Introduction
Fatty acids are essential nutrients, either obtained
externally from daily meals or derived internally from de
novo synthesis and breakdown of cellular triacylglycerol (TAG)
and phospholipid. As important building blocks of the body, they
also participate in energy metabolism and cellular signaling
pathways to maintain physiological functions. However, deregulated
fatty acid metabolism favoring excess lipid biosynthesis and
deposition eventually predisposes the body to metabolic disorders
and carcinogenesis.
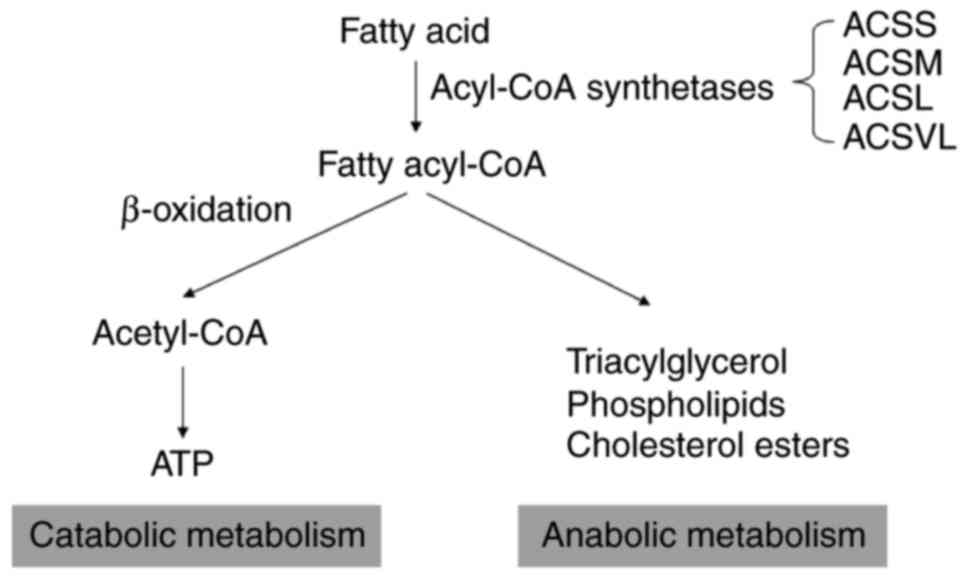
Fatty acids have different turnovers in the body
(Fig. 1). They can break down through
a series of mitochondrial β-oxidation processes into
acetyl-CoA, which then enters the tricarboxylic acid cycle to aid
ATP generation. Alternatively, fatty acids can be incorporated into
TAG, phospholipids or cholesterol esters. It is noteworthy that the
two distinct pathways require a common initial step known as fatty
acid activation by acyl-CoA synthetase (ACS) (1,2).
Long-chain ASCs (ACSLs) are essential enzymes for the activation of
the most abundant long-chain fatty acids (12–20 carbons) (2,3). By
contrast, fatty acids with <6 carbons (e.g., acetate, propionate
and butyrate) are activated by short-chain ACSs (ACSS) and fatty
acids with 6–10 carbons are activated by medium-chain ACSs, while
very long-chain fatty acids (>20 carbons) are activated by very
long-chain ACSs (1–3).
Five isoforms of ACSLs, namely ACSL1, ACSL3, ACSL4,
ACSL5 and ACSL6, are present in mammals, and they have overlapping
but specific roles in the activation of long-chain fatty acids.
Individual ACSL isoforms may have preferred substrates for
activation, dependent on the chain length and saturation status of
fatty acids (1,2). For example, ACSL3 and ACSL4 can activate
polyunsaturated fatty acids (PUFA), but ACSL3 prefers oleic acid,
whereas ACSL4 favors arachidonic acid (AA) and adrenic acid
(1,2).
Recent pathological studies have found the abnormal
expression of ACSLs in cancer tissues in comparison to neighboring
non-cancerous tissues (2,3), as discussed below. Furthermore, their
oncogenic roles and certain involved molecular mechanisms have been
unveiled. The present review thus focuses on ACSLs, evaluating the
recent evidence of the pathological functions of ACSLs in
carcinogenesis and cancer development, and discussing the
chemotherapeutic potential of targeting ACSLs.
Subcellular localization and physiological
functions of ACSLs
ACSL-mediated fatty acid activation contributes to
the two distinct pathways of anabolic lipid biosynthesis and
catabolic fatty acid oxidation. The manner in which cells adapt to
channel fatty acids into the distinct anabolic or catabolic
pathways is currently poorly understood. Certain early hypotheses
suggested that the subcellular localization of ACSLs and specified
fatty acid transportation system may contribute to channeling fatty
acids into different turnovers (1,2,4). Fig. 2
depicts a mitochondrion, where ACSL1, ACSL4 and ACSL5 localize and
support fatty acid synthesis and β-oxidation, and a peroxisome,
where ACSL1 and ACSL4 are involved in alkyl lipid biosynthesis and
β-oxidation. ACSL1, ACSL3 and ACSL4 have also been found to reside
in the endoplasmic reticulum (ER), facilitating glycerolipid
synthesis and ω-oxidation (a minor pathway for medium-chain fatty
acids in the normal physiological condition, but an alternative
pathway when β-oxidation is defective). In addition, ACSL3 in lipid
droplets may aid neutral lipid synthesis and lipid droplet
formation (5).
