Introduction
Ovarian endometrioid carcinomas account for only 10%
of all ovarian carcinomas (1). The
majority of ovarian endometrioid carcinomas are thought to be type
I, low-grade tumors with good prognosis (1). The precursor lesions of ovarian
endometrioid carcinomas have been described as endometrioid
borderline tumors (EBTs); however, EBTs are extremely rare and
constitute only 0.2% of all epithelial ovarian tumors. EBTs exhibit
two major growth patterns: Adenofibromatous and intracystic
(1). The adenofibromatous type arises
from benign ovarian adenofibroma, whereas the intracystic types
arise from the transformation of endometriosis, as revealed by
their frequent coexistence in surgical specimens (2). The process underlying the molecular
pathogenesis of EBT remains unclear, whereas a number of molecular
alterations have been described in ovarian endometrioid carcinomas,
including mutations in β-catenin (16–38%) (3–7),
phosphatase and tensin homolog (PTEN; 14–21%) (8,9), AT-rich
interaction domain 1A (ARID1A; 30%) (10), phosphoinositide-3-kinase catalytic α
polypeptide (PIK3CA; 0%) (11)
and tumor protein p53 (TP53; 60%) (6,12).
Mutations in the tumor-suppressor gene PTEN
have been reported in a relatively high percentage of endometrial
and ovarian cancer cases, particularly in the endometrioid subtype
(9,13,14). The
majority of these mutations were observed in stage I tumors,
indicating that PTEN inactivation represents an early event
during ovarian tumorigenesis (9).
However, PTEN mutations in EBTs have not previously been
described, possibly due to their rarity. PTEN, located at
chromosome 10q23, is a tumor-suppressor gene encoding a lipid
phosphatase that dephosphorylates phosphatidylinositol
3,4,5-trisphosphate [PI(3,4,5)P3], performing opposing actions to
phosphoinositide 3-kinase (PI3K) (15,16).
PI(3,4,5)P3 is converted to PI(3,4)P2, which activates the
proto-oncogene protein kinase B (PKB; also known as AKT). When
phosphorylated, AKT becomes activated and antagonizes apoptotic
pathways via the activation of mechanistic target of rapamycin or
the inactivation of the members of the forkhead family, whereas the
dephosphorylation of PI(3,4,5)P3 by PTEN induces the activation of
the apoptotic pathway (17–19). Therefore, the fundamental role of
PTEN is the inhibition of the PI3K/AKT signaling pathway.
PTEN mutations may disrupt this inhibitory role, thereby
inducing the antiapoptotic pathway.
BAF250a, the protein encoded by ARID1A
(20,21), is one of the accessory subunits of the
SWItch/Sucrose Non-Fermentable complex (SWI/SNF). SWI/SNF is a
chromatin-remodeling complex that is found in all eukaryotes and is
responsible for regulating gene expression during numerous cellular
processes, including differentiation, development, DNA repair,
proliferation, and tumor suppression (22). Using adenosine triphosphate, SWI/SNF
mobilizes nucleosomes, thereby modulating the accessibility of
promoters for transcriptional activation or repression.
Rearrangement of ARID1A has been demonstrated in a breast
cancer cell line, while a lung cancer cell line has been shown to
exhibit ARID1A deletion, indicating that ARID1A is a
tumor-suppressor gene (22). The
nature and pattern of ARID1A mutations in ovarian
endometrioid carcinoma also provide evidence of its role as a tumor
suppressor (10). In a previous
study, ARID1A mutations were revealed to frequently co-occur
with PIK3CA gene mutations and PI3K/AKT signaling pathway
activation, as demonstrated by the increased AKT1 activation in
endometrial cancer (23). However, to
the best of our knowledge, the roles of ARID1A mutation in
EBTs have not previously been described. Therefore, the majority of
these genetic alterations, which are prevalent in ovarian
endometrioid carcinomas, induce the activation of the PI3K/AKT
signaling pathway, and may serve major roles in carcinogenesis as
well as disease progression.
The aim of the present study was to investigate the
PI3K/AKT signaling pathway status in EBTs and the role of
PTEN and ARID1A mutations. The present study examined
the differences in PTEN and ARID1A mutations between
benign and borderline patient tissue samples in order to gain
insight into the molecular pathogenesis involved in the initiation
of EBT.
Materials and methods
Tissue samples
Formalin-fixed, paraffin-embedded (FFPE) tissue
samples isolated from two cases of EBT of the ovaries were analyzed
in the present study. Samples were obtained from the Department of
Obstetrics and Gynecology at the Shimane University Hospital
(Izumo, Japan) between March 2013 and December 2015. The patients
were a 61-year-old female (case 1) and a 43-year-old female (case
2) who had presented with abdominal distension as the chief
complaint. The patients had undergone laparoscopic bilateral
salpingo-oophorectomy, and had no significant previous medical or
family history of disease.
