Introduction
Hepatocellular carcinoma (HCC) is the fifth-most
common malignancy worldwide, and the fastest-increasing cause of
cancer-associated mortality in the USA (1,2). Despite
substantial improvements to the therapeutic strategies used to
treat HCC, the overall prognosis of patients with HCC remains poor,
with a 5-year survival rate of 12% (3). Therefore, the identification of novel
molecular targets for early diagnosis and effective treatment of
HCC is urgently required.
Evidence supports a hypothesis that chronic stress
can affect tumor growth and progression (4); activation of the adrenergic system
increases tumor growth and has been shown to mediate stress-induced
augmentation of tumor progression (4,5). The
β2 adrenergic receptor (ADRB2) is a member of the
superfamily of adrenergic G-protein-coupled receptors (GPCRs), and
is widely expressed in the majority of cell types (6); it is the primary target of the
catecholamine epinephrine during the stress response (7). Previous studies have demonstrated that
the activation of ADRB2 can stimulate several signaling pathways,
including the Ras-mediated Raf proto-oncogene
serine/threonine-protein kinase (Raf)/dual specificity
mitogen-activated protein kinase kinase (MEK)/extracellular
signal-regulated kinase (ERK), phosphoinositide 3-kinase (PI3K)/RAC
serine/threonine-protein kinase (Akt) and cAMP/protein kinase
A/mitogen-activated protein kinase pathways that promote cellular
proliferation and invasion, and suppress apoptosis in cancer cells,
which can enhance tumor growth and facilitate metastasis (8). β-adrenergic blockers have been used
clinically to reduce the rates of progression of several types of
solid tumor (9). The use of
β-adrenergic blockers resulted in a 57% reduction in the risk of
metastasis and a 71% reduction in the 10-year mortality rate in
patients with breast cancer (10,11). In
addition, ICI118551, an ADRB2 blocker, significantly synergized
with the anti-proliferative and pro-apoptotic effects of
gemcitabine to inhibit the proliferation of pancreatic cancer cells
(12). These results indicate that
ADRB2 blockade may serve a role in cancer treatment.
On the basis of previous results concerning the
association between tumor growth and ADRB2, the present study set
out to examine whether ADBR2 antagonism could suppress HCC growth.
The present study tested the inhibitory effect of ADRB2 blockade on
the growth of human HCC cells and examined the potential molecular
mechanism by which ADRB2 blockade causes this growth
inhibition.
Materials and methods
Cell culture and reagents
The human HCC SMMC-7721, Huh7 and Hep3B cell lines,
and the normal liver L02 cell line were purchased from American
Type Culture Collection (Manassas, VA, USA) and were cultured in
Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and maintained
at 37°C in a humidified incubator containing 5% CO2. The
β-adrenergic antagonists, metoprolol and ICI118,551 were purchased
from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The total RNA concentration was assessed
by measuring absorbance at 260 nm using a NanoDrop
spectrophotometer (ND-1,000; Thermo Fisher Scientific, Inc.). Next,
2 µg of total RNA was reverse-transcribed to cDNA using the
PrimeScript RT Reagent kit (Takara Bio, Inc., Tokyo, Japan).
Gene-specific amplification was performed using ABI 7,500 fast
real-time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) and SYBR Green PCR Master Mix (Applied Biosystems; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
Thermocycling conditions for PCR were as follows: First step at
95°C for 10 min followed by 40 cycles at 95°C for 15 sec and 60°C
for 1 min. For the analysis of the melting curve at the end of PCR,
the reaction mixture was heated to 95°C, followed by complete
annealing at 60°C and then followed by a gradual increase in
temperature up to 95°C. The following gene-specific primers were
used in the present study: ADRB2 forward,
5′-GCCTGTGCTGATCTGGTCAT-3′ and reverse,
5′-AATGGAAGTCCAAAACTCGCA-3′; β-actin forward,
5′-GTGGACATCCGCAAAGAC-3′ and reverse, 5′-AAAGGGTGTAACGCAACTA-3′.
The relative expression level of ADRB2 was normalized to the
expression of the housekeeping gene β-actin using the comparative
threshold cycle (2−ΔΔCq) method (13). Each sample was analyzed in triplicate
and the mean expression level was calculated.