Tissue-specific loss-of-function studies are
particularly useful to unveil the physiological function of
individual ACSL isoforms. The majority of studies have focused on
ACSL1, the founding member of the ACSL family. Liver-specific
knockout of ACSL1 reduced TAG synthesis and fatty acid oxidation
(6). In contrast, adipose-specific
(7), skeletal muscle-specific
(8) or heart-specific (9) knockout of ACSL1 exhibited common
phenotypes of decreased fatty acid oxidation alone. In addition,
transgenic mice with all-tissue ACSL5-knockout exhibited decreased
adiposity and increased energy expenditure, indicating its major
role in fatty acid biosynthesis and deposition (10). Thus, the specific role of ACSL5 may be
different from that of ACSL1. However, for the remaining ACSL
isoforms, there are no such loss-of-function mouse models
available.
Taken together, these results suggest that ACSLs are
important for fatty acid metabolism. Whether individual ACSL
isoforms channel fatty acids toward anabolic metabolism or
catabolic metabolism may be dependent on their subcellular
localization and interaction with specific fatty acid
transportation systems. It is also possible that certain
compositions of fatty acids could affect the expression and
localization of ACSL isoforms in physiological and pathological
conditions.
Deregulated expression of ACSLs in clinical
cancer
Accumulating findings have demonstrated that almost
all ACSL members are deregulated in clinical cancer and that a
number are associated with poor patient survival. The expressional
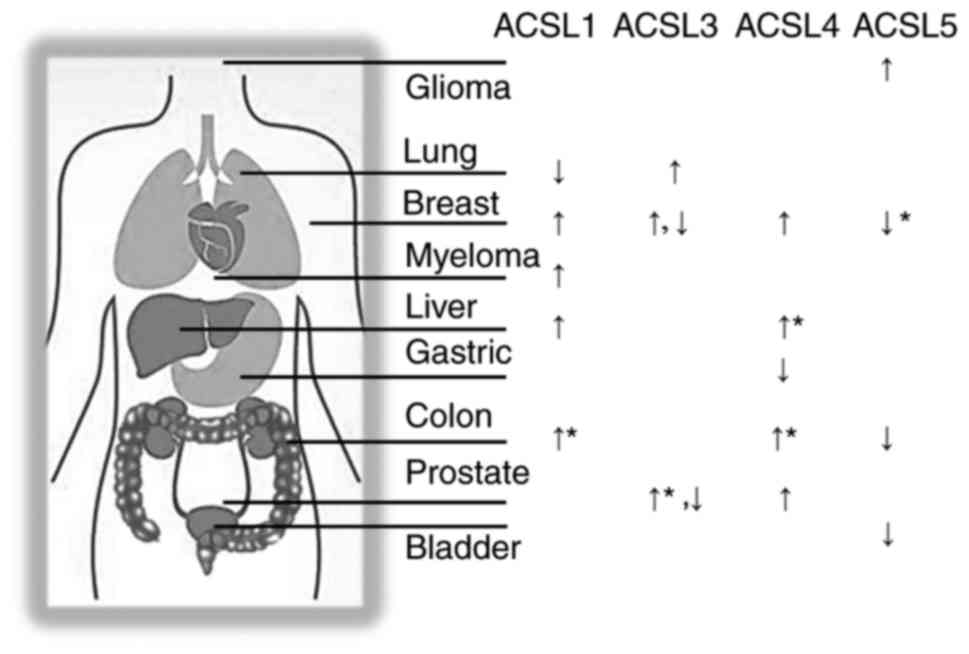
changes of ASCLs in different types of cancer are summarized in
Fig. 3, with the asterisk marks
indicating statistically significant associations with patient
prognosis. Upregulation of ACSL1 was found in multiple types of
cancer, including colon (11,12), breast (13) and liver (14,15)
cancer, and myeloma (16), while
downregulation was found in lung squamous cell carcinoma (17). More importantly, ACSL1 overexpression
was associated with a poor clinical outcome in colon cancer
patients (18). Like those of ACSL1,
the majority of studies of ACLS4 favor an oncogenic role. ACSL4 has
been shown to be overexpressed in multiple cancer types, including
colon (11,19), breast (20), liver (21–23) and
prostate (24) cancer, while another
study has shown its downregulation in gastric cancer (25). In terms of patient survival outcome,
ACSL4 overexpression predicted poorer patient survival in stage II
colon cancer [together with upregulated stearoyl-CoA desaturase
(SCD)] (12) and in liver cancer
(together with downregulated growth arrest and DNA damage inducible
β) (21).
By contrast, ACSL5 appears to exhibit the opposite
function in cancer. ACSL5 was downregulated in colon (26) and breast (27) cancer, where ACSL1 and ACSL4 were
upregulated. ACSL5 was also decreased in bladder cancer (28). More importantly, lower expression of
ACSL5 in breast cancer was associated with a worse prognosis
(27). An exception was reported in
glioma, where ACSL5 was upregulated (29). One recent study also demonstrated that
fibroblast growth factor receptor 2 (FGFR2)-ACSL5 chimera RNA
rendered clinical gastric cancer cells resistant to treatment with
FGFR inhibitors (30).
The role of ACSL3 in cancer is comparatively
complex. ACSL3 was found to be overexpressed in lung cancer
(31), prostate cancer (32) and estrogen receptor-negative breast
cancer (33). Furthermore, its
upregulation predicted an unfavorable prognosis in prostate cancer
(32). However, opposing results were
also determined in studies conducted in prostate cancer and breast
cancer. The expression of ACSL3 was decreased in high-grade and
metastatic prostate cancer (34).
Homozygous deletion of ACSL3 was found in breast cancer following
chemotherapy, and was associated with increased risks of recurrence
and distant metastasis (35). These
contradicting results may suggest that the roles of ACSL3 vary
among the different stages of cancer. Lastly, few studies have
reported on ACSL6 in cancer. Only one case report found a
t(5;12)(q23-31;p13)/ETV6-ACSL6 gene fusion in chronic leukemia,
which rendered cancer cells resistant to treatment using a tyrosine
kinase inhibitor (36).