The resected specimens were sectioned (section
thickness, 3 µm), stained with hematoxylin and eosin and reviewed
by a pathologist. Subsequently, the tissue specimens were
immunohistochemically stained with the following antibodies
overnight at 4°C: Monoclonal anti-PTEN antibody (cat. no. ABM-2052;
1:100; Cascade BioScience, Inc., Winchester, MA, USA), anti-ARID1A
(cat. no. sc-32761; 1:50; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA), anti-p53 (cat. no. 760-2542; 1:200; Roche Diagnostics,
Indianapolis, IN, USA), anti-β-catenin (cat. no. ab32572; 1:1,000;
Abcam, Cambridge, UK) and anti-phosphorylated (p)-AKT (cat. no.
4060; Ser473; 1:50; Cell Signaling Technology, Inc., Danvers, MA,
USA) antibodies. The following day, slides were washed three times
with PBS prior to the detection of antigens using the two-step DAKO
EnVision+ Peroxidase System (DAKO; Agilent Technologies, Inc.,
Santa Clara, CA, USA), according to the manufacturer's protocol, at
room temperature for 30 min. After rinsing with PBS, the sections
were incubated at room temperature for 5 min in 0.05%
diaminobenzidine in PBS with 0.03% H2O2.
Acquisition of tissue specimens was approved by the
Ethics Committee of Shimane University School of Medicine (approval
no. 2004-0381). Written informed consent was provided by all
participants prior to enrollment in the present study. This study
was conducted in accordance with the Declaration of Helsinki and
Title 45, U.S. Code of Federal Regulations, Part 46, Protection of
Human Subjects, effective December 13, 2001.
Scoring of immunohistochemical
staining
All samples were evaluated by two pathologists in
Shimane University Hospital, who were blinded to the present study.
The samples were scored negative for PTEN when complete loss of
expression in the tumor sample was observed, with stromal cells and
normal fallopian tube epithelial cells used as the positive
controls. PTEN staining was considered positive when strong, weak
or heterogeneous staining was observed.
Absence of nuclear staining of ARID1A is referred to
as a ‘clonal loss’ pattern and corresponds to mutations in ARID1A
(10); therefore, such tissues were
scored as ‘loss of expression’. Surrounding stromal cells and
normal fallopian epithelial cells served as positive controls.
p53 was scored ‘wild-type-like’ when <50% of the
tumor cells demonstrated positive nuclear staining. p53 was scored
‘mutant-like’ if >50% of the tumor cells exhibited strong
positive nuclear staining or when discrete geographical patterns
exhibited >50% tumor cell positivity.
DNA extraction
Borderline regions of both cases contained
sufficient tumor tissue to extract DNA and perform sequence
analysis. Benign epithelial regions were not observed in case 1.
Tissue sections were placed on membrane slides and counterstained
with hematoxylin for 1 min at room temperature. Selected tumor
tissues were dissected, and after 48 h of digestion with Proteinase
K solution (cat. no. 25530-049; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 56°C, DNA was extracted from
the dissected samples using a QIAamp DNA Micro kit (Qiagen Inc.,
Valencia, CA, USA), according to the manufacturer's protocol.
Sequence analysis
PCR amplification was performed for all 9 exons of
PTEN using primers (Table I)
and genomic DNA from dissected FFPE tissue. PCR amplification was
performed in 20 µl reaction volumes that contained 75 mM Tris-HCl,
1.5 mM MgCl2, 50 mM KCl, 20 mM
(NH4)2SO4, 0.2 mM of each primer, and 1 U
Taq DNA polymerase (Takara Bio, Inc., Otsu, Japan). The PCR
experiments were conducted under the following conditions: An
initial 5 min denaturation at 95°C; 35 cycles of 1 min each at 94,
57, and 72°C; and a single final extension step for 10 min at 72°C.
Amplified PCR products were sequenced at Beckman Coulter, Inc.
(Brea, CA, USA) and analyzed using the Mutation Surveyor DNA
Variant Analysis Software (version 4.0.6; SoftGenetics LLC, State
College, PA, USA).
 | Table I.Primers used for PCR amplification of
the PTEN gene. |
Table I.