Cell viability assay
Cells were harvested and seeded into 96-well plates
at a density of 2×103 cells per well and cultured in an
environment with 5% CO2 at 37°C. During the experiments,
cells were incubated with metoprolol (0, 5, 10, 20, 50, 100 and 200
µM) or ICI118,551 (0, 5, 10, 20, 50, 100 and 200 µM); Dimethyl
sulfoxide (0.05%) was used as vehicle control. Next, 10 µl Cell
Counting kit-8 solution (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan) was added into the culture medium in each well at
the indicated time (12, 24, 48, 72 and 96 h). After a 1-h
incubation at 37°C, optical density values were obtained using a
microplate reader at a 450-nm wavelength. The experiment at each
time point was repeated in triplicate and the experiment was
independently performed three times.
Cell apoptosis assay
Cell apoptosis was evaluated by flow cytometry using
an Annexin V-Fluorescein Isothiocyanate (FITC) Apoptosis Detection
kit (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China). Briefly,
1×105 cells were treated with metoprolol (50 µM) or
ICI118,551 (50 µM) for 48 h. Next, cells were harvested, washed,
resuspended in the binding buffer. A volume of 5 µl Annexin V-FITC
and 5 µl propidium iodide (PI) was added and mixed gently, and the
cells were stained in the dark for 10 min at room temperature. The
cells were analyzed immediately by flow cytometry (BD FACSCalibur;
BD Biosciences, San Diego, CA, USA) and analyzed using FlowJo
software (version 7.6; FlowJo, LLC, Ashland, OR, USA). The
experiment was repeated three times.
Hoechst 33342 staining
A total of 2×105 Cells were exposed to
metoprolol (50 µM), or ICI118,551 (50 µM) for 48 h. Cells were
fixed in 4% paraformaldehyde for 10 min at room temperature and
permeabilized with 0.1% Triton X-100 for 5 min at room temperature,
then stained with 10 µl Hoechst 33342 (Sigma-Aldrich; Merck KGaA)
for 10 min at room temperature. Next, cells were viewed with a
confocal microscope (Olympus Corporation, Tokyo, Japan).
Quantitative analysis was performed by counting the blue
fluorescent (apoptosis-positive) cells from three independent
fields at magnification, ×400. Values were expressed as the
percentage of apoptotic cells relative to the total number of cells
per field.
Cell cycle analysis
Cells were exposed to metoprolol (50 µM), or
ICI118,551 (50 µM) for 48 h. Cells were harvested and fixed in 70%
ethanol and stored at −20°C for 6 h, then washed twice with
ice-cold PBS and incubated with RNase A and PI (both Sigma-Aldrich;
Merck KGaA) for 30 min in the dark at room temperature. Cell cycle
assay was performed using a flow cytometer (BD FACSCalibur; BD
Biosciences). Cell cycle phase analysis was performed using the
Modifit cell cycle analysis software (version 2.0; BD Biosciences).
The experiment was independently performed for three times.
Western blot analysis
Cells were exposed to metoprolol (50 µM), or
ICI118,551 (50 µM) for 48 h. Following this, total proteins from
treated cells was lysed in Radioimmunoprecipitation Assay buffer
(Beyotime Institute of Biotechnology, Shanghai, China). The protein
concentration was quantified using a Bicinchoninic Acid Assay kit
(Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. A total of 30 µg total protein were
separated by 10% SDS-PAGE and the protein was transferred onto
polyvinylidene fluoride membranes. The membrane was then blocked
with TBST (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20)
containing 5% w/v non-fat dry milk for 2 h at room temperature and
then incubated with the primary antibodies to ADRB2 (1:1,000;
ab182136), caspase-9 (1:1,000; ab32539), B-cell lymphoma-2 (Bcl-2;
1:1,000; ab32124), Bcl-2 associated X (Bax; 1:1,000; ab32503),
cyclin B1, cyclin-dependent kinase 1 (CDK1; 1:10,000; ab133327) or
β-actin (1:3,000; ab8226; all from Abcam, Cambridge, MA, USA) at
4°C overnight. The membrane was washed three times and was then
incubated with the horseradish peroxidase-conjugated goat
anti-rabbit secondary antibody (1:5,000; ab7090) or goat anti-mouse
secondary antibody (1:5,000; ab97040; all from Abcam, Cambridge,
MA, USA) for 2 h at room temperature. The bands were visualized
using the electrogenerated chemiluminescence detection system (EMD
Millipore, Billerica, MA, USA).
