mitogen-activated protein kinase kinase (MEK)1/2, AKT
serine/threonine kinase 3 and phosphatase and tensin homolog]
(5). Mutations in the BRAF, NRAS and
KIT genes are more active compared with other genes (6).
BRAF kinases have been demonstrated to be associated
with melanoma and other types of cancer (7,8). The
substitution of glutamic acid for valine at position 600
(BRAFV600E) accounts for >80% of BRAF mutations and
~66% of melanomas. These mutations lock the BRAF kinase enzyme into
a constitutively activated state, which promotes tumorigenesis by
hyperactivating the extracellular signal-regulated kinase (ERK)
signaling pathway without stimulation by RTKs (9). Therefore, the BRAFV600E
mutation has been considered as an attractive therapeutic target
for melanoma, leading to the development of specific
BRAFV600E inhibitors. In 2011 and 2013, respectively,
vemurafenib and dabrafenib were approved by the US Food and Drug
Administration (FDA) for single agent or combinational treatment of
metastatic and unresectable BRAF-mutated melanomas. Although
administration of these drugs leads to significant levels of tumor
shrinkage in patients who harbor the BRAFV600E mutation,
≤25% of patients develop cutaneous squamous cell carcinomas
(primarily karatoacanthomas) and drug resistance. The underlying
molecular mechanisms mediating resistance to BRAF inhibitors in
melanoma are complex (10). The
paradoxical activation of the mitogen activated protein kinase
(MAPK) pathway in BRAF wild-type cells by homo- or
hetero-dimerization of RAF isoforms or the upregulation of RTKs,
including PDGFβR or insulin-like growth factor 1 receptor, are
mechanisms of drug resistance as well as mutations in KRAS, NRAS
(Q61K and Q61R) (11), MEK1 (C121S
and P124L) or MEK2 (Q60P) (12).
However, previous studies have demonstrated that ATP
competitive RAF inhibitors are not only poor inhibitors of
wild-type BRAF but also increase Raf-1 proto-oncogene
serine/threonine kinase activity to activate the ERK signaling
pathway in BRAF wild-type cells (13–16).
Compared with small molecular inhibitors, gene therapy targeting of
disease-associated genes demonstrates high efficiency, high
specificity and relatively low toxicity to healthy tissue. Small
interfering (si)RNA targeted to active oncogenic
BRAFV600E kinase could provide a promising treatment for
BRAF-mutant melanomas (17).
Cancer cells constantly adapt to promote survival
and escape the immune system, thus ensuring tumor growth. The
phosphoinositide-3-OH kinase (PI3K)/AKT/mammalian target of
rapamycin (mTOR) signaling pathway has an important role in cell
proliferation, survival and apoptosis. In melanoma, the PI3K/RAC-α
serine-threonine-protein kinase (AKT)/mTOR and Raf/MEK/ERK
signaling cascades are interconnected with multiple points of
convergence, cross-talk, and feedback loops. Inhibition of the two
pathways could be more effective than inhibiting either pathway
alone (18). At present, multiple
clinical trials are underway with combined inhibition of the ERK
and PI3K pathways, including Vemurafenib with PX-866 (PI3K
inhibitor) (trial no. NCT01616199), or Dabrafenib with GSK2141795
(AKT inhibitor) (trial no. NCT01902173) to determine if this
strategy can stop tumors from developing drug resistance in
BRAF-mutant melanoma (19).
BRAFV600E siRNA in combination with PI3K
signaling pathway inhibition may benefit patients with
BRAFV600E-positive tumors and act with high efficiency
and low toxicity. The present study aimed to examine the effects of
BRAFV600E siRNA combined with different types of PI3K
signaling pathway inhibitors on BRAFV600E mutant
melanoma cell lines.
Materials and methods
Reagents
Antibodies used in the present study were as
follows: Anti-BRAF (catalog no., Ab33899; Abcam, Cambridge, UK);
anti-MEK1 (catalog no., 9146), anti-MEK2 (catalog no.,),
anti-ERK1/2 (catalog no., 4695), anti-phospho-AKT (Ser473) (catalog
no., 4060s), anti-phospho-MEK1/2 (Ser217/211) (catalog no., 9154),
anti-phospho-ERK1/2 (Thr202/Tyr204) (catalog no., 4377),
anti-phospho-S6 (Ser240/244) (catalog no., 5364), anti-GAPDH
(catalog no., 2118) and anti-β-actin (catalog no., 3700) (Cell
Signaling Technology, Inc., Danvers, MA, USA); and horseradish
peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G (IgG)
(catalog no., ZDR-5306) and HRP-conjugated anti-mouse IgG (catalog
no., ZDR-5307) (ZSGB-Bio, Beijing, China). The PI3K/AKT/mTOR
signaling pathway inhibitors PI-103 (catalog no., S1038), GSK690693
(catalog no., S1113), ZSTK474 (catalog no., S1072) and AZD8055
(catalog no., S1555) were purchased from Selleck Chemicals
(Houston, TX, USA). All siRNA sequences were synthesized and
purified with high performance liquid chromatography by GenePharma
Co., Ltd. (Shanghai, China). All other reagents were of analytical
grade and obtained from commercial sources.
Cell culture
Human embryonic kidney HEK293A and melanoma A375
cell lines were obtained from the Cell Resource Center, Institute
of Basic Medical Sciences, Chinese Academy of Medical
Sciences/Peking Union Medical College (Beijing, China). The
melanoma cell line WM115 was obtained from the American Type
Culture Collection (Manassas, VA, USA). All cells were cultured in
Dulbecco's modified Eagle's medium (Macgene, Beijing, China). All
cells were supplemented with 10% fetal bovine serum (FBS; Corning
Incorporated, Corning, NY, USA) at 37°C with 5% CO2.
Cell transfection
For siRNA silencing, cells were seeded at
2×105 cells/35-mm2/well one day prior to
transfection. Cells were transfected using
Lipofectamine® RNAiMAX transfection reagent (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) in 0.5 ml of GenOpti
(Macgene) with siRNAs as follows: siMB3 (siBraf-mu; targeted to
BRAFV600E; antisense, 5′-AUCGAGAUUUCUCUGUAGCdtdt-3′ and
sense, 5′-GCUACAGAGAAAUCUCGAUdtdt-3′); siWTM (siBraf-wtm; targeted
to wild-type BRAF and BRAFV600E; antisense,
5′-AUGAUCCAGAUCCAAUUCUdtdt-3′ and sense,
5′-AGAAUUGGAUCUGGAUCAUdtdt-3′); siMEK1 (antisense,
5′-AGCAUGAACCAUGAGUUGCdtdt-3′ and sense,
5′-GCAACUCAUGGUUCAUGCUdtdt-3′); siMEK2 (antisense,
5′-TGCTGTGAGGCTCTCCTTCdtdt-3′ and sense,
5′-GAAGGAGAGCCUCACAGCAdtdt-3′) or negative control siRNA
(siControl; Guangzhou Ribo Bio, Co., Ltd., Guangzhou, China) at
different concentrations, as stated in the appropriate figure
legends, into 1.5 ml medium containing 10% FBS, according to the
manufacturer's protocol. Cells were treated and harvested following
the transfection as described in the figure legends.
Gene expression evaluation by reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from cells with TRIzol
(Thermo Fisher Scientific, Inc.), then chloroform was added and the
sample was mixed and centrifuged (12,000 × g) for 15 min at 4°C.
