Introduction
Pancreatic cancer is the fourth leading cause of
cancer-associated mortality in the United States (1). Despite recent advances in treatment, the
5-year overall survival rate for pancreatic cancer remains <5%
(2). This poor prognosis is
considered to be due to the highly aggressive invasion and early
metastasis that is typical of pancreatic cancer, with the majority
of patients presenting with extrapancreatic dissemination at
diagnosis (3).
Therefore, further understanding of the biological
behavior, and molecular and genetic alterations, in the stepwise
progression of pancreatic intraepithelial neoplasia (PanIN) in the
development of pancreatic ductal adenocarcinoma (PDAC) is required.
A previous study associated the MET proto-oncogene receptor
tyrosine kinase (c-MET), which functions in
epithelial-mesenchymal transition (EMT), with PDAC. c-MET
encodes for a membrane-bound receptor tyrosine kinase that is
predominantly expressed by epithelial cells (4). Activation of c-Met occurs following its
phosphorylation in response to the binding of its ligand,
hepatocyte growth factor (HGF; also referred to as scatter factor),
which results in the induction of a downstream signaling cascade
(4). c-Met activating ligands are
secreted by cells of mesenchymal origin (4). The resulting HGF/c-Met pleiotropic
signaling pathway activates mediators of cell proliferation and
motility (4). This signaling cascade
has been associated with tumorigenesis following the identification
of amplification, activating mutation and/or overexpression of
c-MET in the majority of solid organ neoplasms (4). Histopathological evaluation of clinical
samples from patients with PDAC has demonstrated that c-MET
expression levels are increased by ~5-7-fold compared with those of
normal pancreatic tissue samples, with this being proportional to
the tumor grade and correlated with an increased
tumor-node-metastasis stage (4). In
addition, HGF protein secreted by surrounding stromal tissue has
been correlated with c-MET overexpression in patients with
pancreatic cancer, and is associated with a poorer overall survival
(5).
A growing body of evidence suggests that a hierarchy
exists in cancer cell populations, a theory initially made in the
study of hematopoietic malignancies (6). Cancer stem cells (CSCs) comprise a small
minority of tumor cells; however, they appear to be the only cancer
cell type capable of unlimited self-renewal and formation of
xenografts. Interestingly, CSCs appear to have a limited potential
for further differentiation (6). The
majority of patients with PDAC are only candidates for palliative
chemotherapy, which has been proven to be largely ineffective at
halting tumor progression (1). One
proposed mechanism of pancreatic cancer cell resistance to
palliative chemotherapy involves the signaling pathway of the
EMT-associated protein c-Met, a signaling pathway that is essential
for cancer cell proliferation and migration (4). In the present study, the role served by
c-Met in the tumorigenesis and chemoresistance of KRAS
proto-oncogene GTPase (KRAS)G12D-induced PDAC was
investigated, in addition to its potential as a therapeutic target
for the treatment of PDAC.
Materials and methods
Mouse model of PDAC
All animal studies were approved by the Animal
Experiments Committee of Osaka University (Osaka, Japan; approval
no. 24-122-022). Pdx-1Cre/+, KrasLSL-G12D/+ and
Metflox/flox adult transgenic mating pairs of male and
female mice (2 of each type) from a BL6 background (~6 weeks old;
weight unknown) were obtained from the National Cancer Institute
(National Institutes of Health, Bethesda, MD, USA). All experiments
used co-housed littermates to ensure the consistency of microflora;
the temperature was ~22°C in 12 h-cycle of light and dark, and food
and water were added in ad libitum. The Pdx-1Cre/+ mice were
crossed with KrasLSL-G12D/+ mice or Metflox/flox mice
to generate Pdx-1Cre/+/KrasLSL-G12D/+ (Km) and
Pdx-1Cre/+/Metflox/flox (MΔ) mouse strains,
respectively. The Km mice were crossed with MΔ mice to generate the
Pdx-1Cre/+/KrasLSL-G12D/+/Metflox/flox (KmMΔ)
strain. Pdx-1Cre/+, KRASLSL-G12D/+ and
Metflox/flox were present in the pancreases, but not tails,
of compound mutant mice.
Genotyping
Mouse genomic DNA was isolated from tail biopsies.