Statistical analysis
Data are represented as the mean ± standard
deviation. Data were analyzed using GraphPad Prism 5 software for
Windows (GraphPad Software, Inc., La Jolla, CA, USA). Differences
between groups were assessed using one-way analysis of variance
with post hoc Dunnet's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
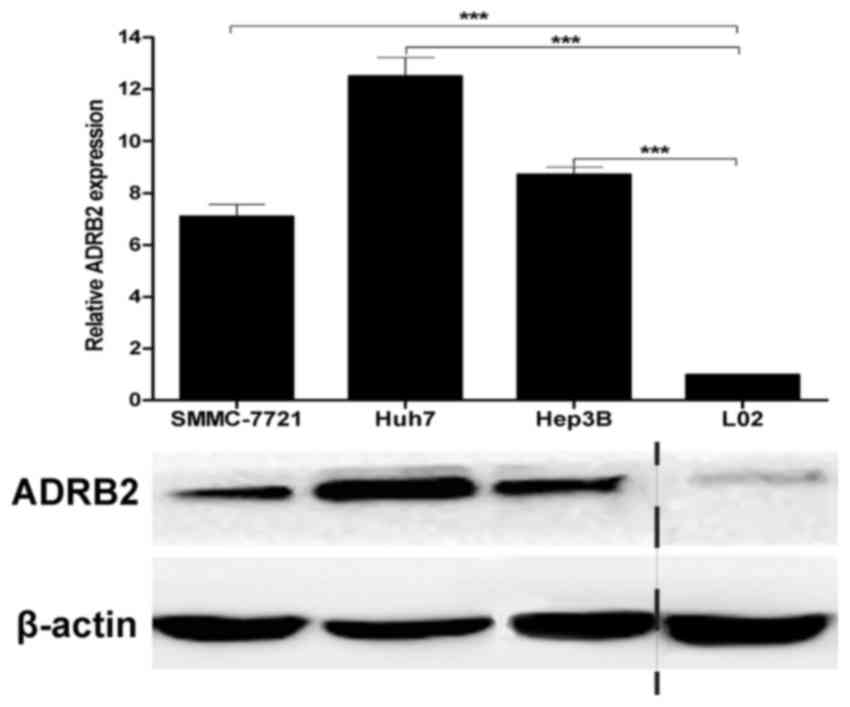
Expression of ADRB2 in HCC cell
lines
A previous study has shown that ADRB2 is in general
expressed at higher levels in HCC tissues than in benign nontumor
liver tissues (14). To assess basal
ADRB2 levels in HCC cell lines, ADRB2 expression was examined in
the HCC SMMC-7721, Huh7 and Hep3B cell lines and the normal liver
L02 cell line by RT-qPCR and western blot analysis. ADRB2 was
highly expressed in all three HCC cell lines assessed compared with
normal liver cell line L02 (Fig.
1).