Total RNA was extracted from the supernatant and purified according
to the manufacturer's protocol. Total RNA was reverse transcribed
using the Reverse Transcription System (Promega Corporation,
Madison, WI, USA). Reverse transcription conditions were as
follows: 42°C for 15 min, 95°C for 5 min, and 0–5°C for 5 min. The
complementary DNA was subjected to PCR with GoTaq® Green
Master Mix (Promega Corporation) using the Techne TC-5000 PCR
Thermal Cycler (GMI, Ramsey, MN, USA) according to the
manufacturer's protocol. The primers used were as follows: β-actin
forward, 5′-CCAACCGCGAGAAGATGA-3′ and reverse,
5′-CCAGAGGCGTACAGGGATAG-3′); Total-braf forward,
5′-CTGCCTCATTACCTGGCTCACTA-3′ and reverse,
5′-CACCATGCCACTTTCCCTTGT-3′; and Braf(T1599A) forward,
5′-TGGTGTGAGGGCTCCAGCTTGT-3′ and reverse,
5′-ATGGGACCCACTCCATCGAGATTTCT-3′. PCR was performed as follows:
95°C for 5 min (1 cycle), 95°C for 30 sec, 60°C for 45 sec, 72°C
for 45 sec (25 cycles for β-actin and 31 cycles for
Total-braf and Braf(T1599A)) and 72°C for 5 min (1
cycle). The reaction solution was analyzed by 1% agarose gel
electrophoresis with ethidium bromide staining. β-actin was used as
a control. Finally, the products were analyzed by Image
Lab™ software 6.0 (ChemiDoct XRS System; Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Western blot analysis
Treated cells were harvested with cell lysis buffer
(20 mM TrisCl pH 7.5, 150 mM NaCl, 1% Triton X-100; catalog no.,
P1003; Beyotime Institute of Biotechnology, Haimen, China)
supplemented with a protease inhibitor cocktail (Pierce; Thermo
Fisher Scientific, Inc.). Cell extracts were normalized for protein
content using a BCA protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Total protein (8 µg) was separated by 10%
SDS-PAGE, transferred to polyvinylidene fluoride membranes (catalog
no., IPVH00010; EMD Millipore, Billerica, MA, USA) and blocked with
5% (w/v) milk in TBS-Tween. Western blot analysis was performed
following standard protocols using the indicated antibodies
[anti-BRAF, anti-MEK1, anti-MEK2, anti-ERK1/2, anti-phospho-AKT
(Ser473), anti-phospho-MEK1/2 (Ser217/211), anti-phospho-ERK1/2
(Thr202/Tyr204), anti-phospho-S6 (Ser240/244), anti-GAPDH,
anti-β-actin, HRP-conjugated anti-rabbit IgG and HRP-conjugated
anti-mouse IgG]. The primary antibodies were incubated at 25°C for
1 h, and the secondary antibodies were incubated at 25°C for 3 h.
The membrane was developed using the chemiluminescent HRP substrate
kit followed by the procedure provided by the manufacturer (EMD
Millipore). The densitometric quantification of the protein bands
was determined using the ChemiDoc™ XRS+System (Bio-Rad
Laboratories, Inc.).
Cell viability assay
Cells (A375 or WM115) in 96-well plates (4,000
cells/well) were transfected with siRNA or treated with gradient
concentrations of the compounds at 37°C for 48 or 72 h. Cell
proliferation was determined using a Cell Counting Kit-8 (CCK-8;
Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to
the manufacturer's protocol. Briefly, CCK-8 was added 1.5 h prior
to detection of the optical density (OD) at 450 nm with a
micro-plate reader. Cell viability was calculated as follows: Cell
viability=(ODsample-ODblank)/(ODNC-ODblank).
ODNC indicated the OD value of culture medium of the
cells that were transfected by negative control siRNA, which was
used as a control. ODblank indicated the OD value of
culture medium at 450 nm. The half-maximal inhibitory concentration
(IC50) was calculated using GraphPad Prism software
(version 5; GraphPad Software, Inc., La Jolla, CA, USA) and
presented as the mean with 95% confidence limits.
5-ethynyl-2′-deoxyuridine (EdU)
proliferation assay
Cell proliferation was determined using the
Cell-Light™ EdU Apollo®567 In Vitro Imaging kit
(Guangzhou RiboBio, Co., Ltd.). A375 cells were seeded in 96-well
plates (4,000 cells/cell) one day prior to treatment. Following
treatment, cells were incubated with 50 µM EdU at the indicated
times as stated in the figure legends for 2 h prior to fixation,
permeabilization and staining. Cell nuclei were stained with 1X
Hoechst 33342 for 30 min. The images were obtained with a High
Content Screening machine Operetta™ (PerkinElmer, Inc.,
Waltham, MA, USA) and the images were analyzed using Harmony 3.5.1
(PerkinElmer, Inc.). The border cells with irregular nuclei were
considered, which were removed with a common filter (using the
‘Select Population’ function, with ‘Nuclei’ as ‘Population’, the
‘Common Filter’ selected as the ‘Method’ and ‘hoechst’ selected as
the selective objects), and cells whose intensity in the Cy3
channel was 1.5 times higher than the background were defined as
EdU-positive cells. The percentage of EdU-positive cells was
calculated from 12 randomly selected fields/well. The data were
normalized to the control cells and presented as percentages.