Briefly, to extract DNA, tail biopsy samples were incubated at 95°C
in 25 mM 10 N NaOH and 0.2 mM EDTA buffer (pH 12.0), followed by
neutralization with 1 M Tris-HCl (pH 5.0) at 20°C. After
centrifuging at 300 × g for 15 min at 4°C, a total of 2 µl of each
sample supernatant underwent a polymerase chain reaction (PCR)
using the PrimeSTAR Max PCR kit (Takara Bio, Ltd., Shiga, Japan)
according to the manufacturer's protocol. PCR reaction was
performed on the GeneAmp® PCR System 9600 (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Thermocycling conditions for Pdx-1Cre/+ and
KrasLSL-G12D/+ samples were as follows: 94°C for 2 min; 35
cycles of 94°C for 30 sec, 60°C for 30 sec and 72°C for 30 sec; and
72°C for 5 min. Thermocycling conditions for Metflox/flox
samples were as follows: 94°C for 2 min; 45 cycles of 94°C for 30
sec, 54°C for 30 sec and 72°C for 30 sec; and 72°C for 5 min. PCR
was performed using the following primer pairs (https://www.jax.org/): (https://www.jax.org/): Pdx-1Cre/+ forward (F),
5′-GCGGTCTGGCAGTAAAAACTATC-3′ and reverse (R),
3′-GTGAAACAGCATTGCTGTCACTT-5′; KrasLSL-G12D Y-116 F:
5′-TCCGAATTCAGTGACTACAGATG-3′; KrasLSL-G12D Y-117 F:
5′-CTAGCCACCATGGCTTGAGT-3′; KrasLSL-G12D Y-118 R:
5′-ATGTCTTTCCCCAGCACAGT-3′; and Metflox/flox F:
5′-TTAGGCAATGAGGTGTCCCAC-3′ and R: 3′-CCAGGTGGCTTCAAATTCTAAGG-5′.
An 8 µl aliquot of each PCR product was separated on 2% agarose gel
using electrophoresis and visualized with ethidium bromide
staining.
Gemcitabine (GEM) treatment
Mice were injected with GEM (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) intra-peritoneally on days 1, 8 and 15.
Mice were given 125 mg/kg GEM, or PBS as control, as previously
described (7). Survival of the mice
was observed until 400 days post-injection.
Immunohistochemistry (IHC)
IHC was performed as previously described (8). Briefly, the pancreas was fixed with 10%
formaldehyde, embedded in paraffin and cut into 4-µm-thick
sections. The sections were subsequently deparaffinized in xylene,
boiled for antigen retrieval and incubated with anti-c-Met (mouse
monoclonal; 1:100 dilution) and anti-proliferation marker protein
Ki-67 (Ki-67; rabbit polyclonal; 1:50 dilution) (both Abcam,
Cambridge, UK) primary antibodies overnight at 4°C. Following
incubation with the rabbit (cat. no. PK4001) and mouse (cat. no.
PK4002) secondary antibodies (both Vector Laboratories, Inc.,
Burlingame, CA, USA; dilution 1:1,000) for 2 h at 22°C, these were
then visualized with avidin-biotin complex reagents (ABC-HRP kit;
Vector Laboratory, Inc., Burlingame, CA, USA) and
3,3′-diaminobenzidine. Serial sections were evaluated for each
antibody under the light microscope. The alcian blue staining was
performed using a kit (cat. no. H-3501; Vector Laboratories, Inc.)
for 30 min at 22°C according to the manufacturer's protocol.
