Introduction
Follicular lymphoma (FL) accounts for ~30% of all
non-Hodgkin's lymphomas (1). FL is
usually indolent, and patients have long survival rates (2). However, in 25–60% of all patients with
FL, FL undergoes transformation into diffuse large B-cell lymphoma
(DLBCL), which results in rapid progression, treatment resistance
and mortality (2). DLBCL is a
malignancy with a high mortality rate due to the lack of biomarkers
for early diagnosis and efficient therapeutic strategies (3). Therefore, it is critically important to
identify biomarkers for DLBCL and to investigate the biological
mechanisms of DLBCL.
Accumulating evidence reveals that lncRNAs serve key
functions in tumorigenesis, cancer progression and metastasis
(4,5).
Long non-coding RNAs (lncRNAs), a major class of non-coding RNAs,
are RNA polymerase II transcripts that are >200 bp and do not
encode proteins (6). Multiple reports
have revealed that lncRNAs may regulate the expression of
protein-coding genes through transcriptional, post-transcriptional,
post-translational and/or epigenetic regulation (7,8).
Previously, a number of studies had revealed that lncRNA expression
may be deregulated in various types of human cancer (9,10). For
example, prostate cancer associated 3 (non-protein coding) was
significantly upregulated in prostate cancer, compared with health
tissues (11). Additionally, it was
indicated that H19, imprinted maternally expressed transcript
(non-protein coding) was overexpressed in hepatocellular carcinoma
and that this overexpression was disease-associated (12). According to the competing endogenous
RNA (ceRNA) hypothesis (13), ceRNAs
may compete for the same micro RNA (miRNA) response elements to
regulate each other (14).
Previously, studies revealed that the ceRNA network may serve
prognostic or diagnostic functions in cancer. For example, Zhou
et al (15,16) identified dysregulated
lncRNA-associated ceRNA networks as biomarkers for pancreatic and
ovarian cancer. Previously, a number of studies indicated that
altered expression of certain lncRNAs may be an important mechanism
of DLBCL progression. A number of lncRNAs, including HOX transcript
antisense RNA (HOTAIR) (17), tumor
protein p53 pathway corepressor 1 (lincRNA-p21) (18), paternally expressed 10 (PEG10)
(19), MEF2C antisense RNA 2, SACS
antisense RNA 1, RP11-25K19.1, MME antisense RNA 1, RP11-360F5.1
and CSMD2 antisense RNA 1 (20) which
were significantly associated with the survival outcomes of DLBCL.
Peng et al (21,22) reported that hepatocellular carcinoma
upregulated long non-coding RNA and leukemia-associated non-coding
IGF1R activator RNA 1 were associated with cell proliferation in
DLBCL. Zhou et al (23)
identified a 17-lncRNA signature for subtype classification and
prognosis prediction by analyzing differentially expressed lncRNAs
between germinal center B-cell-like and activated B-cell-like
subtypes. However, the molecular mechanisms and functions
underlying the involvement of lncRNAs in the transformation of
DLBCL remain largely unknown.
In the present study, the aim was to identify
differentially expressed lncRNAs and mRNAs involved in the
transformation of DLBCL by analyzing a cohort of previously
published datasets from the Gene Expression Omnibus (GEO). In an
attempt to provide novel information on the molecular mechanisms
and functions of lncRNAs, a bioinformatics analysis was conducted
to identify the lncRNA-miRNA-mRNA regulatory axis in DLBCL.
Subsequently, Gene Ontology and Kyoto Encyclopedia of Genes and
Genomes analysis was performed in order to investigate the
potential functions of dysregulated lncRNAs.
Materials and methods
Microarray data and data
preprocessing
Microarray data was downloaded from a previous study
by Brodtkorb et al (24),
which was referenced in the GEO database (accession no. GSE53820;
www.ncbi.nlm.nih.gov/geo/). In this
dataset, preprocessed usingt the limma package in R (version,
3.34.2; www.r-project.org/), expression profiles
were obtained from a total of 81 biopsies, which were taken from 41
patients diagnosed with FL using the Affymetrix HG U133 Plus 2.0
Gene Chip (Affymetrix; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Of these, 49 biopsies (43 with a histological diagnosis of FL
and 6 with DLBCL) were sourced from 24 patients with a subsequent
transformation to a higher-grade lymphoma (namely, DLBCL) and 32
biopsies were sourced from 17 patients without any sign of
transformation. The cut-off values used for selecting
differentially expressed mRNAs were fold change ≥2 and
P<0.05.
lncRNA classification pipeline
In order to evaluate the expression of lncRNAs in
microarray data, a pipeline, previously described by Zhang et
al (25), was employed to
identify the probe sets uniquely mapped to lncRNAs from the
Affymetrix array by using the following criteria: i) For the probe
sets with Refseq IDs, those labeled as ‘NR’ (where NR indicates
non-coding RNA in the Refseq database) were retained; ii) for the
probe sets with Ensembl gene IDs, those annotated with ‘lncRNA’,
‘processed transcripts’, ‘non-coding’ or ‘misc_RNA’ in Ensembl
annotations were retained; iii) the probe sets obtained were
refined by filtering pseudogenes, ribosomal RNAs, microRNAs,
transfer RNA (tRNA)s, small nuclear RNAs and small nucleolar RNAs.
A total of 2,448 annotated lncRNA transcripts with corresponding
Affymetrix probe IDs were obtained. The cut-off values used for
selecting differentially expressed lncRNAs were fold change ≥2 and
P<0.05.