A recent bioinformatics study (37) utilizing online databases Oncomine
(https://www.oncomine.org/resource/login.html) and
PrognoScan (http://www.abren.net/PrognoScan/) stated certain novel
associations between ACSL expression and cancer survival outcomes.
On the one hand, which was in line with previous findings (12,18,27), the
study showed that the overexpression of either ACSL1 or ACSL4 was
associated with a poorer prognosis in patients with colon cancer,
and that ACSL5 downregulation was associated with poor survival in
breast cancer patients. On the other hand, the study suggested
certain novel links. High ACSL1 expression was associated with
worse survival in lung cancer patients and high ACSL3 was
associated with worse survival in patients with melanoma. However,
there are also opposing results (37)
linking higher ACSL expression with improved patient survival in
multiple types of cancer, including breast, ovarian, brain and lung
cancer, and acute myeloid leukemia. The dry-lab bioinformatic
approach enabled fast analyses and novel associative findings, but
the obtained results may require further validations using datasets
from the other databases or sources.
Taken together, the results from the majority of
studies supported the fact that ACSL1 and ACSL4 serve oncogenic
roles in the majority of cancer types, whereas ACSL5 may suppress
tumor development. However, the function of ACSL3 in cancer may be
dependent on the stages of prostate and breast cancer. The
aforementioned ACSL-knockout transgenic mice models, alone or
cross-bred with other transgenic mice with known oncogenes [e.g.,
KRAS proto-oncogene, GTPase and HBx (hepatitis B virus
X-interacting protein), etc.], will be useful to shed light on the
cause-effect function of ACSLs in carcinogenesis and cancer
development.
Molecular mechanisms to deregulate ACSL
expression in cancer
ACSLs serve essential roles in fatty acid activation
and metabolism, and their expression is also readily controllable
by intracellular fatty acids in physiological and pathological
conditions. Physiologically, diet-derived fatty acids can activate
several transcription factors, including peroxisome
proliferator-activated receptors α and δ (PPARα and PPARδ), liver X
receptors α and β (LXRα and LXRβ), cAMP response element-binding
and specificity protein 1 (Sp1) (13,15,38,39).
These transcription factors can then activate different ACSLs
through direct binding to conserved responsive elements in the ACSL
promoter regions. Pathologically, cancer cells could hijack these
transcriptional mechanisms to upregulate ACSLs, as evidenced by the
following three examples. Firstly, cancer cells can utilize an
epigenetic approach to activate ACSL indirectly. PPARα increases
ACSL1 transcription, but this action could be repressed by
microRNA-9 (miR-9) in the normal liver. However, liver cancer
overexpressed the long non-coding RNA (lncRNA) known as highly
upregulated in liver cancer. This lncRNA could dampen miR-9 and
relieve its suppression of PPARα expression and consequently
turn on ACSL1 transcription (15).
Secondly, oncogenes can activate ACSL transcription indirectly
through modulation of the aforementioned transcription factor. In
breast cancer cells, HBx was reported to act as a co-activator to
sensitize Sp1-mediated transcription of ACSL1 (13). Thirdly, hepatitis B virus mutant large
surface protein also increased ACSL3 expression through induction
of ER stress, and this action could promote lipid biosynthesis and
deposition (40). Glycogen synthase
kinase 3β can also activate ACSL3 in an ER stress-dependent manner,
but the exact mechanism remains unclear.
Apart from direct regulation of ACSL transcription,
cancer cells can manipulate the mRNA stability of ACSLs by
targeting their 3′-untranslated regions (3′UTR). Previous studies
reported that miR-205 was downregulated in liver cancer (41) and lung squamous cell carcinoma
(17) to augment ACSL1 mRNA
stability, since miR-205 can directly bind to the 3′UTR of ACSL1
and facilitate its degradation. A recent study further showed that
DNA polymorphism (rs8086, T/T genotype) in the 3′UTR of the ACSL1
gene enabled higher ACSL1 expression compared with the
corresponding C/T or C/C genotype. It was suggested that the
genotype difference in 3′UTR may affect the binding affinity of
microRNA and the resultant ACSL1 mRNA stability. More importantly,
those patients with the T/T genotype and colon cancer had a poor
disease-free survival outcome (18).
Note that, in liver cancer, HBx repressed miR-205 expression and
consequently increased ACSL4 mRNA stability (41). It is thus rational to propose that HBx
can activate ACSL1 using the same mechanism.
Alternatively, protein ubiquitination and the
involved proteasome degradation pathway is another mechanism to
regulate ACSL expression. Fatty acid AA treatment could enhance
ACSL4 protein ubiquitination and shorten its protein half-life in
HepG2 cells via a negative feedback loop (42), as AA is the preferred substrate of
ACSL4. By contrast, in breast cancer, the hormone 17β-estradiol
extended the half-life of the ACSL4 protein, and subsequently
increased cellular uptake of AA and eicosapentaenoic acid (43). This 17β-estradiol-mediated ACSL4
upregulation was found to be essential for the invasiveness of
estrogen receptor-expressing breast cancer cells.
Effects of ACSLs on carcinogenesis and
cancer development
ACSL-mediated lipid anabolism may promote cancer
initiation. Silencing of ACSL3 markedly inhibited HCV secretion,
suggesting that phospholipid generation by the ACSL pathway is
involved in the replication and secretion of the hepatitis B virus
and the hepatitis C virus (44). The
latter two viruses are well-known etiologies of liver cancer. Note
that cancer is characterized by 10 hallmarks, including
self-sufficiency in growth signals, deregulated metabolism, tissue
invasion and metastasis, and evasion of programmed cell death
(45). Next, the effects of ACSLs on
the relevant cancer hallmarks and the possible molecular mechanisms
involved are summarized.