Primers used for PCR amplification of
the PTEN gene.
| Exon | Forward primer
5′-3′ | Reverse primer
5′-3′ |
|---|
| 1 |
TTCCATCCTGCAGAAGAAGC |
CAGCCGCAGAAATGGATAC |
| 2 |
ACTCCAGCTATAGTGGGGAAA |
TTTTCTGTGGCTTAGAAATCTTTT |
| 3 |
TGATTACTACTCTAAACCCATAGAAGG |
TTGTTTTAGAAGATATTTGCAAGC |
| 4 |
AAAGATTCAGGCAATGTTTGTT |
TCTCACTCGATAATCTGGATGAC |
| 5 |
TCCAGTGTTTCTTTTAAATACCTGTT |
GATCCAGGAAGAGGAAAGGAA |
| 6 |
ATATATGTTCTTAAATGGCTACGA |
ACATGGAAGGATGAGAATTTC |
| 7 |
TCATTAAAATCGTTTTTGACAGTTT |
TCTGTCCTTATTTTGGATATTTCTC |
| 8 |
TGTTTAACATAGGTGACAGATTTTCTT |
ACAAGTCAACAACCCCCACA |
| 9 |
TGTTCATCTGCAAAATGGAATAA |
CACAATGTCCTATTGCCATTAAA |
Results
Clinical findings
The tissue samples were obtained from a 61-year-old
female (case 1) and a 43-year-old female (case 2) with abdominal
distension as the chief complaint. The patients underwent
laparoscopic bilateral salpingo-oophorectomy, and had no
significant previous medical or family history of disease. The
serum cancer antigen 125 expression levels were within normal
limits (<35.0 U/ml), and the results of the general examination
were also normal. Following the initial surgical procedure, the
patients underwent total hysterectomy and omentectomy due to the
EBT diagnosis. Follow-up data was available in case 1 for 5 years
and in case 2 for 2 years. The two patients were alive and well at
the last follow-up.
Pathological findings
Case 1
Gross observations of the tumor of the right ovary
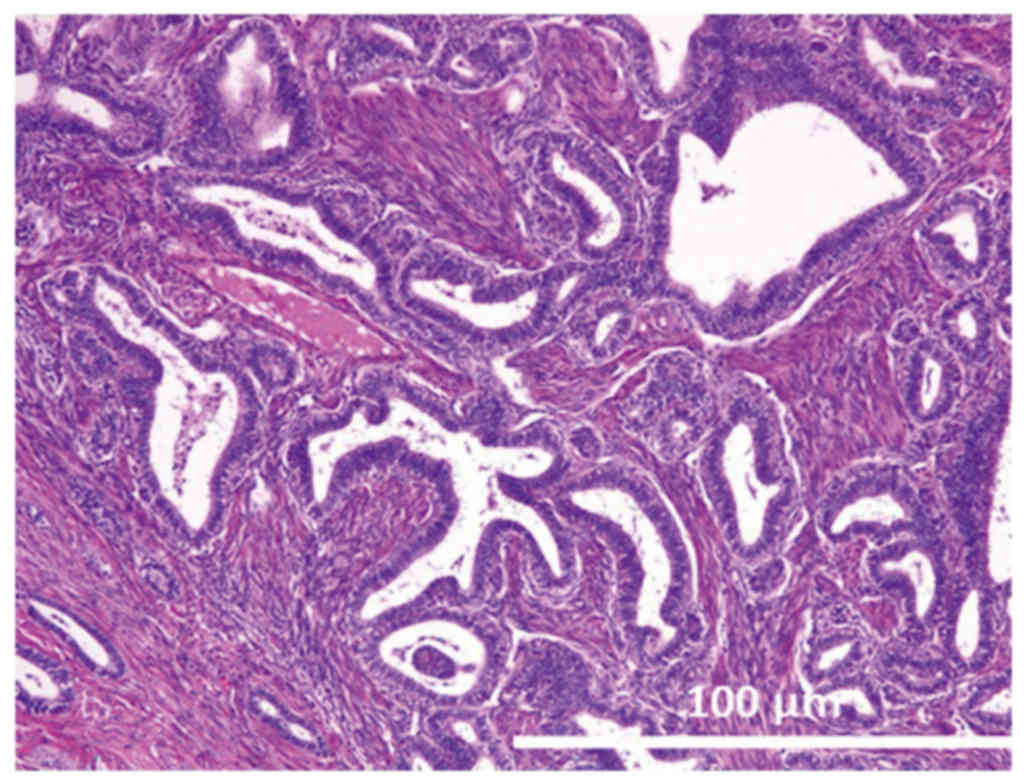
revealed that it had a maximum diameter of 7.5 cm. Microscopically,
the tumor was categorized as having an endometrioid
adenofibromatous growth pattern. Numerous glands with intervening
fibrous stroma were observed and these glands demonstrated varying
degrees of endometrial hyperplasia, ranging from simple hyperplasia
to a more marked proliferation, similar to that observed in complex
atypical hyperplasia (Fig. 1). No
invasion of the ovarian stroma was identified.