Histology was performed using hematoxylin and eosin (H&E)
staining kits (hematoxylin; cat. no. 8650; Sakura Finetech, Tokyo,
Japan] for 5 min and eosin (cat. no. 8659; Sakura Finetech) for 1
min at 22°C according to the manufacturer's protocol, and confirmed
by two pathologists.
Quantification of PanIN
progression
ImageJ software version 1.6.0_24 (National
Institutes of Health; http://rsbweb.nih.gov/ij) software was used for the
manual detection of total tissue or individual acinar, ductal and
parenchymal lesions (including acinar to ductal metaplasia, PanIN
and invasive ductal adenocarcinoma). The total percentage of tissue
surface area occupied by each lesion was calculated for 10 random
fields of view for ≤3 independent slides (8).
Statistical analysis
Results are presented as the mean ± standard
deviation unless otherwise indicated. Statistically significant
differences were determined using a log-rank test (for survival
rate using Kaplan-survival curves) or Student's t-test. P<0.05
was considered to indicate a statistically significantly
difference.
Results
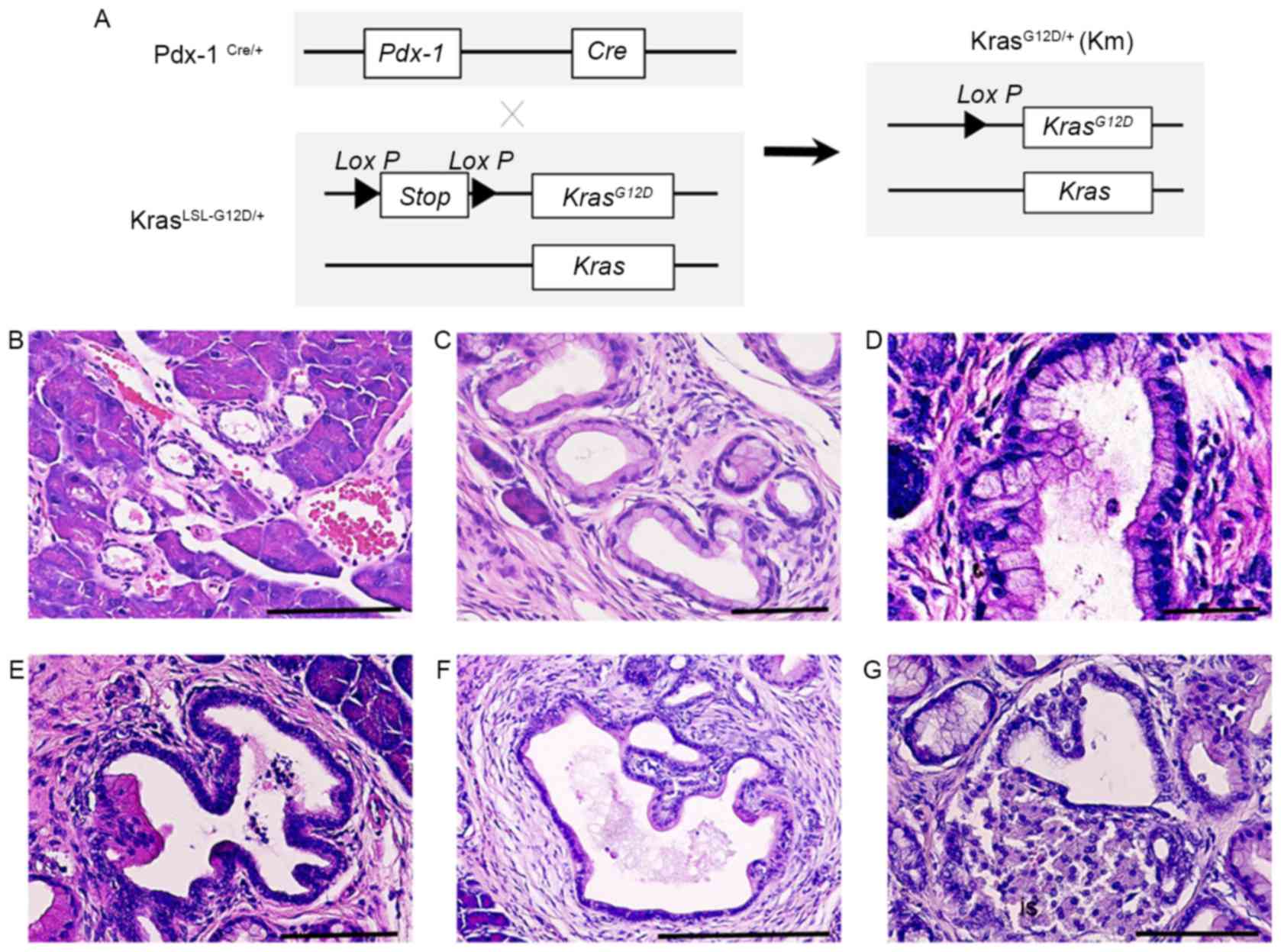
Endogenous Kras G12D expression
induces early and advanced stage PanINs
Previous reports have demonstrated that oncogenic
KrasG12D induces the formation of ductal lesions that
recapitulate the development of human pancreatic neoplastic lesions
in Pdx-1Cre/KrasLSL-G12D/+ mutant mice (9). The pancreas of
Pdx-1Cre/KrasLSL-G12D/+ mutant mice developed ductal
lesions that represented all three stages of human PanIN lesions
(Fig. 1). Normal pancreatic tissues
from wild-type control mice (CTL) revealed a normal cuboidal ductal
epithelium monolayer, islet cells and surrounding acinar tissue
(Fig. 1B). Low-grade PanIN lesions
(PanIN-1) were composed of flat or papillary columnar or cuboidal
cells that retained their nuclear polarity and lacked atypical
nuclei (Fig. 1B). Intermediate-grade
PanIN lesions (PanIN-2), which are more architecturally complex
compared with PanIN-1 lesions, presented nuclear abnormalities,
including loss of polarity, crowding, variable size (pleomorphism),
hyperchromasia and pseudo-stratification; however, the presence of
mitoses were rare (Fig. 1C).
High-grade PanIN lesions (PanIN-3) displayed a widespread loss of
polarity, marked nuclear atypia and prevalent mitoses within the
basement membrane (Fig. 1D).
Histological study revealed that PanIN-2 lesions had a marked loss
of polarity and moderate nuclear atypia (Fig. 1E) and that PanIN-3 lesions had a
complete loss of cellular polarity, marked nuclear atypia and cell
clusters budding into the ductal lumen (Fig. 1F). Histological study also
demonstrated that PDAC had the tendency to invade adjacent
structures, including islets (Fig.
1G). These results indicate that KRAS activating mutations
induce the progression of PanIN.