ACSLs and ungoverned cell
proliferation
Fatty acids promote unchecked cancer cell
proliferation directly, by providing essential biosynthetic and
functional intermediates. Cholesterol supplementation alone
enhanced cell proliferation in liver cancer cells (41). Several studies highlighted that ACSLs
stimulated proliferation in cancer cells. Knockdown of ACSL1, ACSL3
or ACSL4 independently decreased cell proliferation and
anchorage-independent growth in multiple cancer cells and xenograft
tumor growth in nude mice (11,21,31,43).
By contrast, forced overexpression of these ACSLs increased cell
proliferation and tumor growth (15,20,24,46,47).
Furthermore, treatment with a PUFA analogue triacsin C (a potent
inhibitor of ACSL1, ACSL3 and ACSL4, but not ACSL5) or other
inhibitors repressed cancer cell growth in vitro and in
vivo (48). Lastly, it was
reported that blockage of octamer-binding protein 1 binding to
ACSL3 enhancer by the use of pyrrole-imidazole polyamides reduced
ACSL3 expression and castration-resistant tumor growth (32). In general, these findings were in line
with the observations that ACSLs are upregulated in cancer.
However, in a few exceptions, knockdown of ACSL1 or ACSL4 with
specific small interfering RNA promoted lung (37) or gastric (25) cancer cell growth, respectively, and
in vivo xenograft growth of gastric tumors (25).
ACSLs and deregulated cancer
metabolism
It is yet unclear whether the deregulated expression
of ACSLs result from the increasing demands of the cancerous
tissues to metabolize and store oversupplied lipids. Particularly
in those cancer types that originate from tissues with a higher
basal level of lipid metabolism and deposition (e.g. liver, colon,
breast and prostate cancer), ACSL1 (11–15) and
ACSL4 (19–24) were generally upregulated. These two
enzymes participate in lipid anabolism and catabolism. By contrast,
ACSL5 [another ACSL member which drives lipid biosynthesis rather
than fatty acid oxidation (10)] was
downregulated in breast and prostate cancer (26,27). It is
thus tentative to propose that ACSL1- and ACSL4-mediated lipid
catabolism are useful for cancer promotion.
Apart from lipid metabolism, ACSL isoforms can also
affect glucose metabolism. ACSL1 overexpression in cancer was found
to increase intracellular acylcarnitine and lower basal respiration
rates, while ACSL4 upregulation decreased PUFA concentration and
promoted glycolysis (11). Combined
expression of ACSL1 and ACSL4 together with SCD enabled cells to
upregulate phospholipids and urea cycle metabolites. Importantly,
the expression level of ACSLs could be altered when cancer cells
shift from anabolic lipid metabolism to catabolism. ACSL3 was
increased in early carcinogenesis to promote lipid anabolism and
deposition, but deceased in advanced breast and prostate cancer to
increase lipid utilization (34,35). This
shift can promote cancer cell survival and invasiveness, which will
be discussed in detail in the following sections.
ACSLs and tissue invasion and
metastasis
Cancer cell invasion and migration are
characteristic for cancer progression, recurrence and metastasis.
Accumulating data have shown that ACSL1 and ACSL4 could promote
cancer cell invasion and migration in breast and prostate cancer
cells (20,24,37,43). The
common epithelial-mesenchymal transition (EMT) contributes to their
actions in cell migration and invasion. A series of studies
conducted in colon cancer further indicated that ACSL1 and ACSL4
may act synergistically on cancer progression (11). ACSL1 promoted cell invasion, but ACSL4
stimulated cell proliferation and migration in colon cancer cells.
The combined overactivation of ACSLs together with SCD contributed
to EMT, cancer invasion and poor patient survival in patients with
stage II colon cancer (11). Another
study also showed that ACSL4-mediated uptake of fatty acids AA and
eicosapentaenoic acid was essential for 17β-estradiol-induced cell
migration and invasion in breast cancer cells (43). By contrast, homozygous deletion of
ACSL3 promoted distant metastasis in breast cancer patients
following adjunct chemotherapy treatment (35). In another investigation studying how
CUB-domain containing protein 1 mechanically activated high
metastatic potency in triple-negative breast cancer cells (49), ACSL3 was again found to decrease
invasiveness through increased abundance of lipid droplets and
decreased fatty acid oxidation.
Fatty acids are natural ligands of several nuclear
transcription factors, e.g., hepatocyte nuclear factor-4α (HNF-4α),
PPARα and retinoic acid X receptor-α. Long-chain acyl-CoA
thioesters, which are products of ACSL-mediated fatty acid
activation, can directly bind to HNF-4α and modulate its
transcriptional activity (50). ACSLs
can facilitate HNF-4α modulation by converting fatty acids to
acyl-CoA, while acyl-CoA thioesterase acts in the opposite manner.
These modulations are dependent on the chain length and saturation
degree of the fatty acyl-CoA thioesters. For example, PUFA
(α-linolenic acid and eicosatrienoic acid) in the physiological
plasma concentration could suppress HNF-4α transcriptional
activity. As HNF-4α is inversely associated with EMT and β-catenin
signaling activity in liver and colon carcinogenesis (51), the ACSL-mediated modulation of HNF-4α
activity could thus promote cell invasion and migration.
Furthermore, ACSL4 could also upregulate cyclooxygenase-2 (COX-2)
expression and synergize with the latter to activate AA metabolism
and cancer cell invasion (47,52).