Case 2
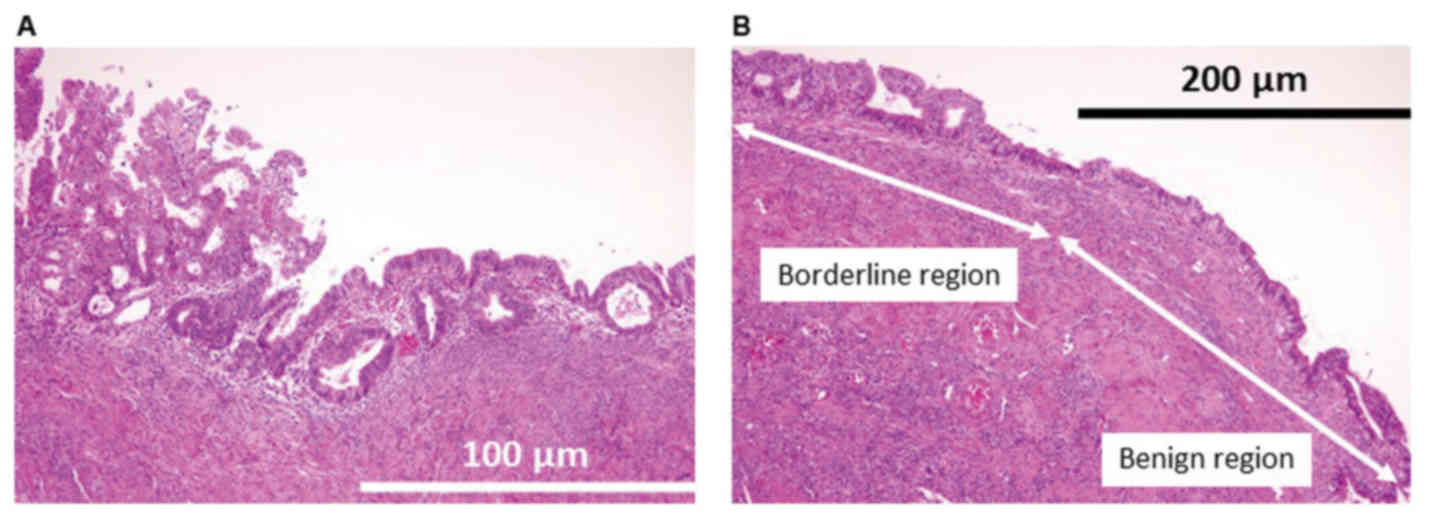
Gross observations of the tumor in the left ovary
revealed that its maximum diameter was 4.5 cm. Microscopically, the
tumor was categorized as having an intracystic growth pattern. The
tumor exhibited a papillary growth pattern protruding into a cystic
space (Fig. 2A). Cells with moderate
cytological atypia were observed. Atypical endometrial glands were
observed in the endometrial background (Fig. 2B). No invasion of the ovarian stroma
was identified.
Immunohistochemical findings
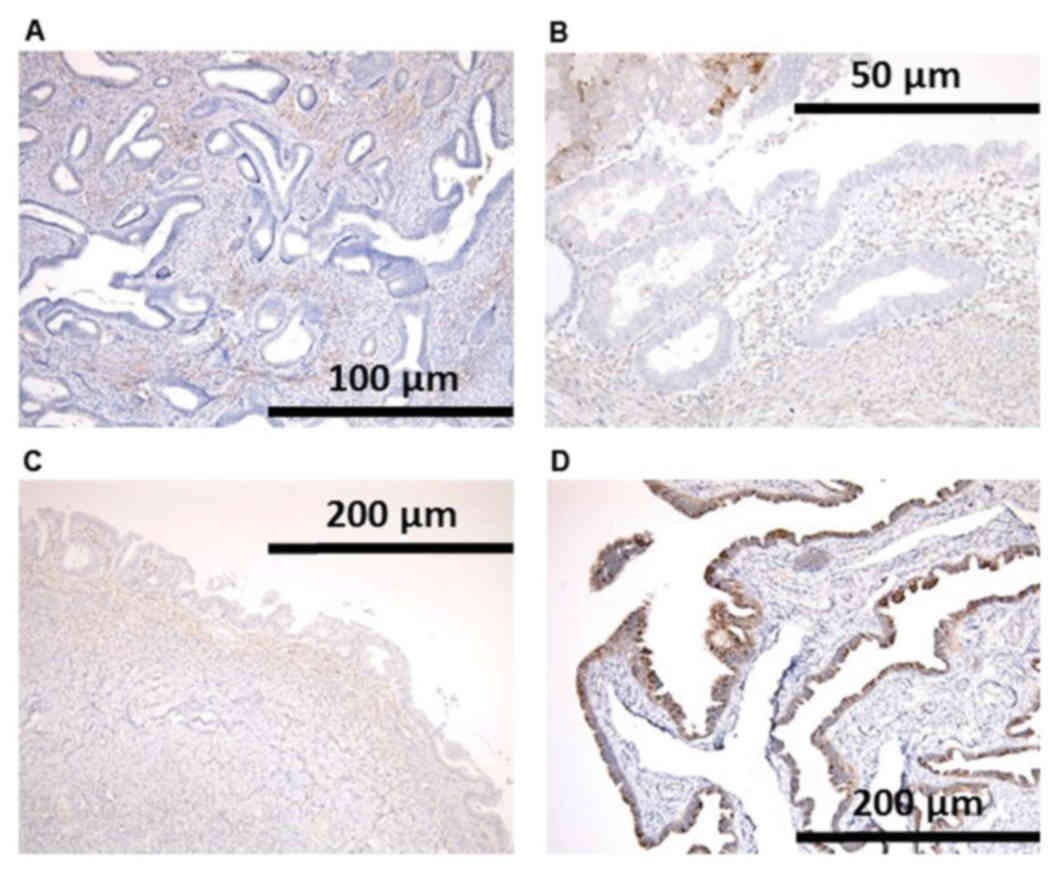