Cell cycle analysis
Treated cells (105-106
cells/plate) were harvested and washed with PBS and then fixed with
pre-cooled 70% ethanol at 4°C overnight. The cell pellets were
washed and suspended in PBS containing 20 µg/ml RNase A at 37°C for
30 min. DNA was stained with 20 µg/ml propidium iodide (M&C)
and 0.1% Triton X-100 (Thermo Fisher Scientific, Inc.). The cells
were analyzed using a FACSCalibur™ flowcytometer (BD Biosciences,
Franklin Lakes, NJ, USA), and cell cycle analysis was performed
using ModFit LT3.2 software (Verity Software House, Topsham, ME,
USA).
Statistical analysis
All data are presented as the mean ± standard
deviation. Statistical analyses were performed using a two-tailed
unpaired t-test with GraphPad Prism software (version 5; GraphPad
Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference. Unless otherwise specified,
all assays were performed in triplicate.
Results
siRNA targeting of mutant
BRAFV600E decreased the viability of
BRAFV600E mutant melanoma cell lines
The specificity and efficiency of siWTM and siMB3 on
the A375 melanoma cell line, which harbors the Braf(T1599A)
mutation, and normal HEK293A cells was investigated. siWTM, which
targets wild-type BRAF and mutant BRAFV600E,
significantly decreased the viability of A375 and HEK293A cells
(P<0.001 and P<0.05, respectively; Fig. 1A and B). The siMB3 siRNA, which
specifically targets BRAFV600E, significantly decreased
the viability of A375 cells (P<0.001), but did not significantly
decrease HEK293A cell viability compared with siWTM. MEK1 silencing
significantly decreased the viability of A375 and HEK293A cells
(P<0.001 and P<0.05, respectively; Fig. 1A and B); however, MEK2 and MEK1/2
combined silencing significantly decreased the viability of A375
cells (P<0.01; Fig. 1A), but not
HEK293A cells.
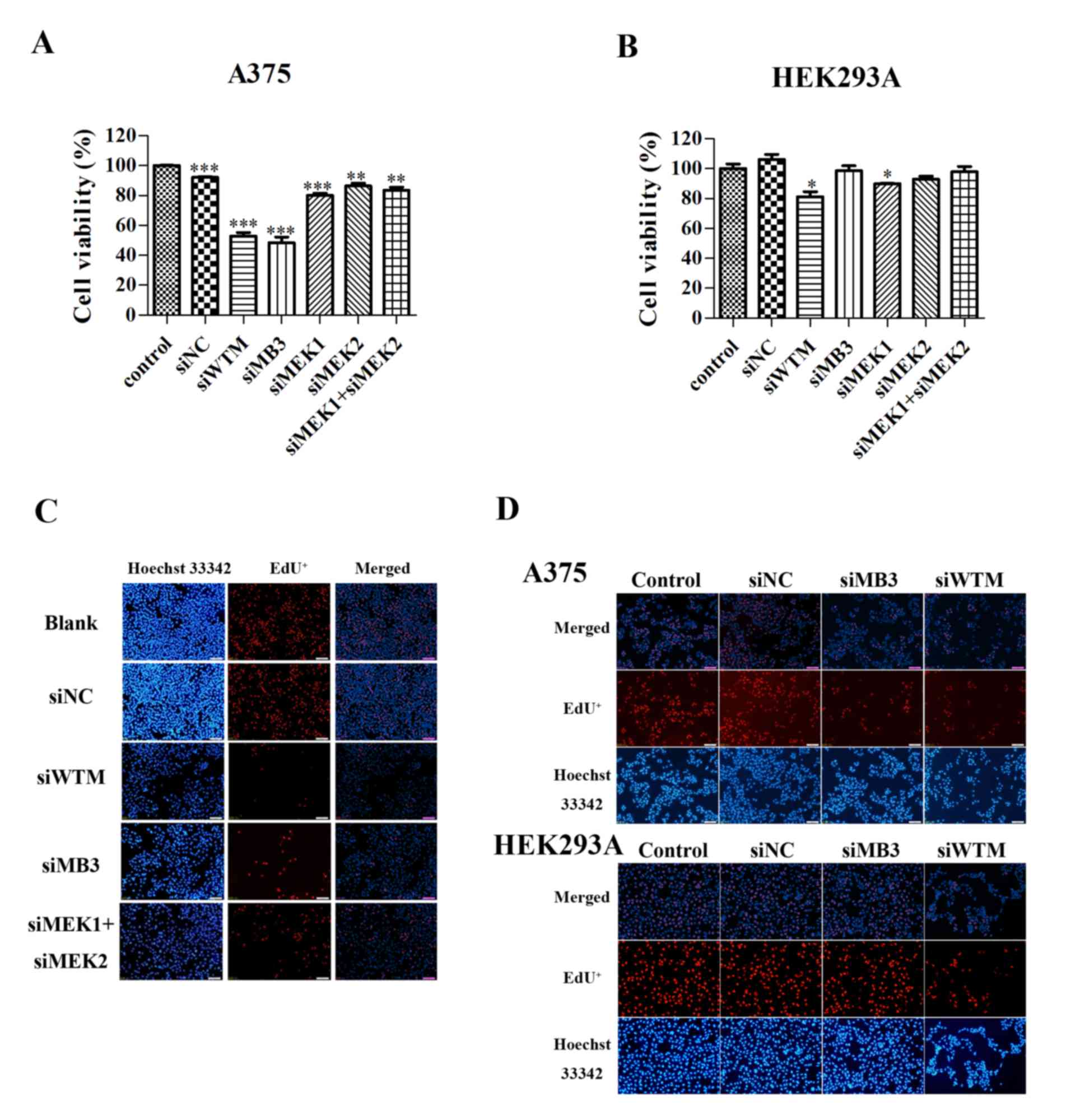 | Figure 1.BRAF and
BRAFV600E-targeted siRNAs significantly decrease A375
cell viability. Viability of (A) A375 and (B) HEK293A cells
following treatment with 30 nmol/l of siWTM, siMB3, siMEK1, siMEK2
or siNC siRNA for 48 h. (C) DNA replication was assessed by the EdU
method in melanoma A375 cells. Cells were treated with or without
30 nM of siWTM, siMB3, siMEK1 +siMEK2, and siControl for 48 h. EdU
was stained cells (red) and Hochest 33342 (blue) was stained the
nuclei of total cells. (D) The DNA replication measurement on A375
and HEK293A cells with 30 nM of siWTM or siMB3. EdU was stained
(red) following 48 h treatment. Data are represented as the mean ±
standard deviation (n=3). *P<0.05, **P<0.01, ***P<0.001,
compared with the corresponding control. siRNA, small interfering
RNA; EdU, 5-ethynyl-2′-deoxyuridine; BRAF, B-Raf proto-oncogene
serine threonine kinase; siWTM, siRNA targeting wild type BRAF and
mutant BRAFV600E; siMB3, siRNA targeting mutant
BRAFV600E; MEK, mitogen-activated protein kinase kinase;
NC, negative control. |
To investigate the molecular mechanisms underlying
the effects of the siRNA treatments on cell viability, EdU
retention assays were performed to examine the regulatory effect of
the two BRAF-targeted siRNAs on DNA replication in A375 and HEK293A
cell lines. A375 and HEK293Acells treated with siMB3 and siWTM
exhibited decreased DNA replication 48 h following treatment
(Fig. 1C and D). In addition, A375
cells treated with siMEK1/2 exhibited decreased DNA replication.
This data demonstrates that targeting BRAFV600E with
siRNA markedly decreases cell viability and inhibits DNA
replication.
Activity of the RAF/MEK/ERK and
PI3K/AKT/mTOR signaling pathways in melanoma cell lines
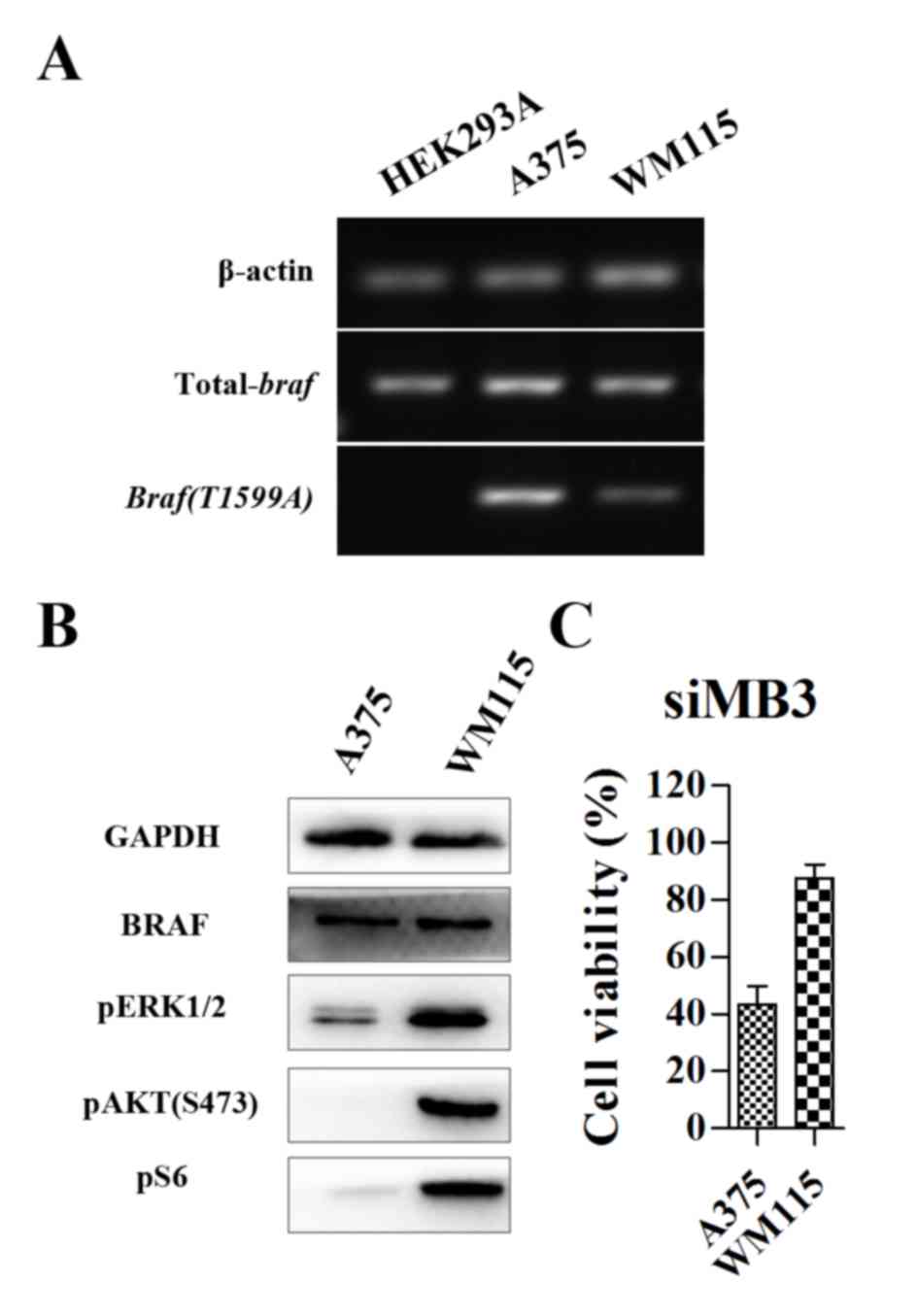
Comparison of the expression of BRAFV600E
in HEK293A, A375 and WM115 cells lines revealed that
BRAFV600E expression in WM115 cells was markedly lower
than that in A375 cells (Fig. 2A).
HEK293A cells were included as a control, as they did not express
the mutant protein BRAFV600E. Western blot analysis
revealed that levels of phosphorylated (p)ERK1/2, pAKT (S473) and
pS6 ribosomal protein [pS6; a surrogate readout for mammalian
target of rapamycin complex 1 (mTORC1) activity] were markedly
increased in WM115 cells compared with A375 cells, indicating that
the activity of the RAF/MEK/ERK and PI3K/AKT/mTOR signaling
pathways was increased in WM115 cells compared with A375 cells
(Fig. 2B). In A375 cells, the
hyperactivation of the ERK pathway by the BRAFV600E
mutation is a major contributor to tumorigenesis (8,9). siMB3
treatment decreased cell viability of the melanoma A375 cell line
(almost 60% inhibition) (Fig. 2C).
Although WM115 cells exhibited increased expression of pERK, pAKT
(S483) and pS6 protein compared with A375 cells (Fig. 2B), they exhibited decreased
expressionof mutant BRAF at the mRNA level (Fig. 2A) and exhibited less sensitivity to
siRNA targeting BRAFV600E compared with the A375
cells.