ACSLs and programmed cell death
Apoptosis and other forms of programmed cell death
are important for cellular non-immune surveillance to eradicate
damaged or mutated cells during carcinogenesis. However, these
defensive actions can be utilized smartly by the evolved cancer
cells to counteract chemotherapy and other death signals. In
general, genetic inhibition of ACSLs could induce lipotoxicity and
cell death in cancer cells (21,23,31,53,54).
Triacsin C treatment reduced overall ACSL activities and induced
cell death in cancer cells, but forced overexpression of the
insensitive ACSL5, markedly compensated ACSL activity and rescued
cell death induced by triacsin C (53,55).
Importantly, the pro-survival function was again replicated in
ACSS2 (which utilizes short-chain acetate for fatty acid activation
and acetyl-CoA production). ACSS2 is upregulated in numerous types
of cancer (e.g., breast and liver cancer, and glioblastoma) and is
capable of utilizing acetate as an alternative energy source to
support cell survival under metabolic stress (56). Taken together, these results
substantiated the role of ACSLs in cancer cell survival.
Fatty acids, particularly AA (a preferred substrate
of ACSL4), are apoptosis inducers. This was identified by a
landmark study investigating the mechanisms involved in COX-2
inhibition-induced cell death in colon cancer (57). Accumulation of unesterified AA, by
either exogenous supplementation in the culture medium or treatment
with triacsin C, resulted in cell death. However, in cancer cells,
the pro-apoptotic activities of unesterified AA could be
neutralized by ACSL4 (which activates AA for esterification into
TAG) and COX-2 (which facilitates AA to convert to prostaglandin).
Inducible expression of ACSL4 or COX-2 dampened AA accumulation and
consequently rescued cell death (57). Note that these two enzymes have been
found to be upregulated in colon cancer and other types of cancer,
and this study therefore disclosed the oncogenic mechanism of ACSL4
in cancer cell survival.
Unexpectedly, two relevant studies recently reported
that ACSL4 was essential for the induction of ferroptosis, another
form of programmed cell death. Ferroptosis is characterized by
ungoverned lipid peroxidation and it can be prevented by iron
chelators and antioxidants (58,59). The
first study (60) screened lipid
species by the use of redox lipidomics approaches in order to
identify potential ferroptosis signals. It was found that the
oxidized form of acyl-AA and acyl-adrenic acid (AdA) were necessary
and essential for ferroptosis. In the process, ACSL4 facilitated AA
and AdA esterification for subsequent lopoxygenase (LOX)-mediated
oxidation. Consistently, genetic or pharmacological inhibition of
ACSL4 could prevent ferroptosis. The other parallel study (61) utilized two different genetics
methodologies to search for essential genes in ferroptosis
execution. The clustered regularly interspaced short palindromic
repeats-based genetic screening approach and the other
transcriptome approach comparing ferroptosis-resistant and
-sensitive cells commonly uncovered ACSL4. ACSL4 was then confirmed
to be necessary and essential in ferroptosis. These two studies
were in line with an earlier report (62) revealing that ACSL4 may mark the
sensitivity of ferroptosis in breast cancer cell lines.
Taken together, ACSLs may generally render
resistance to fatty acid-induced lipotoxicity and cell death in
cancer cells. However, ACSL4 also enables cells to undergo
ferroptosis through oxidized AA and AdA. Why ACSL4 directs AA
metabolism toward these two distinct pathways (cell survival and
ferroptosis) is unresolved. One question to be raised is whether
there is a threshold of AA concentration to differentiate survival
and ferroptosis. In other words, we propose that ACSL4 is necessary
for fatty acid activation; it can cope with a low level of AA
metabolism and esterify AA to the COX-2 pathway to promote cell
survival. However, excess AA induces Fenton reaction-involved
oxidative stress, and the latter may switch off the COX-2
conversion pathway, but turn on the LOX oxidation pathway.
Tentative post-translational modifications (probably oxidation of
sulfides) of the key amino acids in these two enzyme proteins may
affect their activities. Another translational question to be
answered is whether ferroptosis inducers can specifically target
clinical cancer cells overexpressing ACSL4, but safely spare the
non-cancerous tissues with physiological ACSL4 expression in
vivo. Considering that ACSL4 is overexpressed in numerous types
of cancer, specific activation of ferroptosis may be a desirable
anticancer strategy.
Conclusions
In light of the epidemic of overweight and obese
individuals, excess fatty acid metabolism has increasingly been
found to be associated with metabolic disorders and carcinogenesis.
Based on the evidence examined thus far, ACSL1 and ACSL4 are
overexpressed in the majority of cancer types and exhibit oncogenic
activities. These ACSLs can promote unchecked cancer cell
proliferation, switch on deregulated cancer metabolism, facilitate
tumor invasion and metastasis, and evade programmed cell death.
ACSL3 is relatively complex, with varying expression and function
in the different stages of prostate and breast cancer. By contrast,
ACSL5 is generally decreased in those cancer types where ACSL1 and
ACSL4 are upregulated. It is thus imperative to clarify the
molecular mechanisms involved in individual ACSL isoforms and
design targeted therapies in a precision mode. Manipulation of ACSL
activities by genetic or pharmacological approaches (e.g., triacsin
C to inhibit ACSL1 and ACSL4, but spare ACSL5) is theoretically
applicable, but the non-specific toxicity cannot be overlooked due
to the importance of fatty acid activation in physiology. Notably,
ACSL4 marks the sensitivity of ferroptosis, making it a desirable
target in cancer. Future studies are necessary to deepen our
understanding of ACSL-involved fatty acid metabolism and
carcinogenesis.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81672370), the
Natural Science Foundation of Guangxi Province (grant no.