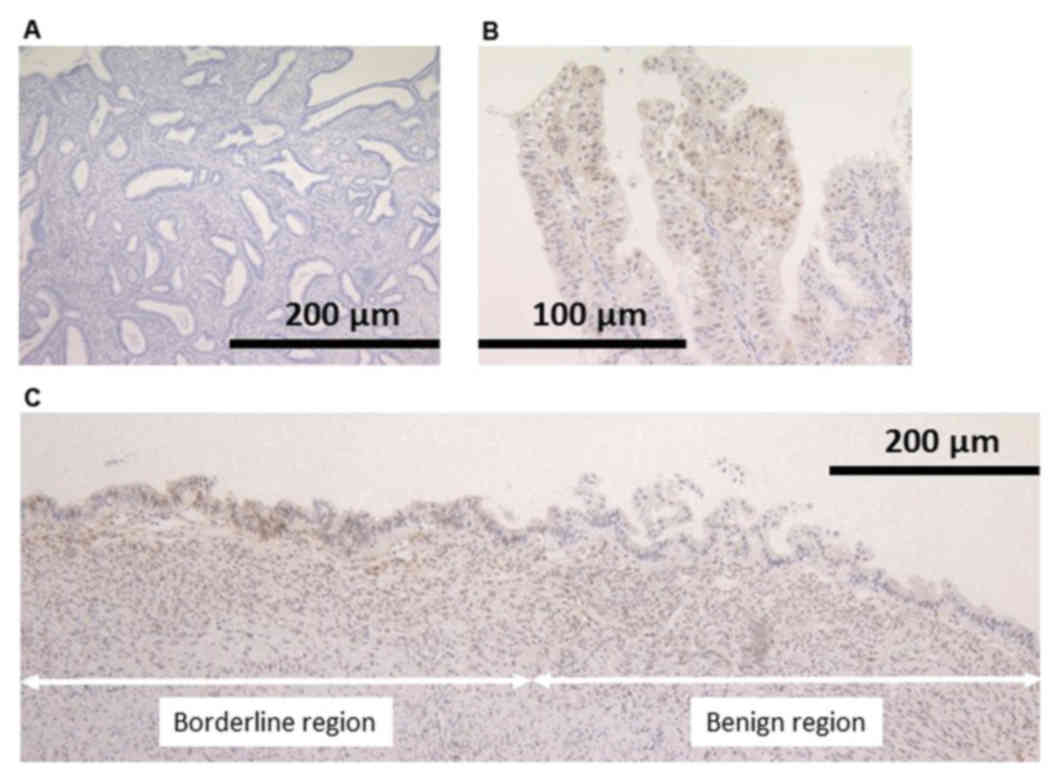
Loss of PTEN protein expression was revealed in the
nuclei of neoplastic cells in both EBTs. In case 2, loss of PTEN
expression was identified not only in the area of endometriosis
with atypia, but also endometriosis without atypia (Fig. 3).
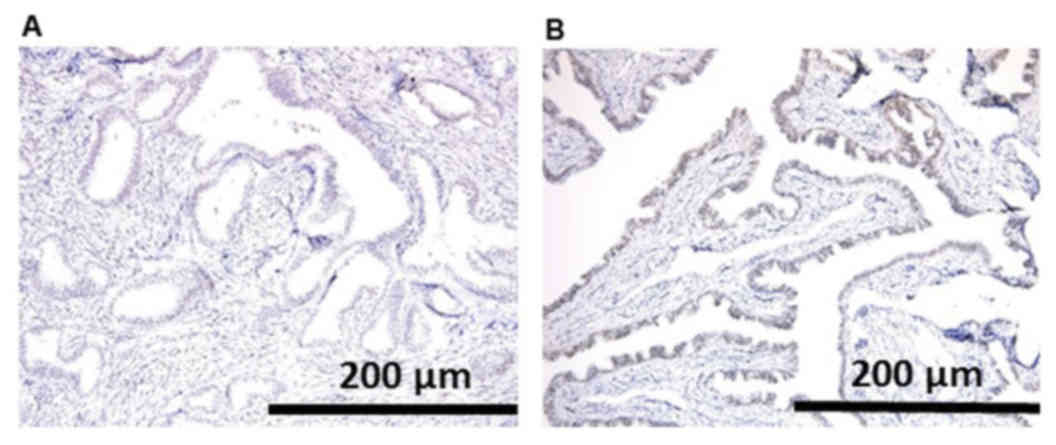
Loss of ARID1A protein expression was observed in
the nuclei of neoplastic cells, whereas strong expression was
maintained in stromal and normal fallopian tube epithelial cells of
the same section in case 1 (Fig. 4).