Effects of BRAFV600E siRNA
combined with PI3K signaling pathway inhibitors on cell
viability
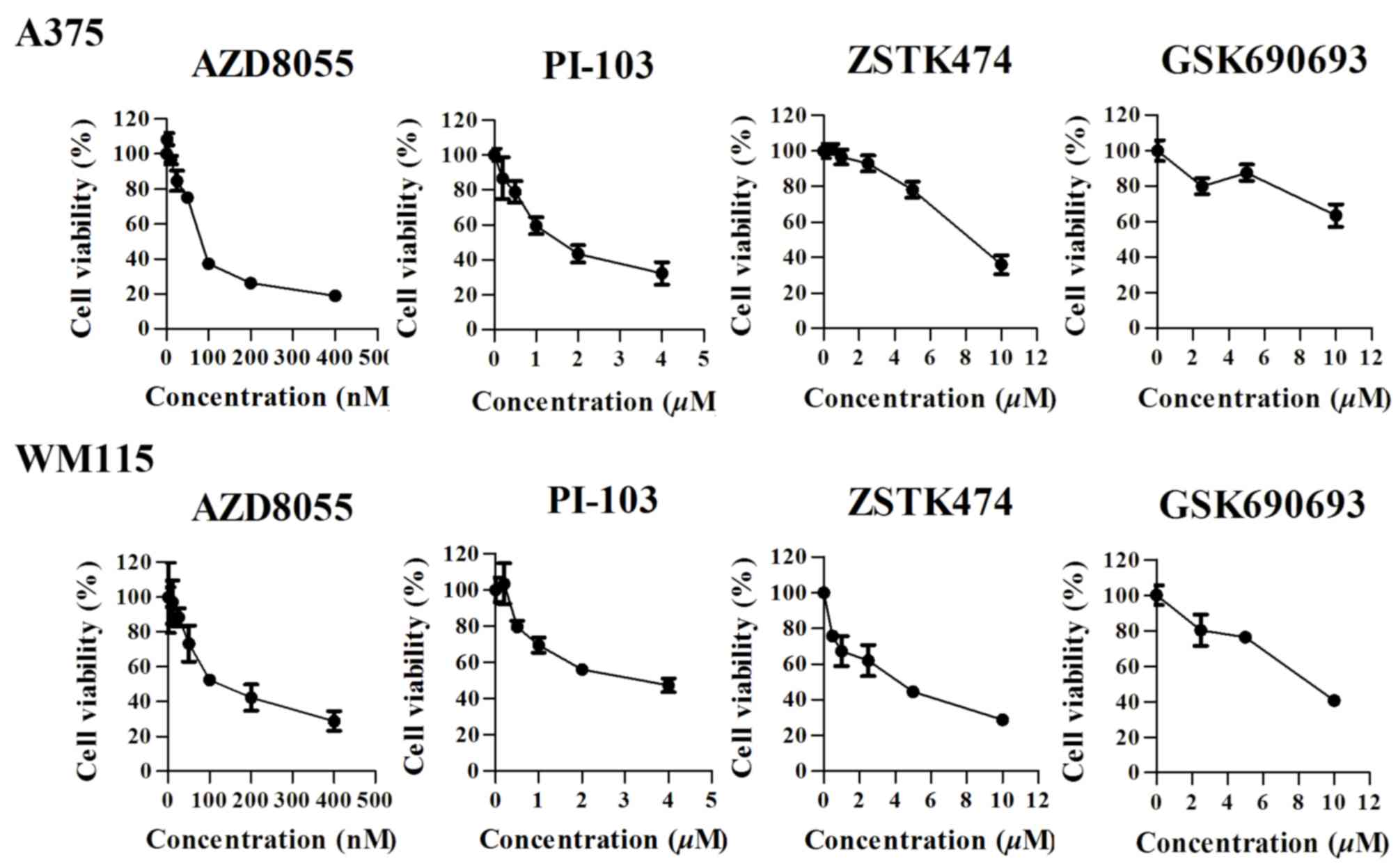
To explore the role of the PI3K signaling pathway in
the regulation of cell growthinBRAFV600-positive cell
lines, A375 and WM115 cells were treated with four types of kinase
inhibitors, including the dual PI3K/mTOR inhibitor PI-103, the mTOR
inhibitor AZD8055, the pen-class I PI3K inhibitor ZSTK474 and the
AKT inhibitor GSK690693. A375 and WM115 cells were treated with
increasing concentrations of the four inhibitors, and cell
viability was measured following treatment for 72 h using a CCK-8
assay. The four drugs exhibited different effects on cell viability
in the two cell lines (Fig. 3).
AZD8055 exhibited a dose-dependent antitumor effect on A375 cells,
with an IC50 value of 91.67 nM (95%CI: 85.22 to 98.11
nM). At a concentration of 100 nM the growth inhibition rate was
63.7±2.69%. The IC50 value for PI-103 was 1.67 µM (95%
CI: 1.333–2.012 µM), and at a concentration of 2 µM, the growth
inhibition rate was 56.7±4.97%. The IC50 value for
ZSTK474 was 8.0343 µM (95% CI: 7.472–8.638 µM). A375 cells were not
sensitive to GSK690693.
The WM115 cell line exhibited decreased expression
of BRAFV600E, but increased PI3K signaling pathway
activity compared with A375 cells (Fig.
2). All four inhibitors exhibited antitumor effects on WM115
cells, with IC50 values of139.9 nM, 2.943, 3.436, and
7.885 µM (AZD8055 >PI-103 > ZSTK474 > GSK690693),
respectively.
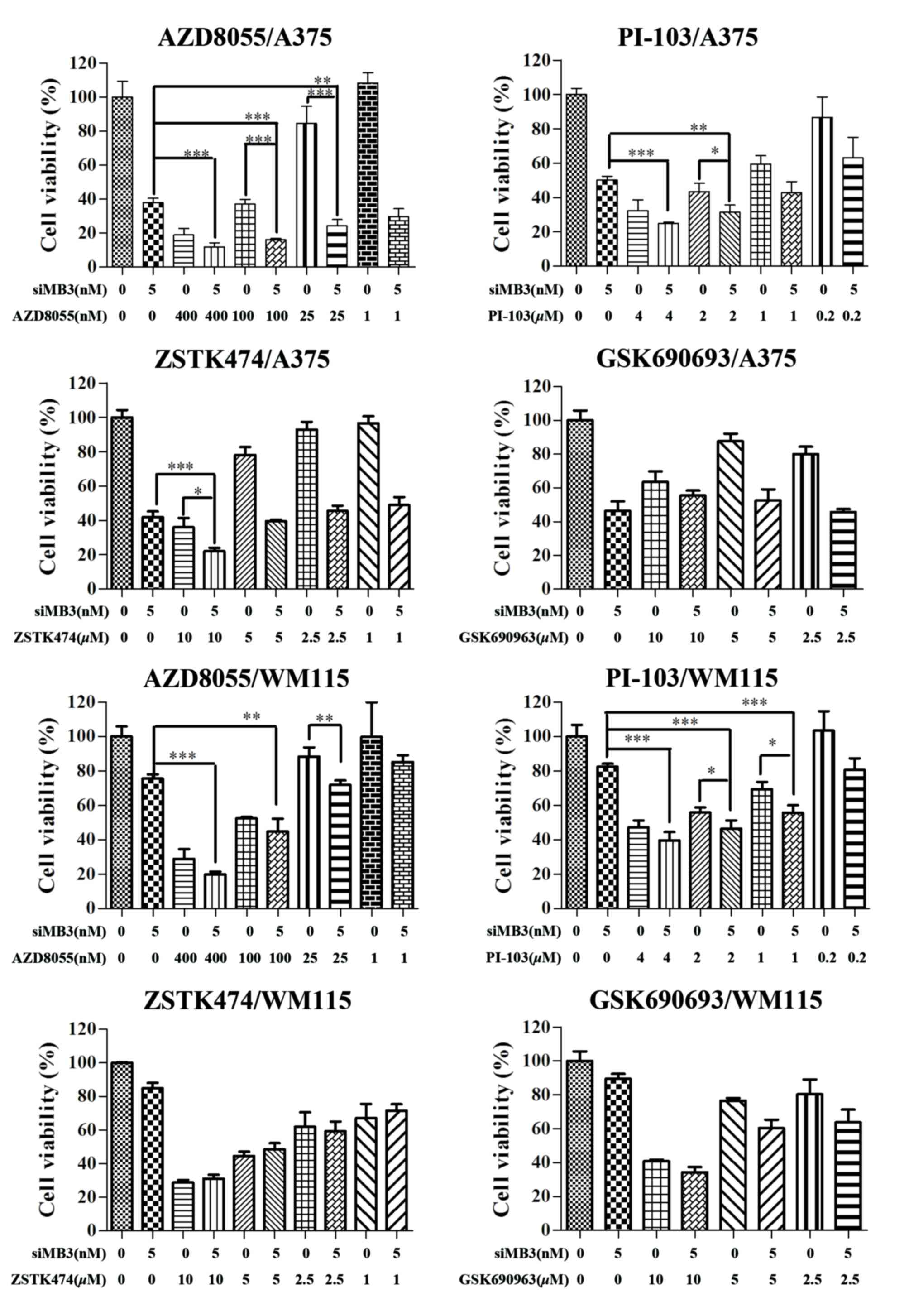
Combination treatment with BRAFV600E
siRNA and PI3K signaling pathway inhibitors may be an effective
treatment for patients with BRAFV600E-positive tumors.