2015GXNSFCB139004), the Guangxi Hundred-Talent Program (awarded in
2016) and the Guangxi Medical University Training Program for
Distinguished Young Scholars (awarded in 2017).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YT and JZ searched the literature and organized the
data. YT drafted the manuscript. SCH provided important
suggestions, and critically revised the manuscript. YMJ and GDL
designed the research, summarized the data and finalized the
paper.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ellis JM, Frahm JL, Li LO and Coleman RA:
Acyl-coenzyme A synthetases in metabolic control. Curr Opin
Lipidol. 21:212–217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grevengoed TJ, Klett EL and Coleman RA:
Acyl-CoA metabolism and partitioning. Annu Rev Nutr. 34:1–30. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Neess D, Bek S, Engelsby H, Gallego SF and
Færgeman NJ: Long-chain acyl-CoA esters in metabolism and
signaling: Role of acyl-CoA binding proteins. Prog Lipid Res.
59:1–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Digel M, Ehehalt R, Stremmel W and
Füllekrug J: Acyl-CoA synthetases: Fatty acid uptake and metabolic
channeling. Mol Cell Biochem. 326:23–28. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fujimoto Y, Itabe H, Kinoshita T, Homma
KJ, Onoduka J, Mori M, Yamaguchi S, Makita M, Higashi Y, Yamashita
A and Takano T: Involvement of ACSL in local synthesis of neutral
lipids in cytoplasmic lipid droplets in human hepatocyte HuH7. J
Lipid Res. 48:1280–1292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li LO, Ellis JM, Paich HA, Wang S, Gong N,
Altshuller G, Thresher RJ, Koves TR, Watkins SM, Muoio DM, et al:
Liver-specific loss of long chain acyl-CoA synthetase-1 decreases
triacylglycerol synthesis and beta-oxidation and alters
phospholipid fatty acid composition. J Biol Chem. 284:27816–27826.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ellis JM, Li LO, Wu PC, Koves TR, Ilkayeva
O, Stevens RD, Watkins SM, Muoio DM and Coleman RA: Adipose
acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and
is required for cold thermogenesis. Cell Metab. 12:53–64. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li LO, Grevengoed TJ, Paul DS, Ilkayeva O,
Koves TR, Pascual F, Newgard CB, Muoio DM and Coleman RA:
Compartmentalized acyl-CoA metabolism in skeletal muscle regulates
systemic glucose homeostasis. Diabetes. 64:23–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ellis JM, Mentock SM, Depetrillo MA, Koves
TR, Sen S, Watkins SM, Muoio DM, Cline GW, Taegtmeyer H, Shulman
GI, et al: Mouse cardiac acyl coenzyme a synthetase 1 deficiency
impairs Fatty Acid oxidation and induces cardiac hypertrophy. Mol
Cell Biol. 31:1252–1262. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bowman TA, O'Keeffe KR, D'Aquila T, Yan
QW, Griffin JD, Killion EA, Salter DM, Mashek DG, Buhman KK and
Greenberg AS: Acyl CoA synthetase 5 (ACSL5) ablation in mice
increases energy expenditure and insulin sensitivity and delays fat
absorption. Mol Metab. 5:210–220. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sanchez-Martinez R, Cruz-Gil S,
García-Álvarez MS, Reglero G and Ramirez de Molina A: Complementary
ACSL isoforms contribute to a non-Warburg advantageous energetic
status characterizing invasive colon cancer cells. Sci Rep.
7:111432017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sánchez-Martínez R, Cruz-Gil S, Gómez de
Cedrón M, Álvarez-Fernández M, Vargas T, Molina S, García B,
Herranz J, Moreno-Rubio J, Reglero G, et al: A link between lipid
metabolism and epithelial-mesenchymal transition provides a target
for colon cancer therapy. Oncotarget. 6:38719–38736. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Cai X, Zhang S, Cui M, Liu FSun B,
Zhan W, Zhang X and Ye L: HBXIP up-regulates ACSL1 through
activating transcriptional factor Sp1 in breast cancer. Biochem
Biophys Res Commun. 484:565–571. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cui M, Wang Y, Sun B, Xiao Z, Ye L and
Zhang X: MiR-205 modulates abnormal lipid metabolism of hepatoma
cells via targeting acyl-CoA synthetase long-chain family member 1
(ACSL1) mRNA. Biochem Biophys Res Commun. 444:270–275. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cui M, Xiao Z, Wang Y, Zheng M, Song T,
Cai X, Sun B, Ye L and Zhang X: Long noncoding RNA HULC modulates
abnormal lipid metabolism in hepatoma cells through an
miR-9-mediated RXRA signaling pathway. Cancer research. 75:846–857.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ivyna Bong PN, Ng CC, Lam KY, Megat
Baharuddin PJ, Chang KM and Zakaria Z: Identification of novel
pathogenic copy number aberrations in multiple myeloma: The
Malaysian. Mol Cytogenet. 7:242014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang W, Jin Y, Yuan Y, Bai C, Wu Y2, Zhu
H and Lu S: Validation and target gene screening of hsa-miR-205 in
lung squamous cell carcinoma. Chin Med J (Engl). 127:272–278.
2014.PubMed/NCBI
|
|
18
|
Vargas T, Moreno-Rubio J, Herranz J, Cejas
P, Molina S, Mendiola M, Burgos E, Custodio AB, de Miguel M, et al:
3′ UTR polymorphism in ACSL1 gene correlates with expression levels
and poor clinical outcome in colon cancer patients. PLoS One.
11:e01684232016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cao Y, Dave KB, Doan TP and Prescott SM:
Fatty acid CoA ligase 4 is up-regulated in colon adenocarcinoma.