In case 2, ARID1A expression was revealed to be present, and was
most likely wild-type protein. Wild-type p53 and β-catenin
expression patterns were observed in both tissue samples.
Furthermore, the expression level of p-AKT in these
two cases was assessed. The tumor cells in case 1 were negative for
p-Akt expression (Fig. 5A); however,
in case 2, p-AKT expression was not identified in the area of
endometriosis without atypia, but was in the endometriosis with
atypia (Fig. 5B and C).
PTEN mutational analysis
PCR amplification and sequence analysis were
performed for the borderline regions in case 1 tissue samples, and
in the benign and borderline regions in the case 2 tissue samples.
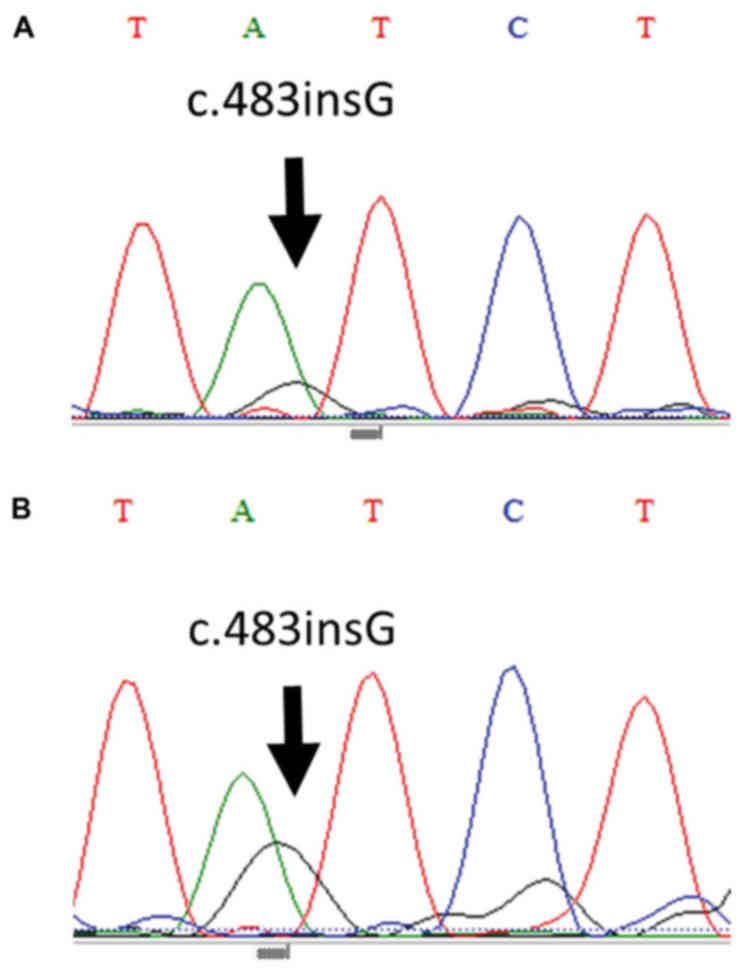
The borderline regions of both samples were revealed to harbor a
c.483insG mutation in exon 8 of the PTEN gene (Fig. 6). Regarding the regions of
endometriosis without atypia, the same mutations were detected.
c.483insG represents a frameshift mutation that results in the
formation of a stop codon (p.ASP485X) for PTEN protein
translation.
Discussion
There are few previous studies investigating EBTs
due to their rarity; to the best of our knowledge, only two series
and one case study have been previously reported (24–26). EBTs
were observed in the right and left ovaries in 2–5% of cases at the
time of diagnosis in these studies, and the predominant growth
pattern was adenofibromatous in both case series, as well as in
case 1 of the present study. Among all of these studies, only case
2 of the present study was demonstrated to be associated with
endometriosis. Additionally, glandular and papillary proliferations
with a high grade of complexity and mild atypia in the lining
epithelial cells were identified in the present study. In all of
the cases mentioned, no squamous differentiation was observed and
there was no stromal invasion; the growth was limited to only one
ovary with the capsule intact. In the long-term follow-up, both of
the case series of EBTs demonstrated no signs of recurrence or
metastasis, whereas 20% of patients with well-differentiated
endometrioid carcinoma were revealed to have recurrence (24). It has been observed that the
endometrioid neoplasms of the ovary are usually carcinomas, whereas
borderline tumors are extremely rare, and are thought to arise from
adenofibroma or endometriosis (1).