Therefore, in the present study, the combinational effects of four
PI3K signaling pathway inhibitors with siMB3 on cell viability were
measured (Fig. 4). Combining mTOR
inhibition (AZD8055), dual PI3K/mTOR inhibition (PI-103), or PI3K
inhibition (ZSTK474) with BRAFV600E siRNA resulted in
significantly greater growth inhibition than inhibitor or siMB3
alone in A375 cells. The combination of AZD8055 with siMB3 had the
greatest effect on cell viability compared with that of siMB3 with
PI-103 or ZSTK474. Treatment of A375 cells with siMB3 and AZD8055
significantly decreased cell viability compared with AZD8055 or
siMB3 alone (84.8±0.62 compared with 62.9±2.69 and 62.1±2.54%,
respectively; P<0.001). Although no significant effect on cell
viability was observed with AZD8055 at a low concentration (1 nM),
the synergistic effect with siMB3 was significant compared with
AZD8055 treatment alone (P<0.005). When cells were co-treated
with 5 nM of siMB3 and 400, 100, 25 or 1 nM AZD8055, the cell
inhibition rates were 75.8±4.45, 75.9±3.62, 73.5±7.00, and
70.3±4.6, respectively.
There was no significant improvement with
combination treatment in WM115 cells compared with siMB3 or
inhibitor treatment alone. WM115 exhibit low levels of
BRAFV600E, therefore the ERK signaling pathway may be
activated by the hyperactivation of growth factor signaling or
other receptor signaling pathways. This may explain why WM115 cells
were not sensitive to BRAFV600E inhibition. Inhibiting
the PI3K/AKT/mTOR and MEK1/2 signaling pathways may provide an
efficient treatment for WM115 cells.
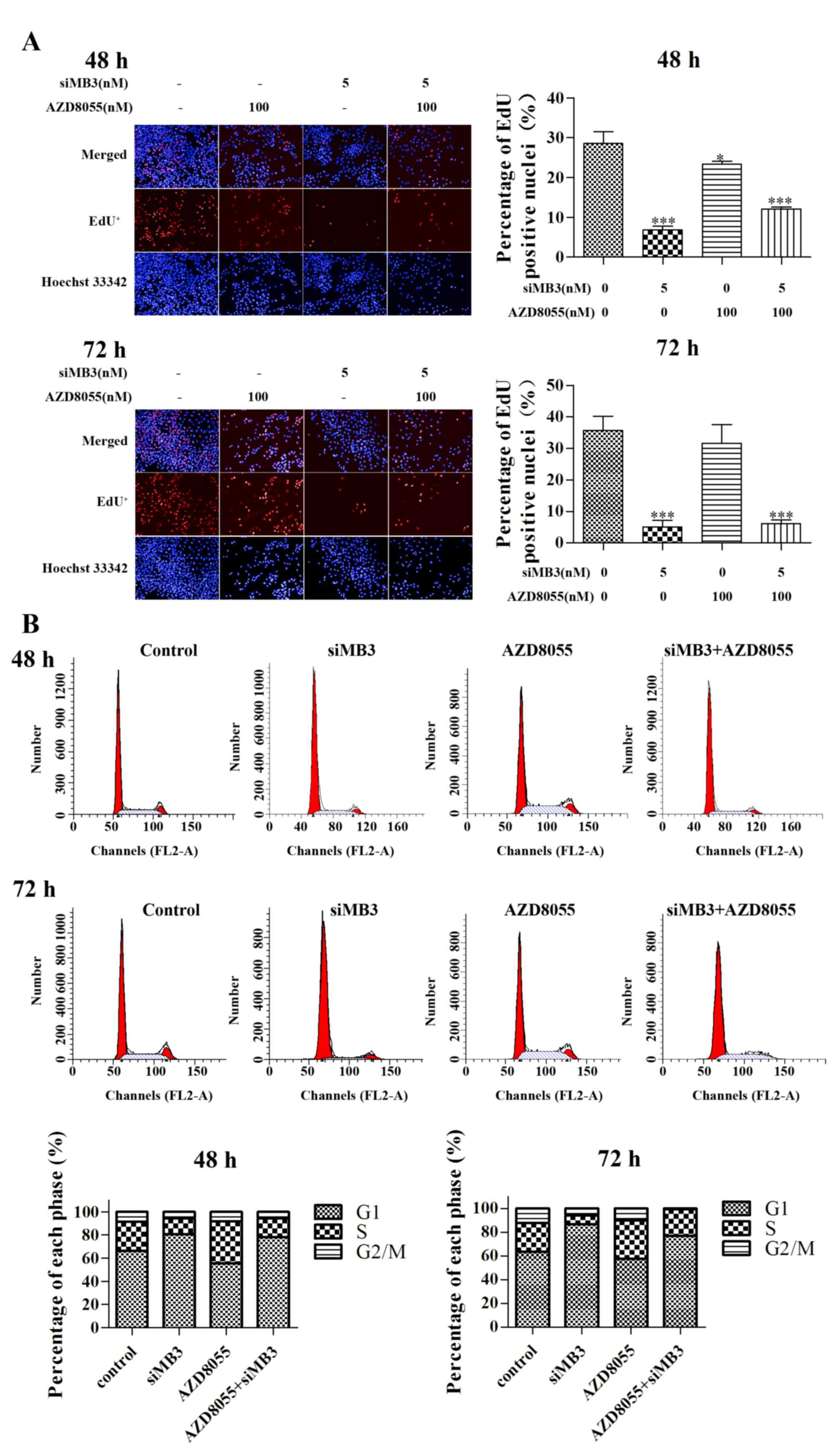
Effect of siMB3/AZD8055 combination
treatment on DNA replication and cell cycle progression
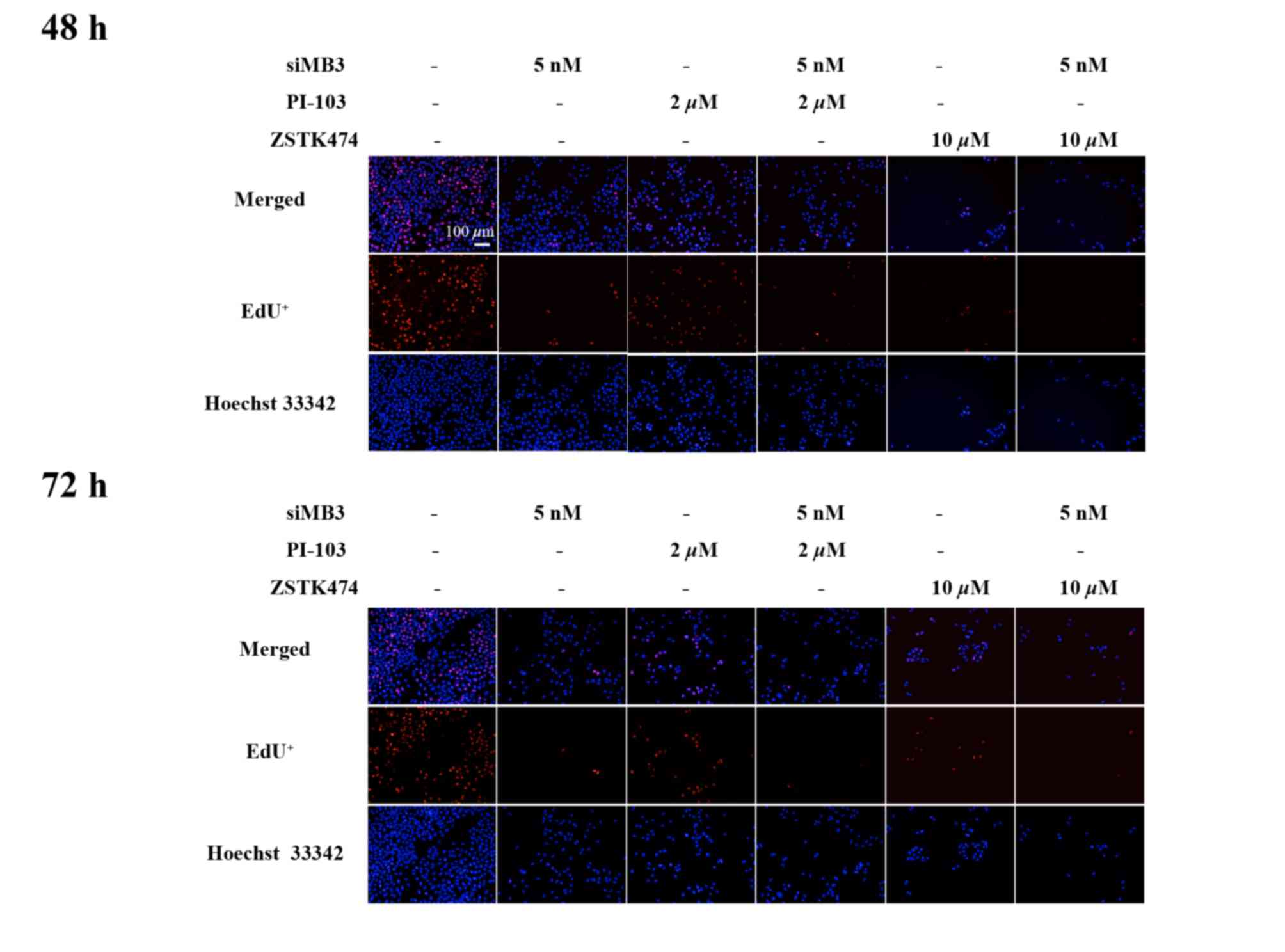
The effect of combined treatment with siMB3 and
AZD8055 on DNA replication was measured using the EdU method. siMB3
alone, AZD8055 treatment alone and siMB3/AZD8055 combination
treatment significantly inhibited DNA replication compared with
control untreated A375 cells (P<0.001, P<0.05 and P<0.001,
respectively; Fig. 5A). The siMB3
alone and siMB3/AZD8055 combination treatments exhibited >80%
inhibition of DNA replication following treatment for 72 h. AZD8055
arrested A375 cells at S phase, which correlated with the observed
inhibition of DNA replication (Fig.