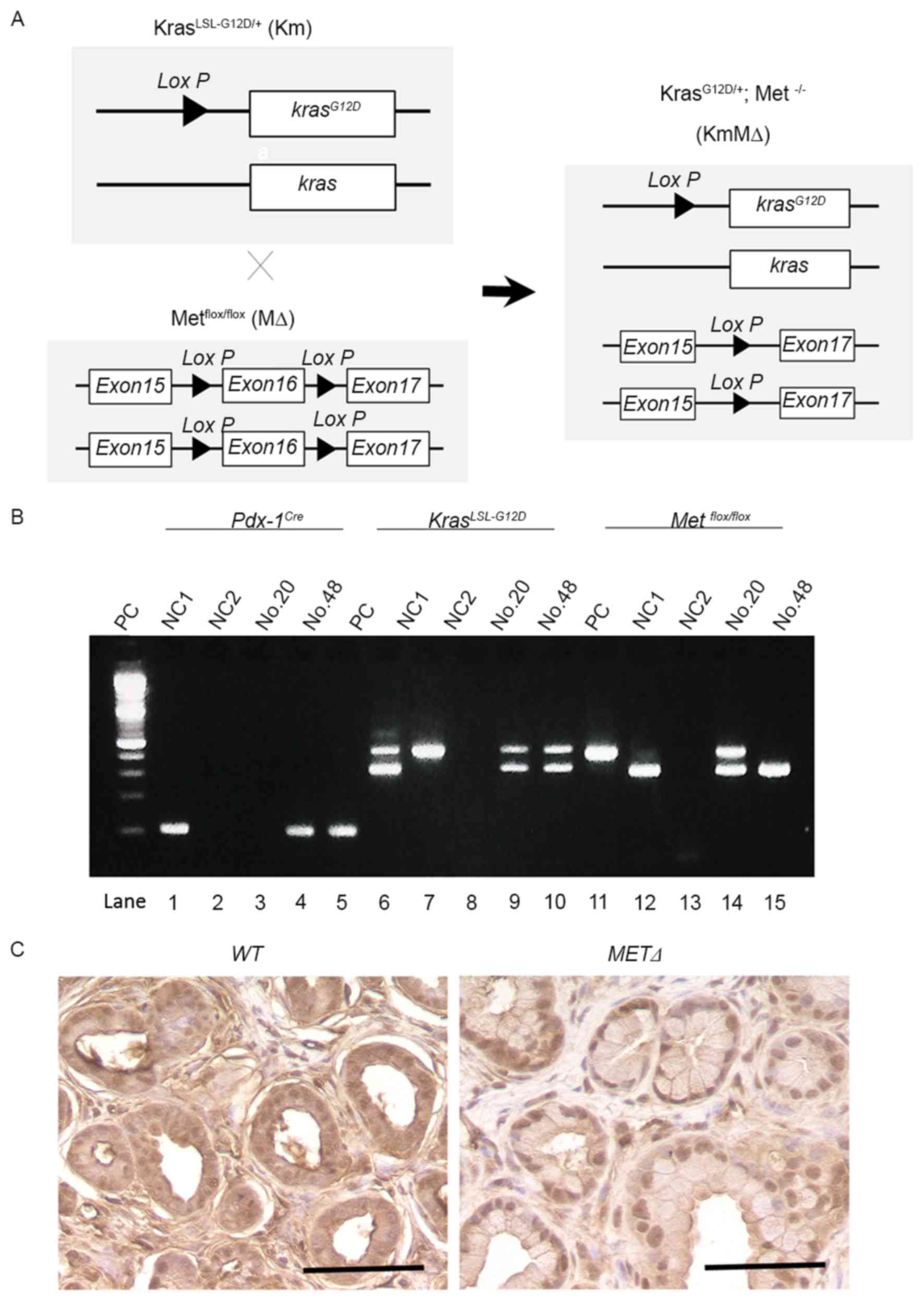
Successful targeted deletion of c-MET
in the pancreas of Km mice
To examine the potential role of c-Met in the
development of PDAC, Km mice were crossed with MΔ mice to generate
KmMΔ mice (Fig. 2A). This excised the
c-MET gene as part of a Cre-mediated silencing cassette and
subsequent recombination generated by a single Lox P site was
detected in all mice. The resulting strain with floxed c-MET
alleles was utilized for breeding with strains harboring the
Pdx-1Cre and the constitutively active KrasLSL-G12D
knock-in allele (Fig. 2B).
Immunostaining identified cytoplasmic c-Met protein in wild-type
(Met+/+) pancreatic tissue (Fig.
2C). However, cytoplasmic c-Met was not detected in the c-MET
knockout MΔ strain (Fig. 2C).
c-MET expression does not influence
the development of pancreatic neoplasia
To examine the development of pancreatic neoplasia,
pancreatic tissues were harvested from CTL, Km and KmMΔ mice at 6
months of age and evaluated by H&E staining (Fig. 3). This revealed that no pancreatic
neoplasia was observed in the pancreatic tissue of CTL mice
(Fig. 3A), whereas the pancreatic
tissues of Km (Fig. 3B) and KmMΔ mice
(Fig. 3C) had numerous regions of
pancreatic neoplasia. The regions of pancreatic neoplasia observed
were positive for Ki-67 (Fig. 3D-F).
These results suggest that the cells in regions of neoplasia were
proliferating markedly. In Km and KmMΔ mice, ducts and stromal
regions were significantly increased and acinar regions were
significantly decreased compared with CTL mice (Fig. 3G). Comparison of the tissue sample
surface areas indicated that the areas of ducts, acini and stroma
were not significantly different between Km and KmMΔ mice (Fig. 3G), suggesting that the absence of
endogenous c-MET alleles does not affect pancreatic carcinogenesis
in pancreas-specific transcription factor Pdx-1-conditional mice
(Pdx-1Cre/+, KrasLSL-G12D/+ or Metflox/flox).
Survival rates were significantly decreased in Km and KmMΔ mice
compared with the CTL mice (Fig. 3H).
These results suggest that c-MET has little or no impact on the
development of pancreatic neoplasia.