Functional group analysis
GO analysis and KEGG analysis were employed to
determine the biological functions of the identified differentially
expressed mRNAs, based on the freely available online MAS 3.0
system from CapitalBio Corporation (http://bioinfo.capitalbio.com/mas3/; Beijing China).
The P-value (hypergeometric P-value) denotes the significance of
the pathway associated with the conditions. P<0.05 was
considered to indicate a statistically significant difference.
Construction of the lncRNA-miRNA-mRNA
network
To predict the functions of the differentially
expressed lncRNAs, co-expression networks of differentially
expressed lncRNAs were constructed for further bioinformatics
analysis, as previously described by Guttman and Rinn
(26) and Shen et al (27). The StarBase dataset (27) was used to identify potential
dysregulated lncRNA-miRNA pairs. StarBase and TargetScan (28) databases were also used to identify
miRNA-mRNA pairs. Finally, a co-expression network based on
association analysis between the differentially expressed lncRNAs
and mRNAs was constructed. The lncRNA-mRNA interaction was
integrated into the co-expression networks according to positive
regulation and only gene pairs with |R| >0.5 were selected.
Hierarchical clustering analysis
To generate an overview of lncRNA and mRNA
expression profiles between FL and DLBCL, hierarchical clustering
analysis was performed based on the expression values. Cytoscape
3.0 was applied to plot the lncRNA-miRNA-mRNA ceRNA networks
involved in the transformation of DLBCL.
Statistical analysis
All numerical data (log-transformed) are presented
as the mean ± standard deviation of at least 3 determinations.
Statistical comparisons between groups of normalized data were
performed using a Student's t-test (unpaired) or Mann-Whitney
U-test according to the test conditions. P<0.05 was considered
to indicate a statistically significant difference with a 95%
confidence level. All of the above statistical analyses are
analyzed with R software, version 3.2.4 (https://www.r-project.org/).
Results
Systematic comparison of
differentially expressed mRNAs and lncRNAs between FL and
DLBCL
In order to compare differentially expressed mRNAs
and lncRNAs between FL and DLBCL, a publicly available gene
expression database (accession no. GSE53820) was utilized. This
database includes 75 FL samples and 6 DLBCL samples. Differentially
expressed mRNAs in the GSE53820 database were analyzed, and it was
identified that 1,884 genes were upregulated and 814 genes were
downregulated in DLBCL compared with FL.
Based on the NetAffx annotation of the probe sets
and the Refseq and Ensemble annotations of lncRNAs, a total of
2,448 lncRNA transcripts (corresponding to 1,970 lncRNA genes) were
identified in the GSE58320 database. lncRNA expression patterns
between FL and DLBCL were compared, and a total of 123 lncRNAs were
significantly upregulated and 192 lncRNAs were significantly
downregulated (P<0.05) in DLBCL compared with FL.
GO and KEGG analysis of differentially
expressed mRNAs
To identify the potential functions of
differentially expressed mRNAs, GO and KEGG analysis were performed
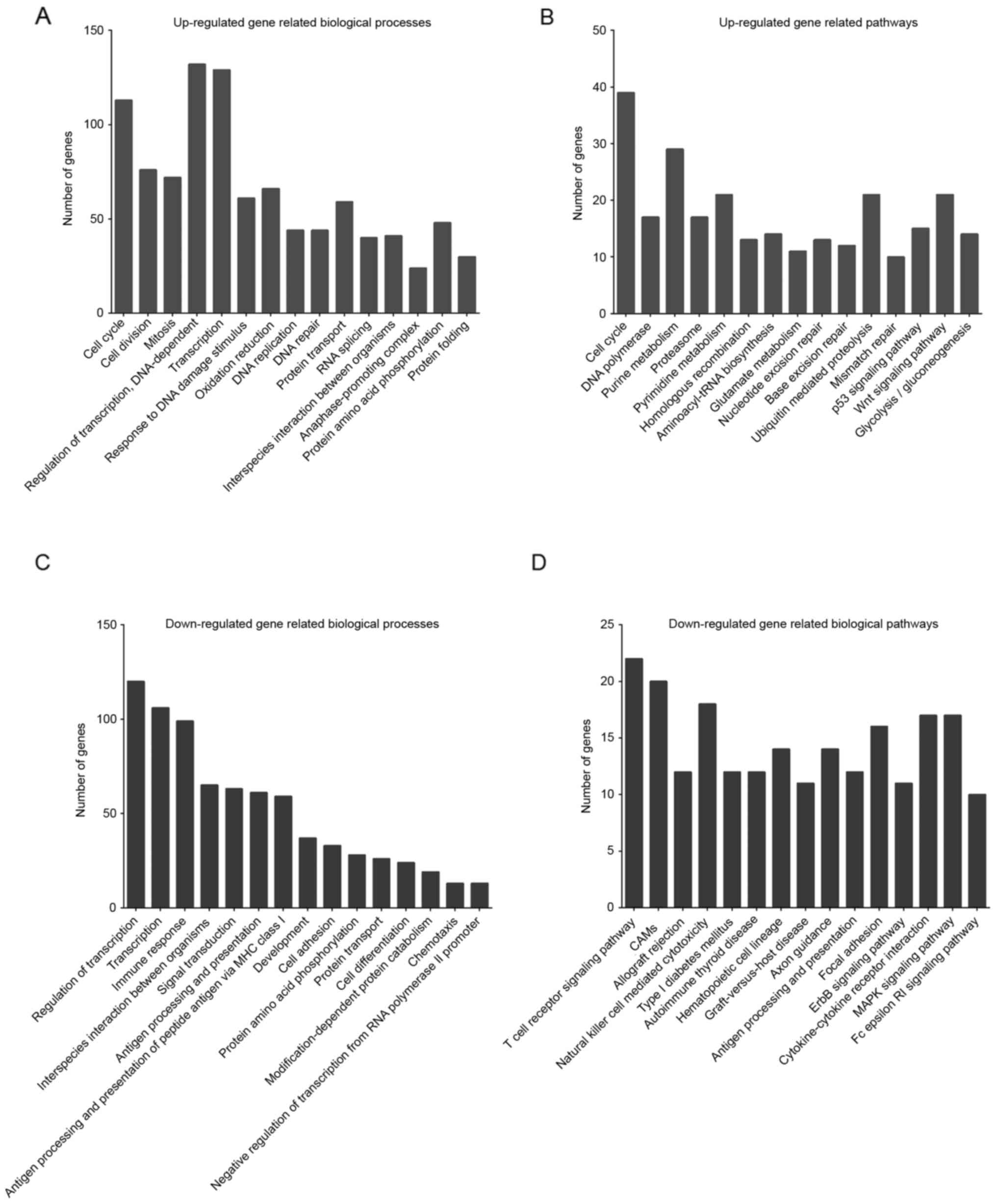
using MAS 3.0 software. GO analysis indicated that the upregulated
genes were primarily involved in the regulation of cell cycle, cell
division, mitosis, DNA-dependent regulation of transcription and
DNA replication, which are mainly associated with cell
proliferation (Fig. 1A). KEGG pathway
analysis revealed that upregulated genes were primarily enriched in
pathways associated with cell cycle, pyrimidine metabolism,
ubiquitin mediated proteolysis and Wnt signaling pathway (Fig. 1B).