5B). At 72 h following treatment with AZD8055 and siMB3, all
cells were arrested in either the G1 or S phase.
Effects of siMB3/AZD8055 combination
treatment on ERK and PI3K signaling pathways
AZD8055 is a potent and specific ATP-competitive
inhibitor, which has been demonstrated to inhibit the
phosphorylation of the mTORC1 substrates p70S6K and pS6, in
addition to inhibiting the phosphorylation of the mTORC2 substrate
AKT (S483) and downstream proteins in multiple cancer clinical
studies (20). The expression of BRAF
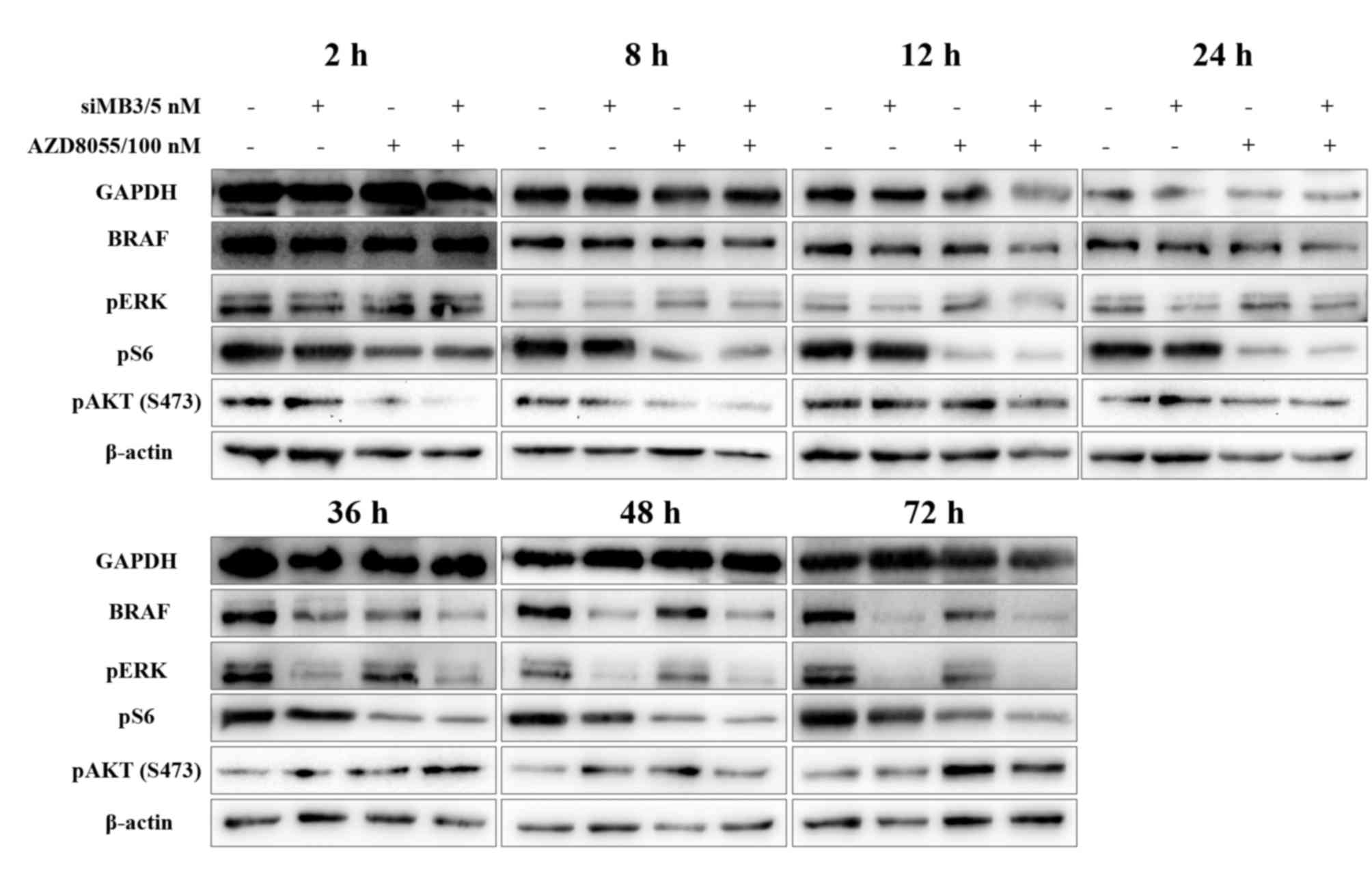
in cells treated with siMB3 alone and siMB3/AZD8055 was markedly
reduced at 12 h following treatment and was completely knocked down
at the 36 h time point (Fig. 6). In
addition, ERK signaling pathway activity was markedly inhibited in
a time-dependent manner under the same conditions. pS6 expression
was reduced from the 2 h time point following AZD8055 alone or
siMB3/AZD8055 treatment; however, pS6 expression levels in the
combination group were decreased compared with the AZD8055 alone
group. Phosphorylation of AKT(S473) in the AZD8055 alone and
siMB3/AZD8055 combination groups was markedly decreased at the 2 h
time point; however, the expression increased again following the
36 and 72 h time points.
Therefore, down regulation of BRAF in
BRAFV600E-positive cells with siRNA leads to a decrease
in pERK, while treatment with AZD8055 leads to a decrease in pS6.
However, continuous administration of AZD8055 would be required to
inhibit pAKT levels and further decrease cell viability. Therefore,
the combination treatment with siMB3 and AZD8055 leads to a
decrease in pERK and pS6 levels, which may contribute to their
combined effect on the viability of A375 cells.