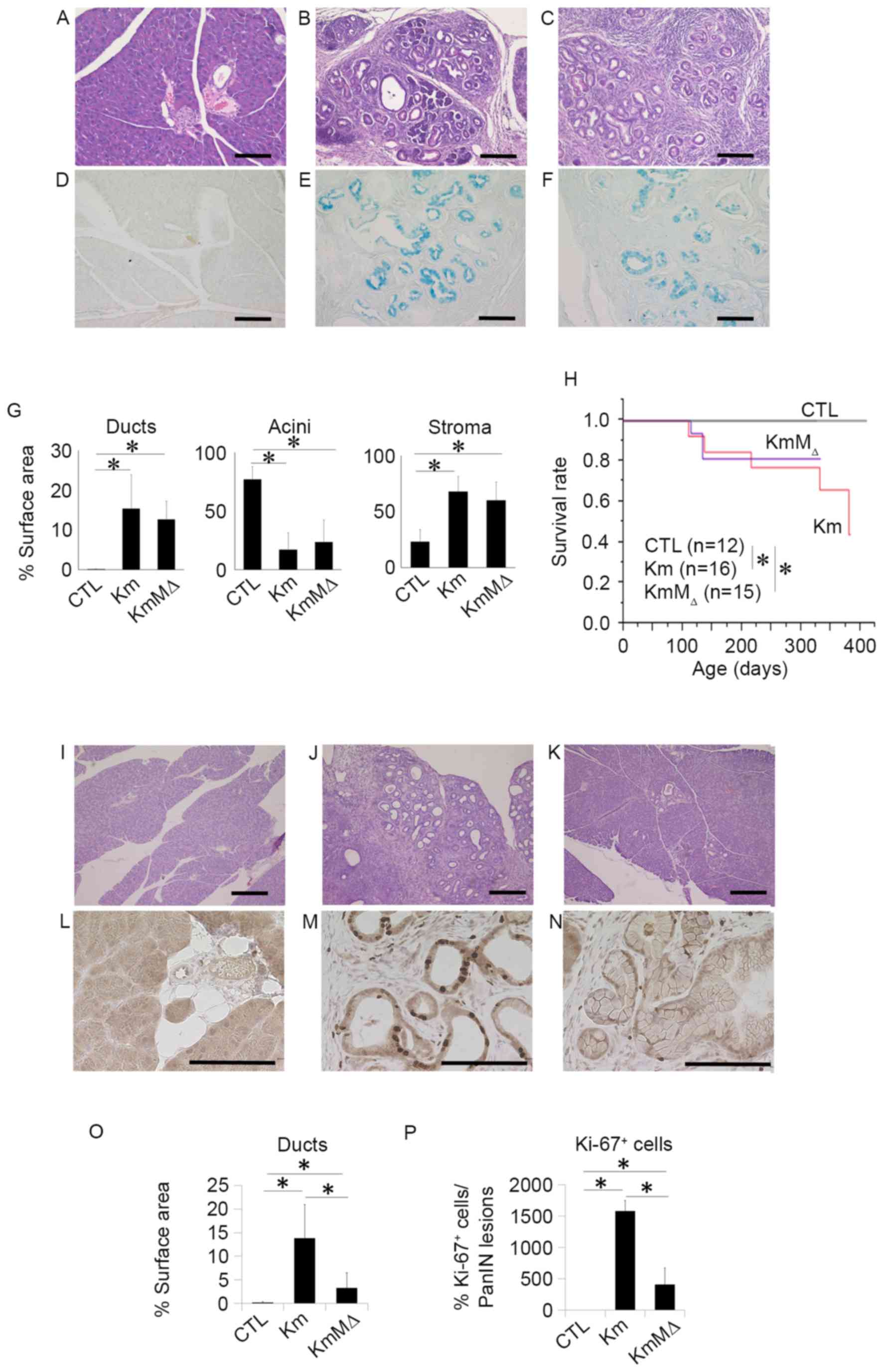 | Figure 3.c-MET expression does not
influence the development of pancreatic neoplasia, and c-MET
deletion in pancreatic neoplasia enhances chemosensitivity to GEM.
Representative hematoxylin and eosin staining of pancreatic tissue
sections from 9-month-old (A) wild-type, (B) Km and (C) KmMΔ mice.
Representative images of alcian blue staining in (D) wild-type, (E)
Km and (F) KmMΔ mice. (G) Quantification of fractional cross
sections occupied by ductal lesions, acinar lesions or stroma in
wild-type, Km and KmMΔ mice. (H) Kaplan-Meier survival rate of
wild-type (n=12), Km (n=15), and KmMΔ mice (n=16). Histological
analysis of the pancreas of (I) wild-type, (J) Km and (K) KmMΔ mice
following 125 mg/kg GEM administration. Representative
immunohistochemistry images of Ki-67 stained (L) wild-type, (M) Km
and (N) KmMΔ mice. (O) Quantification of fractional cross sections
occupied by ductal lesions, in wild-type, Km and KmMΔ mice. (P)
Quantification of fractional cross sections occupied by Ki-67
positive lesions in wild-type, Km and KmMΔ mice. Scale bar, 100 µm.