Ovarian endometrioid carcinomas are genetically
stable and have been described to arise from EBT (1). They are associated with PTEN, ARID1A,
PIK3CA and TP53 mutations (1). PTEN mutations may represent an
early event in the pathogenesis of endometrioidcarcinoma (27). However, the frequency of PTEN
mutation in EBTs has not been described previously. These prevalent
genetic alterations observed in ovarian endometrioid carcinoma are
known to induce the activation of the PI3K/AKT signaling pathway
(6). However, as EBTs are extremely
rare, the status of genetic mutations in these tumors is yet to be
reported.
The present study analyzed the status of genetic
mutations in EBTs and their association with the activation of the
PI3K/AKT signaling pathway for the first time. In case 1, which was
associated with adenofibroma, PTEN and ARID1A
mutations were observed. However, in case 2, which was associated
with endometriosis, ARID1A mutation was not observed,
whereas PTEN mutation was observed in EBT as well as in the
area of endometriosis without atypia, although the PI3K/AKT
signaling pathway was activated only in the EBT area. The results
of genetic alteration analyses are provided in Table II. The observations suggest that the
PTEN mutation represents an early event in EBT
tumorigenesis, whereas the additional genetic alterations may be
necessary in order for the PI3K/AKT signaling pathway to be
activated and induce the development of EBT to invasive carcinoma.
Additional studies will be required to confirm the findings and
implications of the present study.
| Table II.Genetic analyses of distinct tumor
regions. |
Table II.
Genetic analyses of distinct tumor
regions.
| Case no. | Tissue region | PTEN | ARID1A | β-catenin | p53 | p-AKT |
|---|
| Case 1 | Borderline | Mutant | Mutant | WT | WT | Negative |
| Case 2 | Benign
(endometriosis without atypia) | Mutant | WT | WT | WT | Negative |
|
| Borderline
(endometriosis with atypia) | Mutant | WT | WT | WT | Positive |
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KoN drafted the manuscript and carried out the
molecular genetic studies. MI, TM, TI, ES, KS and HY carried out
the molecular genetic studies and participated in the sequence
alignment. RO, KI, RS, MMH and NI carried out the staining. KeN
participated in the design of the study. SK conceived the study,
and participated in its design and coordination and helped to draft
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Ethics
Committee of Shimane University School of Medicine (Izumo, Japan)
(approval no. 960). All patients provided informed written consent.
The research was conducted in accordance with the Declaration of
Helsinki and Title 45, US Code of Federal Regulations, Part 46,
Protection of Human Subjects, effective December 13, 2001.
Consent for publication
All patients provided written informed consent for
the publication of data in this study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen S, Leitao MM, Tornos C and Soslow RA:
Invasion patterns in stage I endometrioid and mucinous ovarian
carcinomas: A clinicopathologic analysis emphasizing favorable
outcomes in carcinomas without destructive stromal invasion and the
occasional malignant course of carcinomas with limited destructive
stromal invasion. Mod Pathol. 18:903–911. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prowse AH, Manek S, Varma R, Liu J, Godwin
AK, Maher ER, Tomlinson IP and Kennedy SH: Molecular genetic
evidence that endometriosis is a precursor of ovarian cancer. Int J
Cancer. 119:556–562. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moreno-Bueno G, Gamallo C, Pérez-Gallego
L, de Mora JC, Suárez A and Palacios J: Beta-catenin expression
pattern, beta-catenin gene mutations, and microsatellite
instability in endometrioid ovarian carcinomas and synchronous
endometrial carcinomas. Diagn Mol Pathol. 10:116–122. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sagae S, Kobayashi K, Nishioka Y, Sugimura
M, Ishioka S, Nagata M, Terasawa K, Tokino T and Kudo R: Mutational
analysis of beta-catenin gene in Japanese ovarian carcinomas:
Frequent mutations in endometrioid carcinomas. Jpn J Cancer Res.
90:510–515. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wright K, Wilson P, Morland S, Campbell I,
Walsh M, Hurst T, Ward B, Cummings M and Chenevix-Trench G:
Beta-catenin mutation and expression analysis in ovarian cancer:
Exon 3 mutations and nuclear translocation in 16% of endometrioid
tumours. Int J Cancer. 82:625–629. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu R, Hendrix-Lucas N, Kuick R, Zhai Y,
Schwartz DR, Akyol A, Hanash S, Misek DE, Katabuchi H, Williams BO,
et al: Mouse model of human ovarian endometrioid adenocarcinoma
based on somatic defects in the Wnt/beta-catenin and PI3K/Pten
signaling pathways. Cancer Cell. 11:321–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu R, Zhai Y, Fearon ER and Cho KR:
Diverse mechanisms of beta-catenin deregulation in ovarian
endometrioid adenocarcinomas. Cancer Res. 61:8247–8255.