siMB3/PI-103 and siMB3/ZSTK474
combination treatments decrease cell viability, and inhibit the
PI3K/AKT/mTOR and Ras/MEK/ERK signaling cascades
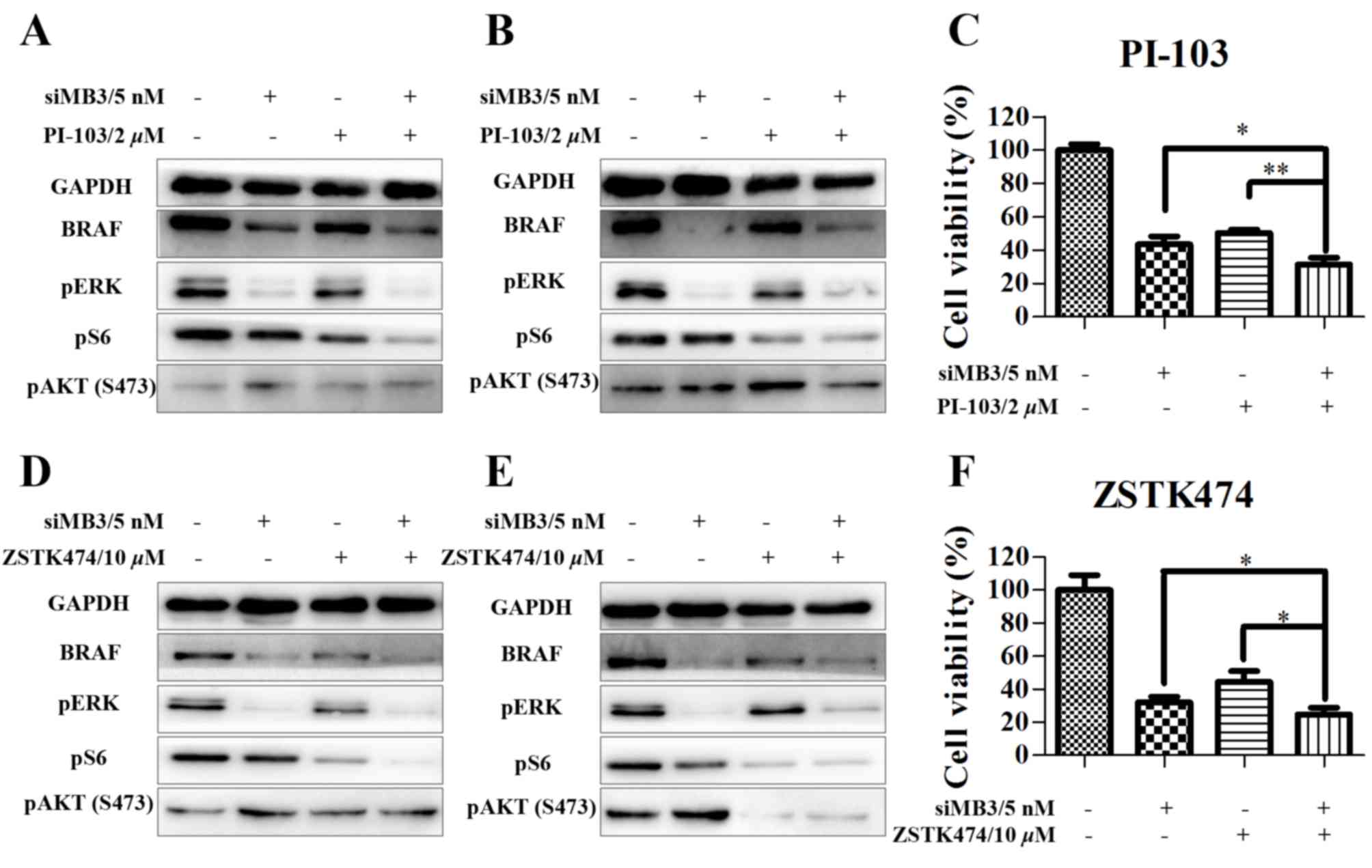
Next, the effects of siMB3/PI-103 combination
treatment were investigated in A375 cells. The phosphorylation of
ERK and S6 was markedly decreased at 48 and 72 h following
combination treatment; however, phosphorylation of AKT (S473) did
not recover at a later time point (Fig.
7A and B). In addition, cell growth inhibition significantly
increased in the combination treatment (5 nM of siMB3 and 2 µM of
PI-103) group compared with the PI-103 treatment alone group
(68.4±4.2% compared with 56.7±4.9%, respectively; P<0.01;
Fig. 7C). Similar effects were
observed with ZSTK474 (Fig. 7D-F). In
addition, compared with siMB3 alone or PI-103 alone groups,
siMB3/PI-103 or siMB3/ZSTK474 combination treatments markedly
inhibited DNA replication and decreased cell number (Fig. 8).
Discussion
Melanoma has become one of the most extensively
studied cancer types in order to develop effective targeted
therapies. Prior to 2011, only two drugs (interferon-α-2b and
interleukin-2) had been previously approved for the treatment of
melanoma (1). By2015, the FDA had
approved 6 first-in-class drugs specific for melanoma (18). Of these, ipilimumab, nivolumab and
pembrolizumab are immunotherapies, whereas the other 3 drugs were
developed for thespecific treatment of BRAF-mutant melanoma
(21–23).
The oncogenic BRAFV600E allele is a
common mutation of the BRAF gene and has been identified in ~50% of
advanced-stage metastatic melanomas (2). Vemurafenib and dabrafenib/trametinib
combination treatments have demonstrated success in patients with
melanoma (24); however,
small-molecule BRAF inhibitors exhibit side effects such as fast
development of drug resistance (19).
Targeting mRNA degradation using siRNA may be a
potential strategy for the treatment of a number of diseases
(25). Although there remain
challenges with siRNA-based treatments in terms of appropriate
delivery vehicles, siRNA could be efficient and potent anticancer
agents (26). Synthetic siRNA or
short hairpin RNAs have been widely used for drug development and a
number of phase I and II clinical trials are in progress (27).
siRNA-based treatments that only deplete the
oncogenic form of BRAF (BRAFV600E) could be a successful
strategy for the treatment for BRAFV600E-positive tumor
types. In the present study, siMB3, a 19mer overlapping the T1799A
mutation site of BRAFV600E, inhibited ERK signaling
pathway activity and decreased the viability of cell lines with
this mutation, including A375 and WM115, but not HEK293A cells, as
these do not harbor the mutation. Previous studies have
demonstrated that a 25mer siRNA (containing the 19mer of siBraf-mu)
targeted to BRAFV600E significantly inhibited melanoma
tumor growth and reduced lung metastases in UACC 903, 1205 Lu, and
C8161 cell lines (17,28,29). In
addition, data from the present study demonstrated that A375 cells
were more sensitive to the effects of siMB3 on cell viability than
WM115 cells due to their increased expression of
BRAFV600E. Preliminary data from a current study from
our group demonstrated that siMB3 treatment may be effective in
vivo; however, the cytotoxic effects of siMB3 on normal cells
limit its application in vivo (Xinmeng Fan et al,
unpublished data).
The RAF/MEK/ERK and PI3K/AKT/mTOR signaling pathways
have been implicated in the tumorigenesis of melanoma (18,30). The
combination inhibitory activity of these two pathways can increase
antitumor activity and specificity and thus reduces side effects on
normal tissues. Previous studies have suggested that combined
targeting of the ERK and PI3K pathways increases antitumor activity
and may serve as a novel treatment for patients with NRAS
mutant-positive melanoma, for which there are currently no
effective therapies (30).
Sanchez-Hernandez et al (17),
demonstrated that combined with BRAFV600E deletion by
siRNA, PI3K/AKT/mTOR signaling pathway inhibitors synergized to
increase apoptosis levels to a greater extent than that achieved by
inhibitor alone in BRAFV600E mutant melanomas, and
suggested that mTOR was a convergence point of BRAF and PI3K
signals in these cells (17,18).
In the present study, PI3K, mTOR and AKT inhibitors
exhibited different antitumor effects on A375 cells. Based on the
cell viability assays, the efficacy decreased in the following
order: AZD8055 (mTOR inhibitor) > PI-103 (dual PI3K/mTOR
inhibitor) > ZSTK474 (pen-class I PI3K inhibitor) >GSK690693
(AKT inhibitor). AZD8055 inhibition of both mTORC1
(IC50=27±3 nM for pAKT473 in MDA-MB-468
cells) and mTORC2 (IC50=24±9 nM for
pS235/236) exhibited potent efficacy and selectivity
compared with rapamycin in vitro and in vivo
(20,31). The inhibition of mTORC1 by rapamycin
results in the release of the negative feedback loop between
ribosomal protein S6 kinase and insulin receptor substrate 1,
leading to hyperactivation of AKT (32).
Silencing of BRAFV600E by siRNA combined
with treatment with AZD8055 significantly decreased the viability
of A375 cells. Western blot analysis demonstrated that AZD8055
inhibited the phosphorylation of S6 (S240/244) and AKT (S473) at 2
h following treatment, whereas the pAKT (S473) expression level
recovered 36 h following treatment.
AKT kinases are important in melanoma tumorigenesis.
Selective activation of AKT3 protein promoted cell survival and
tumor development in ~70% of melanomas (33). In addition, a previous study on drug
resistant cancer types suggested that the hyperactivation of AKT
was associated with a shorter tumor progression time (33). In the present study, AZD8055 was
unable to simultaneously inhibit both mTORC1 and mTORC2 in A375
melanoma cells 36 h following treatment. The pAKT level may be
upregulated by other upstream kinases, including PI3K, or RTKs.