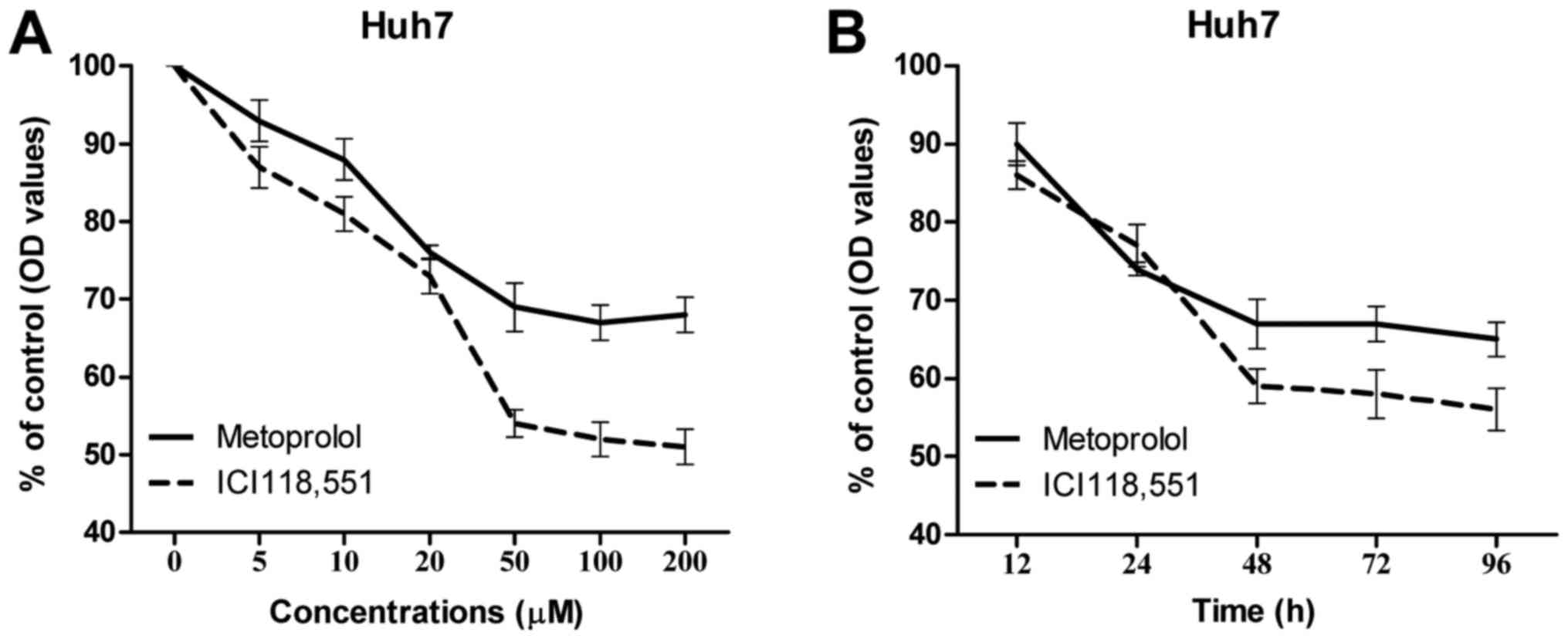
Inhibition of ADBR2 inhibits cell
growth in HCC cells
Next, the growth inhibitory effects of ADBR2
inhibition on the Huh7 HCC cell line were assessed. Various
concentrations of the β-adrenergic antagonists metoprolol and
ICI118,551 between 5 and 200 µM (5, 10, 20, 50, 100 and 200 µM)
were used to treat Huh7 cells for 48 h, cell growth was assessed by
CCK-8 assay. As depicted in Fig. 2A,
treatment with the β-adrenergic antagonists significantly decreased
cell growth compared with the control group with dose dependent
effects; the optimal concentration was 50 µM for the two
β-adrenergic antagonists used. A time-course cell growth assay was
also performed to examine the optimal timing, the results revealed
that metoprolol and ICI118,551 (each at 50 µM) reached the maximum
efficacy at 48 h, with 70 and 60% inhibition of viability; no
further decrease in cell viability at later time points (Fig. 2B).