Meanwhile, downregulated genes were mainly enriched
in categories associated with transcription, immune response,
interspecies interaction between organisms and signal transduction
(Fig. 1C). These results suggest that
these pathways may participate in regulating the transformation of
FL. Downregulated genes were mainly associated with T cell receptor
signaling pathway, cell adhesion molecules, cytokine-cytokine
receptor interaction, MAPK signaling pathway and natural killer
cell mediated cytotoxicity (Fig.
1D).
GO and KEGG analysis of differentially
expressed lncRNAs
Co-expression networks were constructed to identify
the association between differentially expressed mRNAs and lncRNAs
using the GSE53820 database. The cut-off values used for selecting
differentially expressed lncRNAs were a fold change ≥2 and
P<0.05. GO and KEGG analyses were performed for each lncRNA
using the set of co-expressed mRNAs.
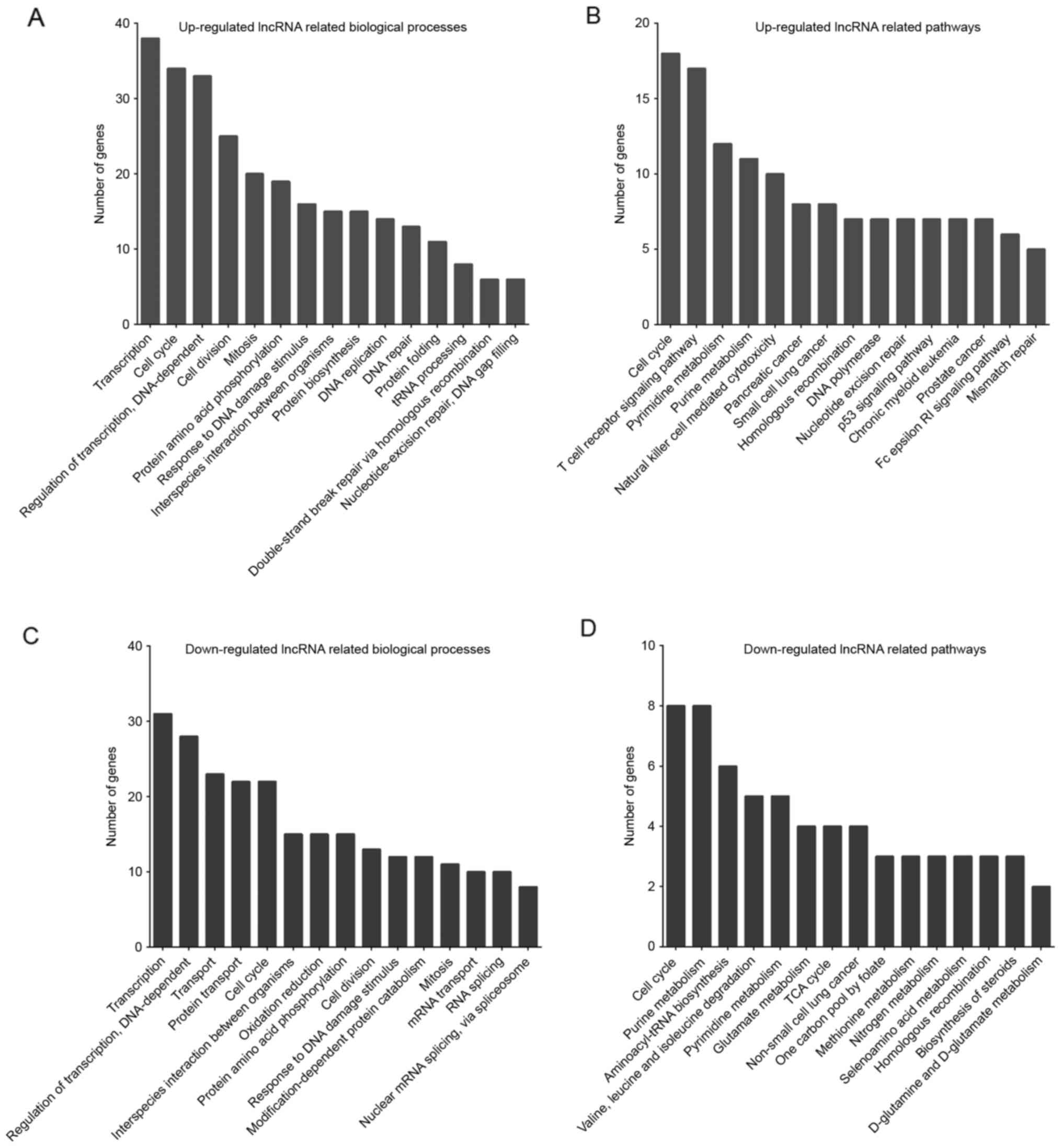
In the present study, the top 500 differentially
expressed lncRNAs and mRNAs were classified according to GO terms
(Fig. 2A and C). GO analysis revealed
that the upregulated lncRNAs were enriched in transcription, cell
cycle, cell division, mitosis, and protein amino acid
phosphorylation (Fig. 2A), while the
downregulated lncRNAs were enriched in transcription, transport,
cell cycle, interspecies interaction between organisms, and
oxidation reduction (Fig. 2C).