This indicated that the enhanced inhibition effect on tumor
progression by AZD8055 in combination with siRNA treatment may not
last in the absence of AKT inhibition. However, siMB3/PI-103 and
siMB3/ZSTK474 combination treatments significantly decreased the
pAKT expression level compared to that of control groups.
The results from the present study demonstrate that
the efficacy of siRNA treatment on mutant genes is dependent on the
percentage of mutation in melanoma cells. siRNA treatment of cells
harboring the BRAFV600E mutation may be a potent
treatment for melanoma cells that harbor high expression and
activating levels of BRAFV600E mutation. However, in
those melanoma cells in which the oncoprotein BRAFV600E
is not the major carcinogenic factor, patients may not be sensitive
to targeted BRAFV600E therapy, including inhibition by
treatment with vemurafenib or elimination of BRAFV600E
expression with siRNA. Targeting the PI3K/AKT/mTOR signaling
pathway, which induces cell apoptosis and decrease cell survival,
may represent an improved treatment strategy. A more comprehensive
understanding of the molecular mechanisms underlying melanoma will
aid with developing, personalized cancer treatments. The data from
the present study may also be relevant for other types of cancer
that harbor the BRAFV600E mutation, including colorectal
carcinomas and non-small cell lung cancer (23).
In conclusion, the present study demonstrated that
silencing BRAFV600E by using siRNA in combination with
PI3K signaling pathway inhibitors improved the effect of PI3K/mTOR
inhibitors on tumor cell viability. Concomitant
BRAFV600E and PI3K inhibition resulted in G1
and S phase arrest. Future studies are required in order to develop
an efficient and safe delivery system for siRNA-based therapy.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the National
Natural Sciences Foundation of China (grant no. 81302626) and the
Specialized Research Fund for the Doctoral Program of Higher
Education (grant no. 20120001120023). The funding agencies had no
role in study design, data collection and analysis, decision to
publish or preparation of the manuscript.
Availability for data and materials
All data generated or analyzed during this study are
included in this published article.
Author's contributions
HH, XN and SL performed the experiments, YW
contributed to the writing of the manuscript. YW, ZY, LiaZ and LihZ
conceived of the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Schadendorf D, Fisher DE, Garbe C,
Gershenwald JE, Grob JJ, Halpern A, Herlyn M, Marchetti MA,
McArthur G, Ribas A, et al: Melanoma. Nat Rev Disease Primers.
1:150032015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Batus M, Waheed S, Ruby C, Petersen L,
Bines SD and Kaufman HL: Optimal management of metastatic melanoma:
Current strategies and future directions. Am J Clin Dermatol.
14:179–194. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gray-Schopfer V, Wellbrock C and Marais R:
Melanoma biology and new targeted therapy. Nature. 445:851–857.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Smalley KS, Haass NK, Brafford PA, Lioni
M, Flaherty KT and Herlyn M: Multiple signaling pathways must be
targeted to overcome drug resistance in cell lines derived from
melanoma metastases. Mol Cancer Ther. 5:1136–1144. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bis S and Tsao H: Melanoma genetics: The
other side. Clin Dermatol. 31:148–155. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rapp UR, Goldsborough MD, Mark GE, Bonner
TI, Groffen J, Reynolds FH Jr and Stephenson JR: Structure and
biological-activity of v-raf, a unique oncogene transduced by a
retrovirus. Proc Natl Acad Sci USA. 80:4218–4222. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wellbrock C and Hurlstone A: BRAF as
therapeutic target in melanoma. Biochem Pharmacol. 80:561–567.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bucheit AD and Davies MA: Emerging
insights into resistance to BRAF inhibitors in melanoma. Biochem
Pharmacol. 87:381–389. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nazarian R, Shi H, Wang Q, Kong X, Koya
RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al: Melanomas
acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS
upregulation. Nature. 468:973–977. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Emery CM, Vijayendran KG, Zipser MC,
Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA,
Karpova MB, et al: MEK1 mutations confer resistance to MEK and
B-RAF inhibition. Proc Natl Acad Sci USA. 106:20411–20416. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Poulikakos PI, Zhang C, Bollag G, Shokat
KM and Rosen N: RAF inhibitors transactivate RAF dimers and ERK
signalling in cells with wild-type BRAF. Nature. 464:427–430. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hatzivassiliou G, Song K, Yen I,
Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor
SL, Vigers G, et al: RAF inhibitors prime wild-type RAF to activate
the MAPK pathway and enhance growth. Nature. 464:431–435. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Heidorn SJ, Milagre C, Whittaker S, Nourry
A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer
CJ, Pritchard C and Marais R: Kinase-dead BRAF and oncogenic RAS
cooperate to drive tumor progression through CRAF. Cell.
140:209–221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Corcoran RB, Dias-Santagata D, Bergethon
K, Iafrate AJ, Settleman J and Engelman JA: BRAF gene amplification
can promote acquired resistance to MEK inhibitors in cancer cells
harboring the BRAF V600E mutation. Sci Signal. 3:ra842010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sharma A, Trivedi NR, Zimmerman MA,
Tuveson DA, Smith CD and Robertson GP: Mutant B-V599E-RAF regulates
growth and vascular development of malignant melanoma tumors.
Cancer Res. 65:2412–2421. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sánchez-Hernández I, Baquero P, Calleros L
and Chiloeches A: Dual inhibition of (V600E)BRAF and the
PI3K/AKT/mTOR pathway cooperates to induce apoptosis in melanoma
cells through a MEK-independent mechanism. Cancer Lett.
314:244–255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fedorenko IV, Gibney GT, Sondak VK and
Smalley KS: Beyond BRAF: Where next for melanoma therapy? Br J
Cancer. 112:217–226. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li S, Li Y, Hu R, Li W, Qiu H, Cai H and
Wang S: The mTOR inhibitor AZD8055 inhibits proliferation and
glycolysis in cervical cancer cells. Oncol Lett. 5:717–721. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sondak VK, Smalley KS, Kudchadkar R,
Grippon S and Kirkpatrick P: Ipilimumab. Nat Rev Drug Discov.
10:411–412. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mullard A: 2014 FDA drug approvals. Nat
Rev Drug Discov. 14:77–81. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mullard A: 2013 FDA drug approvals. Nat
Rev Drug Discov. 13:85–89. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Flaherty KT, Yasothan U and Kirkpatrick P:
Vemurafenib. Nat Rev Drug Discov. 10:811–812. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim DH and Rossi JJ: Strategies for
silencing human disease using RNA interference. Nat Rev Genet.
8:173–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bouchie A: Markets, venture investors and
big pharma interest in RNAi soars. Nat Biotechnol. 32:203–204.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Deng Y, Wang CC, Choy KW, Du Q, Chen J,
Wang Q, Li L, Chung TK and Tang T: Therapeutic potentials of gene
silencing by RNA interference: Principles, challenges, and new
strategies. Gene. 538:217–227. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hingorani SR, Jacobetz MA, Robertson GP,
Herlyn M and Tuveson DA: Suppression of BRAF(V599E) in human
melanoma abrogates transformation. Cancer Res. 63:5198–5202.
2003.PubMed/NCBI
|
|
28
|
Sharma A, Tran MA, Liang S, Sharma AK,
Amin S, Smith CD, Dong C and Robertson GP: Targeting
mitogen-activated protein kinase/extracellular signal-regulated
kinase kinase in the mutant (V600E) B-Raf signaling cascade
effectively inhibits melanoma lung metastases. Cancer Res.
66:8200–8209. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Posch C, Moslehi H, Feeney L, Green GA,
Ebaee A, Feichtenschlager V, Chong K, Peng L, Dimon MT, Phillips T,
et al: Combined targeting of MEK and PI3K/mTOR effector pathways is
necessary to effectively inhibit NRAS mutant melanoma in vitro and
in vivo. Proc Natl Acad Sci USA. 110:4015–4020. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chresta CM, Davies BR, Hickson I, Harding
T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini
P, et al: AZD8055 is a potent, selective, and orally bioavailable
ATP-competitive mammalian target of rapamycin kinase inhibitor with
in vitro and in vivo antitumor activity. Cancer Res. 70:288–298.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
O'Reilly KE, Rojo F, She QB, Solit D,
Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al:
mTOR inhibition induces upstream receptor tyrosine kinase signaling
and activates Akt. Cancer Res. 66:1500–1508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stahl JM, Sharma A, Cheung M, Zimmerman M,
Cheng JQ, Bosenberg MW, Kester M, Sandirasegarane L and Robertson
GP: Deregulated Akt3 activity promotes development of malignant
melanoma. Cancer Res. 64:7002–7010. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shao Y and Aplin AE: Akt3-mediated
resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer
Res. 70:6670–6681. 2010. View Article : Google Scholar : PubMed/NCBI
|