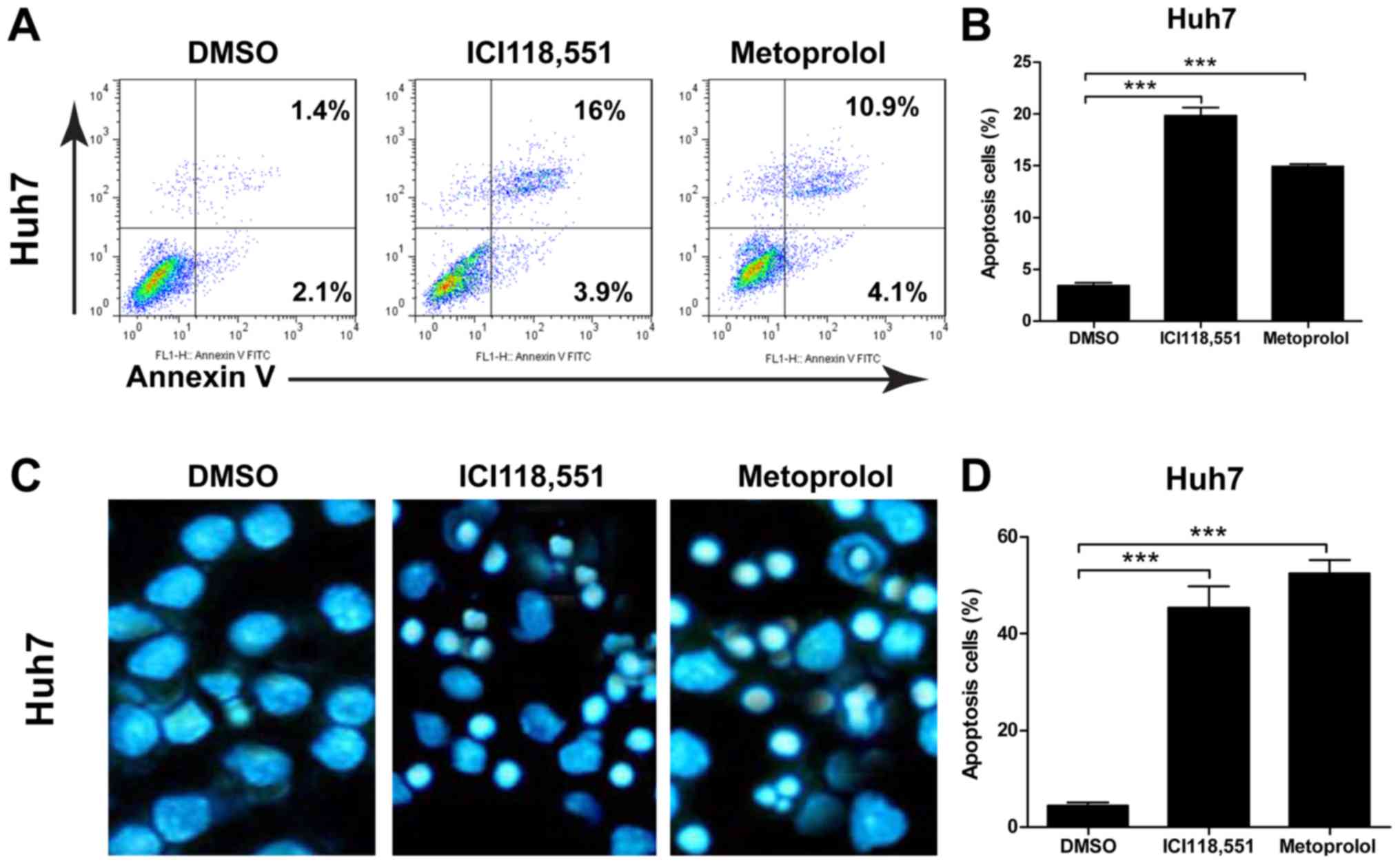
Inhibition of ADBR2 induces cell
apoptosis in HCC cells
To determine whether the effects of ADBR2 inhibition
were associated with induction of apoptosis or reduction of cell
proliferation in HCC cells, an Annexin V/PI apoptosis assay and
Hoechst staining were performed, respectively. To examine the
effect of the antagonists on HCC cell apoptosis, 50 µM metoprolol
or ICI118,551. As shown in Fig. 3A and
B, the percentage of apoptotic cells significantly increased
when cells were exposed to β-adrenergic antagonists for 48 h.
Additionally, Hoechst staining revealed that the cells exhibited
the characteristic appearance of apoptotic cells following
treatment with the two β-adrenergic antagonists for 48 h (Fig. 3C). Treatment with the β-adrenergic
antagonists resulted in a significant increase in the number of
apoptotic cells compared with the basal apoptotic level in the
untreated controls (Fig. 3C). The
percentage of apoptotic cells in the β-adrenergic
antagonists-treated HCC cells were significantly different from the
untreated control cells (Fig.
3D).