Cancer Res. 61:8429–8434. 2001.PubMed/NCBI
|
|
20
|
Wu X, Li Y, Wang J, Wen X, Marcus MT,
Daniels G, Zhang DY, Ye F, Wang LH, Du X, et al: Long chain fatty
acyl-CoA synthetase 4 is a biomarker for and mediator of hormone
resistance in human breast cancer. PloS one. 8:e770602013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xia H, Lee KW, Chen J, Kong SN, Sekar K,
Deivasigamani A, Seshachalam VP, Goh BKP, Ooi LL, Hui KM, et al:
Simultaneous silencing of ACSL4 and induction of GADD45B in
hepatocellular carcinoma cells amplifies the synergistic
therapeutic effect of aspirin and sorafenib. Cell Death Discov.
3:170582017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sung YK, Hwang SY, Park MK, Bae HI, Kim
WH, Kim JC and Kim M: Fatty acid-CoA ligase 4 is overexpressed in
human hepatocellular carcinoma. Cancer Sci. 94:421–424. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu C, Chen L, Jiang Y, Li Y and Wang S:
The effect of fatty acid-CoA ligase 4 on the growth of hepatic
cancer cells. Cancer Biol Ther. 7:131–134. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu X, Deng F, Li Y, Daniels G, Du X, Ren
Q, Wang J, Wang LH, Yang Y, Zhang V, et al: ACSL4 promotes prostate
cancer growth, invasion and hormonal resistance. Oncotarget.
6:44849–44863. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ye X, Zhang Y, Wang X, Li Y and Gao Y:
Tumor-suppressive functions of long-chain acyl-CoA synthetase 4 in
gastric cancer. IUBMB Life. 68:320–327. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gassler N, Schneider A, Kopitz J,
Schnölzer M, Obermüller N, Kartenbeck J, Otto HF and Autschbach F:
Impaired expression of acyl-CoA-synthetase 5 in epithelial tumors
of the small intestine. Hum Pathol. 34:1048–1052. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yen MC, Kan JY, Hsieh CJ, Kuo PL, Hou MF
and Hsu YL: Association of long-chain acyl-coenzyme A synthetase 5
expression in human breast cancer by estrogen receptor status and
its clinical significance. Oncol Rep. 37:3253–3260. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gaisa NT, Reinartz A, Schneider U, Klaus
C, Heidenreich A, Jakse G, Kaemmerer E, Klinkhammer BM, Knuechel R
and Gassler N: Levels of acyl-coenzyme A synthetase 5 in urothelial
cells and corresponding neoplasias reflect cellular
differentiation. Histol Histopathol. 28:353–364. 2013.PubMed/NCBI
|
|
29
|
Yamashita Y, Kumabe T, Cho YY, Watanabe M,
Kawagishi J, Yoshimoto T, Fujino T, Kang MJ and Yamamoto TT: Fatty
acid induced glioma cell growth is mediated by the acyl-CoA
synthetase 5 gene located on chromosome 10q25.1-q25.2, a region
frequently deleted in malignant gliomas. Oncogene. 19:5919–5925.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim SY, Ahn T, Bang H, Ham JS, Kim J, Kim
ST, Jang J, Shim M, Kang SY, Park SH, et al: Acquired resistance to
LY2874455 in FGFR2-amplified gastric cancer through an emergence of
novel FGFR2-ACSL5 fusion. Oncotarget. 8:15014–15022.
2017.PubMed/NCBI
|
|
31
|
Padanad MS, Konstantinidou G,
Venkateswaran N, Melegari M, Rindhe S, Mitsche M, Yang C, Batten K,
Huffman KE, Liu J, et al: Fatty acid oxidation mediated by acyl-CoA
synthetase long Chain 3 is required for mutant KRAS lung
tumorigenesis. Cell Rep. 16:1614–1628. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Obinata D, Takayama K, Fujiwara K, Suzuki
T, Tsutsumi S, Fukuda N, Nagase H, Fujimura T, Urano T, Homma Y, et
al: Targeting Oct1 genomic function inhibits androgen receptor
signaling and castration-resistant prostate cancer growth.
Oncogene. 35:6350–6358. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang J, Scholtens D, Holko M, Ivancic D,
Lee O, Hu H, Chatterton RT Jr, Sullivan ME, Hansen N, Bethke K, et
al: Lipid metabolism genes in contralateral unaffected breast and
estrogen receptor status of breast cancer. Cancer Prev Res (Phila).
6:321–330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marques RB, Dits NF, Erkens-Schulze S, van
Ijcken WF, van Weerden WM and Jenster G: Modulation of androgen
receptor signaling in hormonal therapy-resistant prostate cancer
cell lines. PLoS One. 6:e231442011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jeong HM, Kim RN, Kwon MJ, Oh E, Han J,
Lee SK, Choi JS, Park S, Nam SJ, Gong GY, et al: Targeted exome
sequencing of Korean triple-negative breast cancer reveals
homozygous deletions associated with poor prognosis of adjuvant
chemotherapy-treated patients. Oncotarget. 8:61538–61550. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Su RJ, Jonas BA, Welborn J, Gregg JP and
Chen M: Chronic eosinophilic leukemia, NOS with
t(5;12)(q31;p13)/ETV6-ACSL6 gene fusion: A novel variant of myeloid
proliferative neoplasm with eosinophilia. Hum Pathol (N Y). 5:6–9.