According to KEGG pathway analysis, upregulated
lncRNAs were primarily enriched in pathways associated with cell
cycle, T-cell receptor signaling pathway, pyrimidine metabolism and
purine metabolism (Fig. 2B).
Downregulated lncRNAs were enriched in pathways associated with
cell cycle, purine metabolism, aminoacyl-tRNA biosynthesis and
valine, leucine and isoleucine degradation (Fig. 2D).
Construction of the ceRNA
networks
In order to investigate the molecular mechanisms of
lncRNAs, the lncRNA-miRNA-mRNA axis was predicted in the present
study. Firstly, the interactions between differentially expressed
lncRNAs and their theoretical target miRNAs was predicted using the
StarBase database (27). Then,
TargetScan (28) and the StarBase
database were employed to identify mRNAs targets that are
suppressed by miRNAs. Finally, a co-expression network based on the
association analysis between the differentially expressed lncRNAs
and mRNAs was constructed. The lncRNA-mRNA interaction was
integrated into the co-expression networks according to positive
regulation and only gene pairs with |R| >0.5 were selected.
lncRNA-miRNA-mRNA ceRNA networks involved in the transformation of
DLBCL were constructed using Cytoscape 3.0 (http://www.cytoscape.org/).
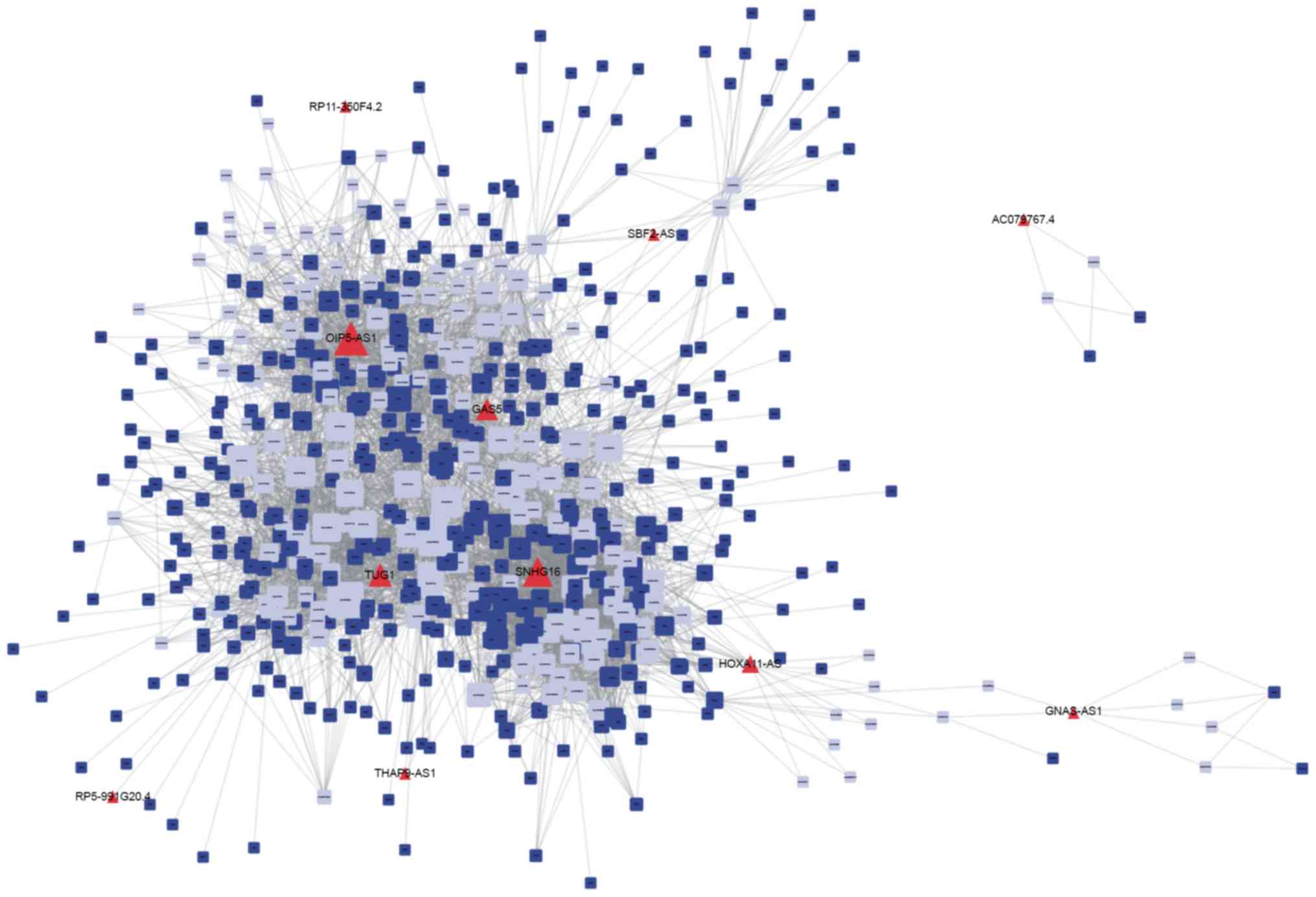
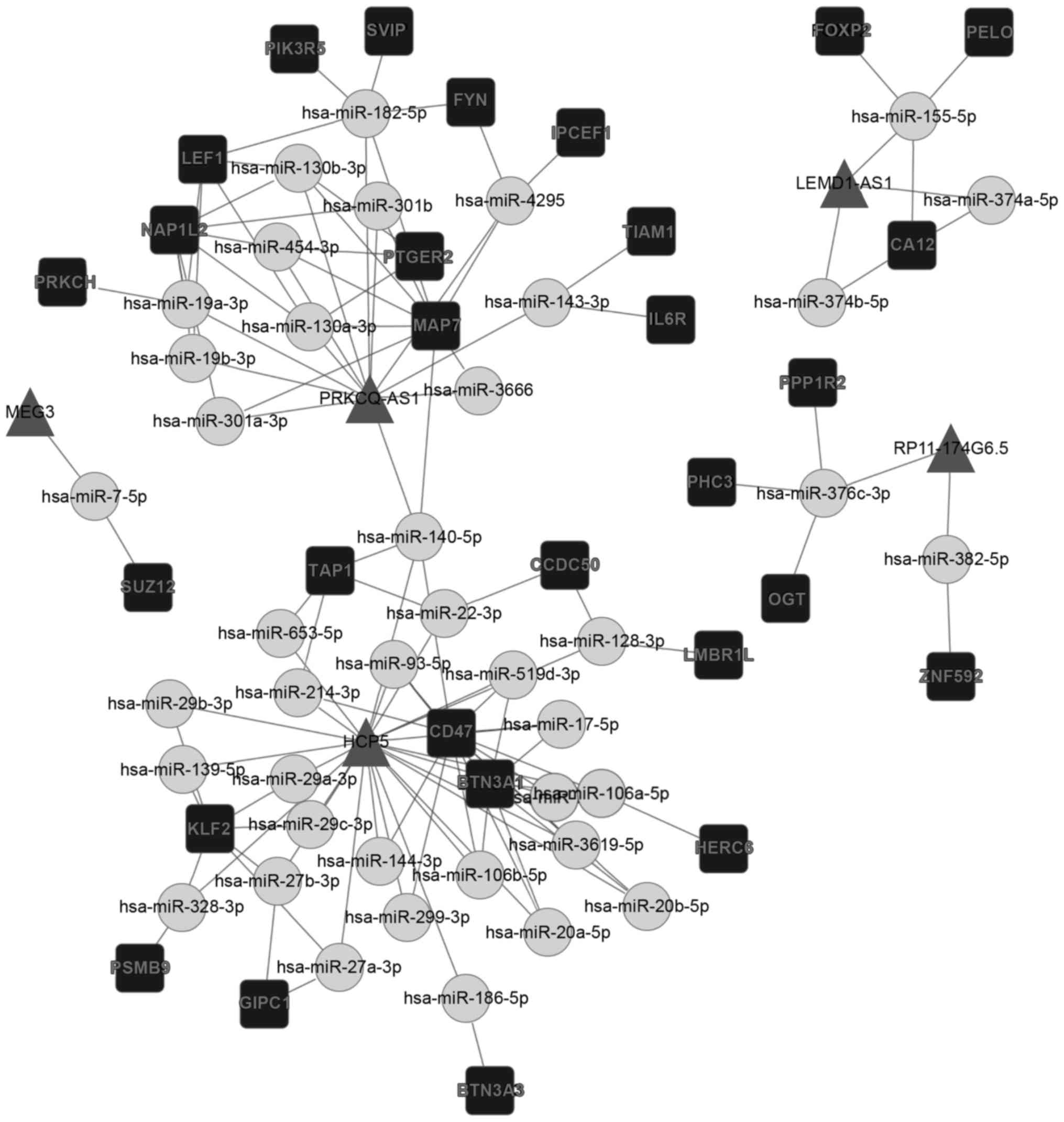
The results of the present study revealed a specific
DLBCL-associated and a specific FL-associated ceRNA network. As
presented in Fig. 3, 14 lncRNAs,
(including OIP5-AS1, SNHG16, HOXA11-AS and NUTM2A-AS1), 198 miRNAs,
and >1,200 mRNAs were involved in the specific DLBCL-associated
ceRNA network. It was revealed that the FL-associated ceRNA network
included 8 lncRNAs, (including HCP5, COX10-AS1, PRKCQ-AS1 and
LEMD1-AS1), 71 miRNAs, and >200 mRNAs (Fig. 4). The networks were constructed using
Cytoscape 3.0.