Results are presented as the mean ± standard deviation (n=5).
*P<0.05. GEM, gemcitabine; CTL, control wild-type mice. |
c-MET deletion in pancreatic neoplasia
enhances chemosensitivity to gemcitabine
Chemoresistance is an important contributory factor
to the high mortality rates of the majority of cancer types,
including pancreatic cancer. A previous report demonstrated that a
high expression of c-MET in pancreatic cancer is correlated with
chemoresistance (10). To examine the
role of c-Met in the chemoresistance of pancreatic neoplasia, 125
mg/kg GEM was administered to CTL, Km and KmMΔ mice. PBS was used
as the control treatment. Histological studies (Fig. 3I-K) revealed that although the
relative occupancy of viable PanIN lesions in Km mice remained at
15%, the amount of PanIN lesions in KmMΔ mice was reduced to <5%
following GEM treatment (data not shown). It has previously been
reported that Ki-67 is a useful predictive marker for
chemotherapeutic responses and clinical prognosis (11). To investigate the cellular
proliferation level in the PanIN lesion, Ki-67 staining was
performed (Fig. 3L-N). Quantification
demonstrated an increase in ductal lesions in KmMΔ mice, which was
more apparent in Km mice (Fig. 3O).
These results demonstrated that >15% of Ki-67 positive cells
were located in PanIN lesions in Km mice (Fig. 3P).
Discussion
CSCs typically exhibit three key characteristics,
which are not mutually exclusive. Firstly, CSCs are highly
tumorigenic and can form tumors in immunodeficient mice through
xenotransplantation, which is not possible for non-CSCs (6). Secondly, CSCs that survive chemotherapy
and radiotherapy generate resistance to such therapies through
regulating intracellular stress; for example, regulating reactive
oxygen species, which non-CSCs cannot (12). Thirdly, CSCs possess metastatic
potential, illustrated by a report that CSCs have the ability to
metastasize through EMT (13).
Previous studies have demonstrated that the
expression of c-Met on the cell surface correlates with the
characteristics of CSCs. Li et al (14) reported that CSCs with high
c-MET expression (c-Methi) and cluster of
differentiation 44 expression had tumorigenic potential in a
NOD/SCID mouse xenograft model. Furthermore, a report has
demonstrated that c-Methi CSCs exhibit resistance to
chemotherapy and radiotherapy, and that c-Met inhibitors are
effective in the killing of pancreatic cancer cells (15). The signaling pathway downstream of
HGF/c-Met serves an essential role in the maintenance of pancreatic
progenitor cells and stem cells (16). Furthermore, the c-Met/HGF signaling
pathway is associated with cancer cell-stroma interactions and the
metastasis of therapy-resistant cancer cells (17). Previous studies are in agreement that
c-Met serves a role in augmenting the pathological functions
associated with the advanced stages of cancer and therapeutic
resistance of pancreatic cancer (4,5,14). However, these studies lack in
vivo evidence demonstrating the role of c-Met in
chemotherapeutic resistance in a pancreatic cancer model. In the
present study, c-Met was demonstrated to induce GEM resistance in a
mouse model of pancreatic cancer through comparing the surface area
of cancerous regions and number Ki-67 positive cells, markers of
proliferating malignant cancer cells (11), in Km and KmMΔ mice. Considering that
CSCs can survive in a unique hypoxic niche, the present in
vivo study of therapeutic resistance in a mouse model has an
advantage over previously studied xenograft models, in terms of the
microenvironment being more similar to that in human cancer.
Our group previously demonstrated (18) that c-Methi pancreatic
cancer cells, which have a high capacity for sphere formation (a
marker of stemness) and exhibit resistance to GEM, are prone to
reprogramming by four transcription factors, proto-oncogene c-Myc,
octamer-binding protein 3/4, leucine-rich repeat protein soc-2 and
Krueppel-like factor 4. This suggests that CSCs are susceptible to
epigenetic reprogramming and that growth factor-dependent
intracellular mechanisms are essential for the determination of
malignant cancer cell behavior. Although a previous study has
demonstrated that the c-Met/HGF signaling pathway induces therapy
resistance in CSCs (19), the
mechanistic roles that c-Methi pancreatic cancer cells
serve in regulating cancer stemness during carcinogenesis remain to
be elucidated.