2016.PubMed/NCBI
|
|
37
|
Chen WC, Wang CY, Hung YH, Weng TY, Yen MC
and Lai MD: Systematic analysis of gene expression alterations and
clinical outcomes for long-chain acyl-coenzyme a synthetase family
in cancer. PLoS One. 11:e01556602016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Weedon-Fekjaer MS, Dalen KT, Solaas K,
Staff AC, Duttaroy AK and Nebb HI: Activation of LXR increases
acyl-CoA synthetase activity through direct regulation of ACSL3 in
human placental trophoblast cells. J Lipid Res. 51:1886–1896. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cao A, Li H, Zhou Y, Wu M and Liu J: Long
chain acyl-CoA synthetase-3 is a molecular target for peroxisome
proliferator-activated receptor delta in HepG2 hepatoma cells. J
Biol Chem. 285:16664–16674. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chang YS, Tsai CT, Huangfu CA, Huang WY,
Lei HY, Lin CF, Su IJ, Chang WT, Wu PH, Chen YT, et al: ACSL3 and
GSK-3β are essential for lipid upregulation induced by endoplasmic
reticulum stress in liver cells. J Cell Biochem. 112:881–893. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cui M, Xiao Z, Sun B, Wang Y, Zheng M, Ye
L and Zhang X: Involvement of cholesterol in hepatitis B virus X
protein-induced abnormal lipid metabolism of hepatoma cells via
up-regulating miR-205-targeted ACSL4. Biochem Biophys Res Commun.
445:651–655. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kan CF, Singh AB, Stafforini DM, Azhar S
and Liu J: Arachidonic acid downregulates acyl-CoA synthetase 4
expression by promoting its ubiquitination and proteasomal
degradation. J Lipid Res. 55:1657–1667. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Belkaid A, Ouellette RJ and Surette ME:
17β-estradiol-induced ACSL4 protein expression promotes an invasive
phenotype in estrogen receptor positive mammary carcinoma cells.
Carcinogenesis. 38:402–410. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yao H and Ye J: Long chain acyl-CoA
synthetase 3-mediated phosphatidylcholine synthesis is required for
assembly of very low density lipoproteins in human hepatoma Huh7
cells. J Biol Chem. 283:849–854. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Migita T, Takayama KI, Urano T, Obinata D,
Ikeda K, Soga T, Takahashi S and Inoue S: ACSL3 promotes
intratumoral steroidogenesis in prostate cancer cells. Cancer Sci.
108:2011–2021. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Orlando UD, Garona J, Ripoll GV, Maloberti
PM, Solano ÁR, Avagnina A, Gomez DE, Alonso DF and Podestá EJ: The
functional interaction between acyl-CoA synthetase 4,
5-lipooxygenase and cyclooxygenase-2 controls tumor growth: A novel
therapeutic target. PLoS One. 7:e407942012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Orlando UD, Castillo AF, Dattilo MA,
Solano AR, Maloberti PM and Podesta EJ: Acyl-CoA synthetase-4, a
new regulator of mTOR and a potential therapeutic target for
enhanced estrogen receptor function in receptor-positive and
-negative breast cancer. Oncotarget. 6:42632–42650. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wright HJ, Hou J, Xu B, Cortez M, Potma
EO, Tromberg BJ and Razorenova OV: CDCP1 drives triple-negative
breast cancer metastasis through reduction of lipid-droplet
abundance and stimulation of fatty acid oxidation. Proc Natl Acad
Sci USA. 114:E6556–E6565. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hertz R, Magenheim J, Berman I and
Bar-Tana J: Fatty acyl-CoA thioesters are ligands of hepatic
nuclear factor-4alpha. Nature. 392:512–516. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ning BF, Ding J, Yin C, Zhong W, Wu K,
Zeng X, Yang W, Chen YX, Zhang JP and Zhang X: Hepatocyte nuclear
factor 4 alpha suppresses the development of hepatocellular
carcinoma. Cancer Res. 70:7640–7651. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Maloberti PM, Duarte AB, Orlando UD,
Pasqualini ME, Solano AR, López-Otín C and Podestá EJ: Functional
interaction between acyl-CoA synthetase 4, lipooxygenases and
cyclooxygenase-2 in the aggressive phenotype of breast cancer
cells. PLoS One. 5:e155402010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mashima T, Sato S, Okabe S, Miyata S,
Matsuura M, Sugimoto Y, Tsuruo T and Seimiya H: Acyl-CoA synthetase
as a cancer survival factor: Its inhibition enhances the efficacy
of etoposide. Cancer Sci. 100:1556–1562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mashima T, Sato S, Sugimoto Y, Tsuruo T
and Seimiya H: Promotion of glioma cell survival by acyl-CoA
synthetase 5 under extracellular acidosis conditions. Oncogene.
28:9–19. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mashima T, Oh-hara T, Sato S, Mochizuki M,
Sugimoto Y, Yamazaki K, Hamada J, Tada M, Moriuchi T, Ishikawa Y,
Kato Y, et al: p53-defective tumors with a functional
apoptosome-mediated pathway: A new therapeutic target. J Natl
Cancer Inst. 97:765–777. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Schug ZT, Peck B, Jones DT, Zhang Q,
Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S1, Blyth K,
et al: Acetyl-CoA synthetase 2 promotes acetate utilization and
maintains cancer cell growth under metabolic stress. Cancer Cell.
27:57–71. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cao Y, Pearman AT, Zimmerman GA, McIntyre
TM and Prescott SM: Intracellular unesterified arachidonic acid
signals apoptosis. Proc Natl Acad Sci USA. 97:11280–11285. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK and
Kagan VE: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kagan VE, Mao GW, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yuan H, Li X, Zhang X, Kang R and Tang D:
Identification of ACSL4 as a biomarker and contributor of
ferroptosis. Biochem Biophys Res Commun. 478:1338–1343. 2016.
View Article : Google Scholar : PubMed/NCBI
|