Investigating the molecular functions
of PRKCQ-AS1, HCP5, OIP5-AS1, growth arrest specific 5 (GAS5) and
taurine upregulated 1 (TUG1)
According to the ceRNA networks, it was revealed
that PRKCQ-AS1, HCP5, OIP5-AS1, GAS5 and TUG1 functioned as key
regulators (Figs. 5 and 6). However, the molecular functions of
PRKCQ-AS1, HCP5, OIP5-AS1, GAS5 and TUG1 in the transformation of
DLBCL remain unknown. By analyzing co-expressed mRNAs, it was
revealed that TUG1, OIP5-AS1 and GAS5 were associated with
anti-apoptosis, cell cycle, DNA repair, mitosis, transcription,
mitosis, G2/M transition of mitotic cell cycle and protein amino
acid phosphorylation functions (Fig.
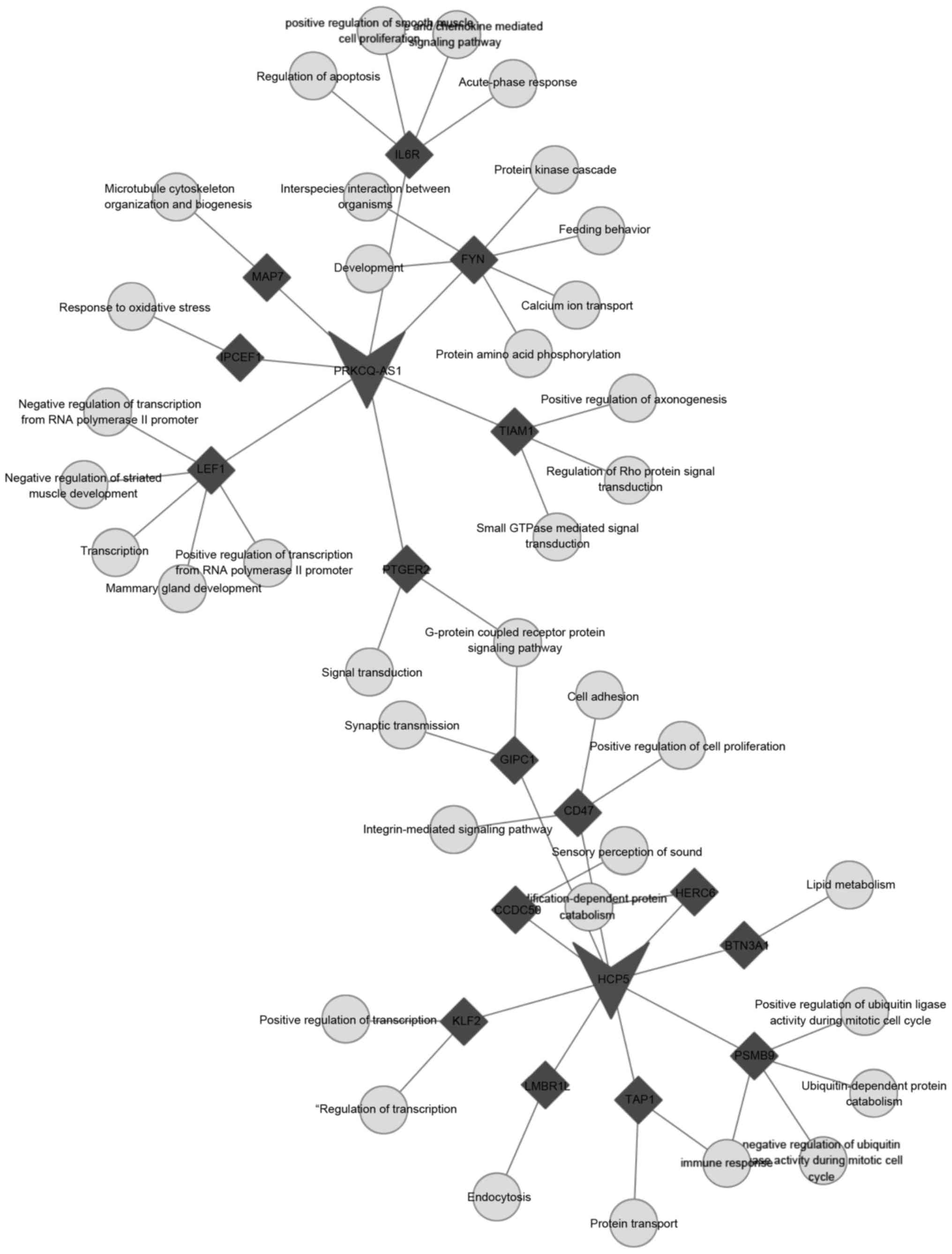
5). PRKCQ-AS1 was associated with the response to oxidative
stress, regulation of smooth muscle cell proliferation and
acute-phase response functions (Fig.
6). HCP5 was associated with transcription, cell adhesion,
lipid metabolism and immune response functions (Fig. 6).
Discussion
The molecular mechanisms involved in the
transformation of DLBCL had previously been unclear. Therefore, it
was critically important to investigate the biological mechanisms
of DLBCL. In the present study, differentially expressed mRNAs and
lncRNAs between FL and DLBCL were identified using the GEO database
accession no. GSE53820. Subsequently, a specific DLBCL-associated
ceRNA network and a specific FL-associated ceRNA network were
constructed. GO and KEGG pathway analyses revealed that
differentially expressed lncRNAs served key functions in regulating
signal transduction, transcription, cell adhesion, development and
protein amino acid phosphorylation.
DLBCL is a malignancy with a high mortality rate due
to a lack of biomarkers for early diagnosis and efficient
therapeutic strategies (3).