The present study demonstrated that pancreatic
tumors in Km and KmMΔ mice were formed at a similar frequency, in
regards to the formation of ducts, acini and stroma, and that
survival rates were not significantly different between Km and KmMΔ
mice. Notably, the data also suggested that the absence of c-Met
made no significant difference to pancreatic tumor formation. This
observation was unexpected, in view of the results of a previous
study on therapy-resistant c-Methi pancreatic CSCs
(20). There are several potential
explanations for this observation. Firstly, it is possible that
selective activation of the c-Met signaling pathway may be
important for the exhibition of characteristic tumor behaviors,
including therapy resistance, because c-MET amplification
and overexpression have been reported to be correlated with worse
prognostic significance in gastric cancer (21). c-MET expression in transgenic
mice may cause carcinogenesis, although the present study used
conditional knockout model. Secondly, as chronic inflammatory
responses coexist with pancreatic cancer formation (22), the present study was designed to
examine the essential role of c-Met. The use of transgenic mice
that had this inflammatory environment present, including the
relevant cytokines and chemokines, may therefore have influenced
carcinogenesis in the pancreas. Thirdly, the present study used a
pancreatic and duodenal homeobox (Pdx)-1 promoter-driven
conditional knockout for KRAS mutant activation in addition
to c-Met knockout. Although Pdx-1 is expressed in stem and
progenitor cells in the pancreas, the function of Pdx-1 appears to
be context-dependent in cancer development, in which an interplay
of multiple transcription factor networks is involved (23). These results suggest that endogenous
c-Met serves an essential role in therapy resistance, which may be
enhanced by the increased expression of endogenous c-Met,
potentially as a result of gene amplification. This appears to be
in contrast to the initiation of pancreatic carcinogenesis, where
multiple factors serve a role. This notion is analogous with
clinical observations in humans, demonstrating a role of c-met in
the early stage of pancreatic carcinogenesis (24). Thus, inhibitors of c-Met may be
beneficial for the control of therapy-resistant CSCs (15).
In conclusion, the present study used conditional
knockout of the c-Met gene in mice carrying a KRAS
mutation to investigate the role of c-Met in pancreatic cancer
development and therapy resistance. c-Met was identified to serve a
role in GEM resistance in vivo. As amplification and altered
expression of the c-MET gene is apparent in gastrointestinal
cancer, the results of the present study suggest that targeting the
c-Met signaling pathway is a potential option for the treatment of
pancreatic cancer and warrants further study.
Acknowledgements
The authors thank all members of our laboratories
for helpful discussion and technical assistance.
Funding
The present study was supported in by the Ministry
of Education, Culture, Sports, Science and Technology of Japan
(http://www.mext.go.jp/english; grant
nos. 23390199, 25112708, 25134711, 30253420 and 26670604); P-DIRECT
(Hideshi Ishii); the Ministry of Health, Labor and Welfare of Japan
(http://www.mhlw.go.jp/english; grant no.
H23-003); the National Institute of Biomedical Innovation
(http://www.nibio.go.jp/english/index.html; grant no.
12-4); and the Osaka University Drug Discovery Fund (http://www.osaka-u.ac.jp/en/index.html)
(Masaki Mori and Hideshi Ishii). Partial support was received from
the Takeda Science and Medical Research Foundation (http://www.takeda-sci.or.jp/index.html)
(Hideshi Ishii), the Princess Takamatsu Cancer Research Fund
(http://www.ptcrf.or.jp/english) (Masaki
Mori, Hideshi Ishii), the Suzuken Memorial Foundation (http://www.suzukenzaidan.or.jp) (Masamitsu
Konno), the Yasuda Medical Foundation (http://www.yasuda-mf.or.jp) (Naohiro Nishida), the
Pancreas Research Foundation (http://www.jprf.or.jp/shoreisho.html) (Koichi
Kawamoto), the Nakatani Foundation (http://www.nakatani-foundation.jp), and the Nakatomi
Foundation, Japan (https://www.nakatomi.or.jp/en/index.html) (Masamitsu
Konno). Institutional endowments (grants) were received from Taiho
Pharmaceutical Co., Ltd. (http://www.taiho.co.jp/english), Evidence Based
Medical Research Center (http://ebmrce.co.jp/index.html), Chugai Co., Ltd.
(http://www.chugai-pharm.co.jp/english/index.html),
Yakult Honsha Co., Ltd. (http://www.yakult.co.jp/english/index.html), and Merck
Co., Ltd. (http://www.merck.co.jp/en/index.html).
Author's contributions
The experiments were performed by KN, MK, KK, NN,
JK, HW and HA. The analysis of data was performed by KN, MK, HE,
KK, RM and TS. The manuscript was written by KN, MK, MM and HI. The
study was designed by TS, SM, HN, YD, MM and HI.
Ethics approval and consent to
participate
All animal studies were approved by the Animal
Experiments Committee of Osaka University (Osaka, Japan; approval
no. 24-122-022).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Howe HL, Wu X, Ries LA, Cokkinides V,
Ahmed F, Jemal A, Miller B, Williams M, Ward E, Wingo PA, et al:
Annual report to the nation on the status of cancer, 1975–2003,
featuring cancer among U.S. Hispanic/Latino populations. Cancer.