2001.PubMed/NCBI
|
|
8
|
Catasus L, Bussaglia E, Rodrguez I,
Gallardo A, Pons C, Irving JA and Prat J: Molecular genetic
alterations in endometrioid carcinomas of the ovary: Similar
frequency of beta-catenin abnormalities but lower rate of
microsatellite instability and PTEN alterations than in uterine
endometrioid carcinomas. Hum Pathol. 35:1360–1368. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Obata K, Morland SJ, Watson RH, Hitchcock
A, Chenevix-Trench G, Thomas EJ and Campbell IG: Frequent PTEN/MMAC
mutations in endometrioid but not serous or mucinous epithelial
ovarian tumors. Cancer Res. 58:2095–2097. 1998.PubMed/NCBI
|
|
10
|
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y,
Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et
al: ARID1A mutations in endometriosis-associated ovarian
carcinomas. N Engl J Med. 363:1532–1543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Campbell IG, Russell SE, Choong DY,
Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB and
Phillips WA: Mutation of the PIK3CA gene in ovarian and breast
cancer. Cancer Res. 64:7678–7681. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schwartz DR, Kardia SL, Shedden KA, Kuick
R, Michailidis G, Taylor JM, Misek DE, Wu R, Zhai Y, Darrah DM, et
al: Gene expression in ovarian cancer reflects both morphology and
biological behavior, distinguishing clear cell from other
poor-prognosis ovarian carcinomas. Cancer Res. 62:4722–4729.
2002.PubMed/NCBI
|
|
13
|
Tashiro H, Blazes MS, Wu R, Cho KR, Bose
S, Wang SI, Li J, Parsons R and Ellenson LH: Mutations in PTEN are
frequent in endometrial carcinoma but rare in other common
gynecological malignancies. Cancer Res. 57:3935–3940.
1997.PubMed/NCBI
|
|
14
|
Risinger JI, Hayes AK, Berchuck A and
Barrett JC: PTEN/MMAC1 mutations in endometrial cancers. Cancer
Res. 57:4736–4738. 1997.PubMed/NCBI
|
|
15
|
Maehama T and Dixon JE: The tumor
suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem.
273:375–378. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Myers MP, Pass I, Batty IH, Van der Kaay
J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP and Tonks NK: The
lipid phosphatase activity of PTEN is critical for its tumor
supressor function. Proc Natl Acad Sci USA. 95:13513–13518. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Franke TF, Kaplan DR and Cantley LC: PI3K:
Downstream AKTion blocks apoptosis. Cell. 88:435–437. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Downward J: Ras signalling and apoptosis.
Curr Opin Genet Dev. 8:49–54. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo
P, Hu LS, Anderson MJ, Arden KC, Blenis J and Greenberg ME: Akt
promotes cell survival by phosphorylating and inhibiting a Forkhead
transcription factor. Cell. 96:857–868. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sif S, Saurin AJ, Imbalzano AN and
Kingston RE: Purification and characterization of mSin3A-containing
Brg1 and hBrm chromatin remodeling complexes. Genes Dev.
15:603–618. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang W, Xue Y, Zhou S, Kuo A, Cairns BR
and Crabtree GR: Diversity and specialization of mammalian SWI/SNF
complexes. Genes Dev. 10:2117–2130. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Reisman D, Glaros S and Thompson EA: The
SWI/SNF complex and cancer. Oncogene. 28:1653–1668. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liang H, Cheung LW, Li J, Ju Z, Yu S,
Stemke-Hale K, Dogruluk T, Lu Y, Liu X, Gu C, et al: Whole-exome
sequencing combined with functional genomics reveals novel
candidate driver cancer genes in endometrial cancer. Genome Res.
22:2120–2129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Roth LM, Emerson RE and Ulbright TM:
Ovarian endometrioid tumors of low malignant potential: A
clinicopathologic study of 30 cases with comparison to
well-differentiated endometrioid adenocarcinoma. Am J Surg Pathol.
27:1253–1259. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bell KA and Kurman RJ: A clinicopathologic
analysis of atypical proliferative (borderline) tumors and
well-differentiated endometrioid adenocarcinomas of the ovary. Am J
Surg Pathol. 24:1465–1479. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jetley S, Khetrapal S, Ahmad A and
Jairajpuri ZS: Atypical proliferative endometrioid tumor of ovary:
Report of a rare case. J Postgrad Med. 62:129–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Levine RL, Cargile CB, Blazes MS, van Rees
B, Kurman RJ and Ellenson LH: PTEN mutations and microsatellite
instability in complex atypical hyperplasia, a precursor lesion to
uterine endometrioid carcinoma. Cancer Res. 58:3254–3258.
1998.PubMed/NCBI
|