Previously, studies had indicated that lncRNAs served key functions
in tumorigenesis, cancer progression and metastasis (3,4). An
increasing number of studies have additionally demonstrated that
the expression of lncRNAs may be deregulated in various types of
human cancer, including DLBCL (9,10,17). In prostate cancer, Crea et al
(29) identified prostate cancer
associated transcript 18 as a novel biomarker and potential
therapeutic target for metastatic prostate cancer. Wan et al
(30) also reported that
androgen-responsive lncRNAs may function as biomarkers for prostate
cancer. In the present study, differentially expressed mRNAs and
lncRNAs between FL and DLBCL were identified using a publicly
available gene expression database, GSE53820. From the microarray
expression profiles, it was identified that 1,654 genes were
upregulated and 1927 genes were downregulated in DLBCL compared
with FL. It was also revealed that 152 lncRNAs were significantly
upregulated, and 37 lncRNAs were significantly downregulated
between the DLBCL and FL groups.
In order to predict the functions of the
differentially expressed lncRNAs, co-expression networks were
constructed and GO and KEGG analysis was performed for each lncRNA
by using a set of co-expressed mRNAs. According to the KEGG pathway
analysis, upregulated lncRNAs were primarily enriched in pathways
associated with the cell cycle, T cell receptor signaling pathway,
pyrimidine metabolism and purine metabolism. Downregulated lncRNAs
were enriched in pathways associated with cell cycle, purine
metabolism, aminoacyl-tRNA biosynthesis and degradation of valine,
leucine and isoleucine. GO analysis revealed that the upregulated
lncRNAs were enriched in transcription, cell cycle, cell division,
mitosis, and protein amino acid phosphorylation, whilst the
downregulated lncRNAs were enriched in transcription, transport,
cell cycle, interspecies interaction between organisms and
oxidation reduction.
Previously, a number of reports had revealed that
the altered expression of certain lncRNAs may be an important
mechanism of DLBCL progression. A number of lncRNAs, including
HOTAIR (17), LincRNA-p21 (18) and PEG10 (19) were significantly associated with the
progression of DLBCL. However, the molecular mechanisms and
functions underlying the involvement of lncRNAs in the
transformation of DLBCL remain largely unknown. In the present
study, in order to investigate the molecular mechanisms involved in
the regulation of DLBCL progression by lncRNAs, lncRNA-miRNA-mRNA
ceRNA networks were constructed based on our analysis. From the
present study, it was revealed that TUG1, PVT1, MALAT1 and HCP5
served key functions in lncRNA-mediated ceRNA networks. According
to GO analysis, the molecular functions of TUG1, PVT1, MALAT1 and
HCP5 in DLBCL were investigated. According to the ceRNA networks
constructed in the present study, it was revealed that PRKCQ-AS1,
HCP5, OIP5-AS1, GAS5 and TUG1 functioned as key regulators.
However, the molecular functions of PRKCQ-AS1, HCP5, OIP5-AS1, GAS5
and TUG1 in the transformation of DLBCL remained unknown. By
analyzing co-expressed mRNAs, it was revealed that TUG1, OIP5-AS1
and GAS5 were associated with anti-apoptosis, cell cycle, DNA
repair, mitosis, transcription, mitosis, G2/M transition of mitotic
cell cycle and protein amino acid phosphorylation. PRKCQ-AS1 was
associated with the response to oxidative stress, regulation of
smooth muscle cell proliferation and acute-phase response. HCP5 was
associated with transcription, cell adhesion, lipid metabolism and
immune response.
In conclusion, differently expressed lncRNAs between
FL and DLBCL were identified for the first time, screened by using
a microarray. Compared with FL, a total of 123 upregulated lncRNAs
and 192 downregulated lncRNAs in DLBCL were identified.
Subsequently, a specific DLBCL-associated ceRNA network and a
specific FL-associated ceRNA network were constructed. GO and KEGG
pathway analyses revealed that differentially expressed lncRNAs
served key functions in regulating signal transduction,
transcription, cell adhesion, development and protein amino acid
phosphorylation. The present study would provide a potential novel
therapeutic and prognostic target for the treatment of DLBCL.
Acknowledgements
The present study is supported by the Program of
Education Department of Zhejiang Province (grant no.
Y201223954).
References
|
1
|
Prochazka V, Papajik T, Jarosova M and
Indrak K: Prognostic factors in follicular lymphoma in the
rituximab era: How to identify a high-risk patient? Biomed Pap Med
Fac Univ Palacky Olomouc Czech Repub. 155:99–108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Martinez-Climent JA, Alizadeh AA, Segraves
R, Blesa D, Rubio-Moscardo F, Albertson DG, Garcia-Conde J, Dyer
MJ, Levy R, Pinkel D and Lossos IS: Transformation of follicular
lymphoma to diffuse large cell lymphoma is associated with a
heterogeneous set of DNA copy number and gene expression
alterations. Blood. 101:3109–3117. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shipp MA, Ross KN, Tamayo P, Weng AP,
Kutok JL, Aguiar RC, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, et
al: Diffuse large B-cell lymphoma outcome prediction by
gene-expression profiling and supervised machine learning. Nat Med.
8:68–74. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li SP, Xu HX, Yu Y, He JD, Wang Z, Xu YJ,
Wang CY, Zhang HM, Zhang RX, Zhang JJ, et al: lncRNA HULC enhances
epithelial-mesenchymal transition to promote tumorigenesis and
metastasis of hepatocellular carcinoma via the miR-200a-3p/ZEB1
signaling pathway. Oncotarget. 7:42431–42446. 2016.PubMed/NCBI
|
|
5
|
Li H, Yu B, Li J, Su L, Yan M, Zhu Z and
Liu B: Overexpression of lncRNA H19 enhances carcinogenesis and
metastasis of gastric cancer. Oncotarget. 5:2318–2329. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hangauer MJ, Vaughn IW and McManus MT:
Pervasive transcription of the human genome produces thousands of
previously unidentified long intergenic noncoding RNAs. PLoS Genet.