107:1711–1742. 2006.
|
|
2
|
Majumder K, Gupta A, Arora N, Singh PP and
Singh S: Premorbid obesity and mortality in patients with
pancreatic cancer: A systematic review and meta-analysis. Clin
Gastroenterol Hepatol. 14:355–368.e. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pandol S, Gukovskaya A, Edderkaoui M,
Dawson D, Eibl G and Lugea A: Epidemiology, risk factors, and the
promotion of pancreatic cancer: Role of the stellate cell. J
Gastroenterol Hepatol. 27(Suppl 2): S127–S134. 2012. View Article : Google Scholar
|
|
4
|
Rizwani W, Allen AE and Trevino JG:
Hepatocyte growth factor from a clinical perspective: A pancreatic
cancer challenge. Cancers (Basel). 7:1785–1805. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ide T, Kitajima Y, Miyoshi A, Ohtsuka T,
Mitsuno M, Ohtaka K and Miyazaki K: The hypoxic environment in
tumor-stromal cells accelerates pancreatic cancer progression via
the activation of paracrine hepatocyte growth factor/c-Met
signaling. Ann Surg Oncol. 14:2600–2607. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lonardo E, Hermann PC, Mueller MT, Huber
S, Balic A, Miranda-Lorenzo I, Zagorac S, Alcala S,
Rodriguez-Arabaolaza I, Ramirez JC, et al: Nodal/Activin signaling
drives self-renewal and tumorigenicity of pancreatic cancer stem
cells and provides a target for combined drug therapy. Cell Stem
Cell. 9:433–446. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Asukai K, Kawamoto K, Eguchi H, Konno M,
Nishida N, Koseki J, Noguchi K, Hasegawa S, Ogawa H, Yamada D, et
al: Prognostic impact of peritumoral IL-17-positive cells and IL-17
axis in patients with intrahepatic cholangiocarcinoma. Ann Surg
Oncol. 22(Suppl 3): S1524–S1531. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bai H, Li H, Zhang W, Matkowskyj KA, Liao
J, Srivastava SK and Yang GY: Inhibition of chronic pancreatitis
and pancreatic intraepithelial neoplasia (PanIN) by capsaicin in
LSL-KrasG12D/Pdx1-Cre mice. Carcinogenesis. 32:1689–1696. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shah AN, Zhang JM, Park SI, Parikh NU and
Gallick GE: Development and characterization of
gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol.
14:3629–3637. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Koh YX, Chok AY, Zheng HL, Tan CS and Goh
BK: A systematic review and meta-analysis of the clinicopathologic
characteristics of cystic versus solid pancreatic neuroendocrine
neoplasms. Surgery. 156:83–96.e2. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu WS: The signaling mechanism of ROS in
tumor progression. Cancer Metastasis Rev. 25:695–705. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li C, Wu JJ, Hynes M, Dosch J, Sarkar B,
Welling TH, Pasca di Magliano M and Simeone DM: c-Met is a marker
of pancreatic cancer stem cells and therapeutic target.
Gastroenterology. 141:2218–2227.e5. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Christensen JG, Schreck R, Burrows J,
Kuruganti P, Chan E, Le P, Chen J, Wang X, Ruslim L, Blake R, et
al: A selective small molecule inhibitor of c-Met kinase inhibits
c-Met-dependent phenotypes in vitro and exhibits cytoreductive
antitumor activity in vivo. Cancer Res. 63:7345–7355.
2003.PubMed/NCBI
|
|
16
|
Oshima Y, Suzuki A, Kawashimo K, Ishikawa
M, Ohkohchi N and Taniguchi H: Isolation of mouse pancreatic ductal
progenitor cells expressing CD133 and c-Met by flow cytometric cell
sorting. Gastroenterology. 132:720–732. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ohuchida K, Mizumoto K, Murakami M, Qian
LW, Sato N, Nagai E, Matsumoto K, Nakamura T and Tanaka M:
Radiation to stromal fibroblasts increases invasiveness of
pancreatic cancer cells through tumor-stromal interactions. Cancer
Res. 64:3215–3222. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Noguchi K, Eguchi H, Konno M, Kawamoto K,
Nishida N, Koseki J, Wada H, Marubashi S, Nagano H, Doki Y, et al:
Susceptibility of pancreatic cancer stem cells to reprogramming.
Cancer Sci. 106:1182–1187. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Awad AJ, Burns TC, Zhang Y and Abounader
R: Targeting MET for glioma therapy. Neurosurg Focus. 37:E102014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Herreros-Villanueva M, Zubia-Olascoaga A
and Bujanda L: c-Met in pancreatic cancer stem cells: Therapeutic
implications. World J Gastroenterol. 18:5321–5323. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Peng Z, Zhu Y, Wang Q, Gao J, Li Y, Li Y,
Ge S and Shen L: Prognostic significance of MET amplification and
expression in gastric cancer: A systematic review with
meta-analysis. PLoS One. 9:e845022014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Inman KS, Francis AA and Murray NR:
Complex role for the immune system in initiation and progression of
pancreatic cancer. World J Gastroenterol. 20:11160–11181. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pedica F, Beccari S, Pedron S, Montagna L,
Piccoli P, Doglioni C and Chilosi M: PDX-1 (pancreatic/duodenal
homeobox-1 protein 1). Pathologica. 106:315–321. 2014.PubMed/NCBI
|
|
24
|
Yu J, Ohuchida K, Mizumoto K, Ishikawa N,
Ogura Y, Yamada D, Egami T, Fujita H, Ohashi S, Nagai E and Tanaka
M: Overexpression of c-met in the early stage of pancreatic
carcinogenesis; altered expression is not sufficient for
progression from chronic pancreatitis to pancreatic cancer. World J
Gastroenterol. 12:3878–3882. 2006. View Article : Google Scholar : PubMed/NCBI
|