9:e10035692013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim T, Cui R, Jeon YJ, Fadda P, Alder H
and Croce CM: MYC-repressed long noncoding RNAs antagonize
MYC-induced cell proliferation and cell cycle progression.
Oncotarget. 6:18780–18789. 2015.PubMed/NCBI
|
|
8
|
Geisler S and Coller J: RNA in unexpected
places: Long non-coding RNA functions in diverse cellular contexts.
Nat Rev Mol Cell Biol. 14:699–712. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li CH and Chen Y: Targeting long
non-coding RNAs in cancers: Progress and prospects. Int J Biochem
Cell Biol. 45:1895–1910. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang JY, Lee JC, Chang YT, Hou MF, Huang
HW, Liaw CC and Chang HW: Long noncoding RNAs-related diseases,
cancers, and drugs. ScientificWorldJournal. 2013:9435392013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dijkstra S, Mulders PF and Schalken JA:
Clinical use of novel urine and blood based prostate cancer
biomarkers: A review. Clin Biochem. 47:889–896. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ariel I, Miao HQ, Ji XR, Schneider T, Roll
D, de Groot N, Hochberg A and Ayesh S: Imprinted H19 oncofetal RNA
is a candidate tumour marker for hepatocellular carcinoma. Mol
Pathol. 51:21–25. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Giza DE, Vasilescu C and Calin GA:
MicroRNAs and ceRNAs: Therapeutic implications of RNA networks.
Expert Opin Biol Ther. 14:1285–1293. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou M, Diao Z, Yue X, Chen Y, Zhao H,
Cheng L and Sun J: Construction and analysis of dysregulated
lncRNA-associated ceRNA network identified novel lncRNA biomarkers
for early diagnosis of human pancreatic cancer. Oncotarget.
7:56383–56394. 2016.PubMed/NCBI
|
|
16
|
Zhou M, Wang X, Shi H, Cheng L, Wang Z,
Zhao H, Yang L and Sun J: Characterization of long non-coding
RNA-associated ceRNA network to reveal potential prognostic lncRNA
biomarkers in human ovarian cancer. Oncotarget. 7:12598–12611.
2016.PubMed/NCBI
|
|
17
|
Yan Y, Han J, Li Z, Yang H, Sui Y and Wang
M: Elevated RNA expression of long non-coding HOTAIR promotes cell
proliferation and predicts a poor prognosis in patients with
diffuse large B cell lymphoma. Mol Med Rep. 13:5125–5131. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peng W, Wu J and Feng J: LincRNA-p21
predicts favorable clinical outcome and impairs tumorigenesis in
diffuse large B cell lymphoma patients treated with R-CHOP
chemotherapy. Clin Exp Med. 17:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peng W, Fan H, Wu G, Wu J and Feng J:
Upregulation of long noncoding RNA PEG10 associates with poor
prognosis in diffuse large B cell lymphoma with facilitating
tumorigenicity. Clin Exp Med. 16:177–182. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun J, Cheng L, Shi H, Zhang Z, Zhao H,
Wang Z and Zhou M: A potential panel of six-long non-coding RNA
signature to improve survival prediction of diffuse large-B-cell
lymphoma. Sci Rep. 6:278422016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Peng W, Wu J and Feng J: Long noncoding
RNA HULC predicts poor clinical outcome and represents
pro-oncogenic activity in diffuse large B-cell lymphoma. Biomed
Pharmacother. 79:188–193. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peng W and Feng J: Long noncoding RNA
LUNAR1 associates with cell proliferation and predicts a poor
prognosis in diffuse large B-cell lymphoma. Biomed Pharmacother.
77:65–71. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou M, Zhao H, Xu W, Bao S, Cheng L and
Sun J: Discovery and validation of immune-associated long
non-coding RNA biomarkers associated with clinically molecular
subtype and prognosis in diffuse large B cell lymphoma. Mol Cancer.
16:162017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brodtkorb M, Lingjaerde OC, Huse K, Trøen
G, Hystad M, Hilden VI, Myklebust JH, Leich E, Rosenwald A, Delabie
J, et al: Whole-genome integrative analysis reveals expression
signatures predicting transformation in follicular lymphoma. Blood.
123:1051–1054. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Sun S, Pu JK, Tsang AC, Lee D,
Man VO, Lui WM, Wong ST and Leung GK: Long non-coding RNA
expression profiles predict clinical phenotypes in glioma.
Neurobiol Dis. 48:1–8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guttman M and Rinn JL: Modular regulatory
principles of large non-coding RNAs. Nature. 482:339–346. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shen X, Xie B, Ma Z, Yu W, Wang W, Xu D,
Yan X, Chen B, Yu L, Li J, et al: Identification of novel long
non-coding RNAs in triple-negative breast cancer. Oncotarget.
6:21730–21739. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Crea F, Watahiki A, Quagliata L, Xue H,
Pikor L, Parolia A, Wang Y, Lin D, Lam WL, Farrar WL, et al:
Identification of a long non-coding RNA as a novel biomarker and
potential therapeutic target for metastatic prostate cancer.
Oncotarget. 5:764–774. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wan X, Huang W, Yang S, Zhang Y, Pu H, Fu
F, Huang Y, Wu H, Li T and Li Y: Identification of
androgen-responsive lncRNAs as diagnostic and prognostic markers
for prostate cancer. Oncotarget. 7:60503–60518. 2016. View Article : Google Scholar : PubMed/NCBI
|