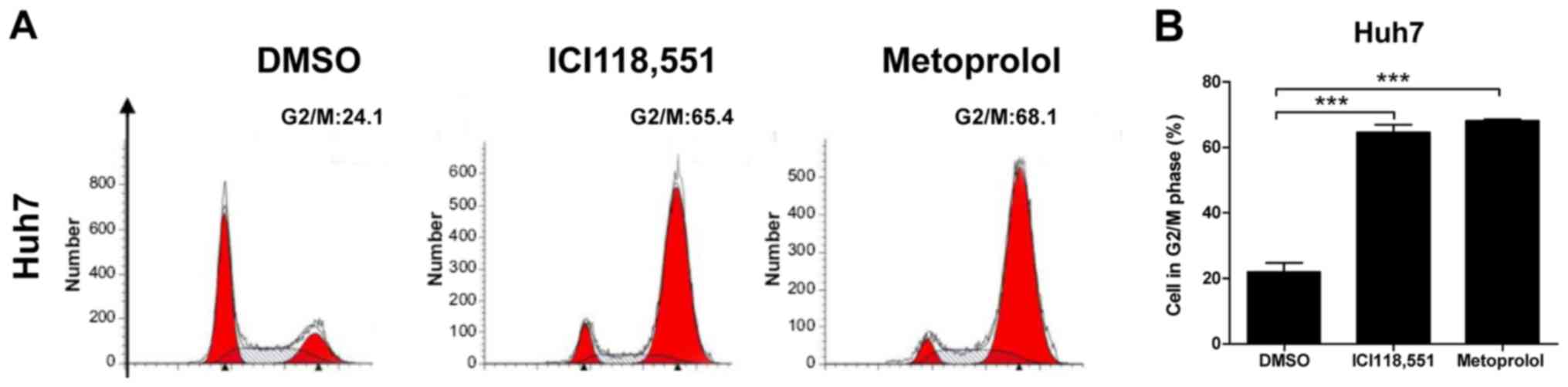
Inhibition of ADBR2 induces
G2/M phase cell cycle arrest in HCC cells
To examine the inhibitory effects of ADBR2
antagonsim on HCC cell growth, the cell cycle distribution was
examined by flow cytometry using the PI staining method. As
depicted in Fig. 4A and B, treatment
of Huh7 cells with the β-adrenergic antagonists for 48 h caused a
significant increase in the proportion of cells in G2/M
phase, when compared with the untreated control cells.
Specifically, the proportion of Huh7 cells in G2/M phase
increased from 24.1 to 65.4% following ICI118,551 treatment, and to
68.1% for metoprolol treatment. These results indicate that
antagonism of ADBR2 leads to arrest in the G2/M phase of
the cell cycle in HCC cells. Taken together, these results indicate
that inhibition of ADBR2 induced cell apoptosis and cell cycle
arrest in HCC cells, which led to cell growth inhibition.
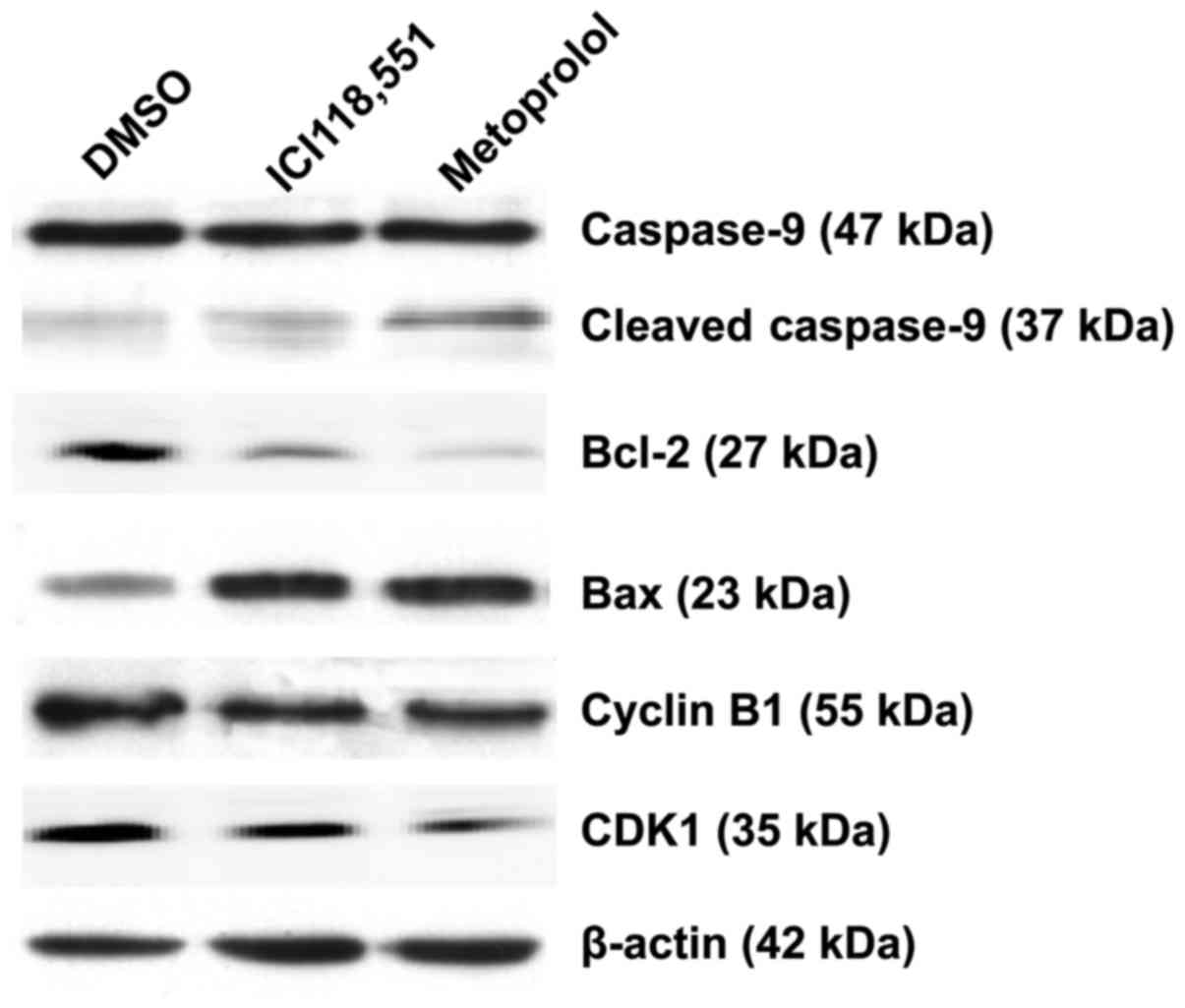
Inhibition of ADBR2 downregulates the
expression of Bcl-2 and cyclin B1 in HCC cells
To determine the molecular mechanisms that were
involved in apoptosis and G2/M phase cell cycle arrest,
several cell apoptosis and cell cycle relevant proteins were
screened for changes in expression level in Huh7 cells following
treatment with ICI118,551 (50 µM) or metoprolol (50 µM). As
depicted in Fig. 5, the expression of
Bcl-2 were decreased following treatment with either antagonist,
whereas the cleavage of caspase-9 and Bax increased, compared with
the control cells. In addition, expression of the G2/M
phase proteins cyclin B1 and CDK1 were downregulated in Huh7 cells
treated with β-adrenergic antagonists.
Discussion
ADRB2 has been studied for more than four decades,
it belongs to the GPCR superfamily, and can regulate cancer
development (15). Recently, emerging
evidence has demonstrated the association between β-adrenergic
antagonism and cancer. β-adrenergic antagonists also exert
antitumor effects via non-genomic mechanisms, including matrix
metalloproteinase, mitogen-activated protein kinase pathways,
prostaglandins, cyclooxygenase-2, oxidative stress and nitric oxide
synthase; thus, treatment with these antagonists may exert a
beneficial clinical effect in patients with cancer (8). The present study demonstrated that ADRB2
was highly expressed in human HCC cells; ADBR2 antagonism
attenuated HCC cell growth by inducing cell apoptosis and
G2/M phase cell cycle arrest. The results of the present
study also indicated that this mechanism may involve elevated
levels of caspase-9 and Bax expression and downregulation of Bcl-2,
cyclin B1 and CDK1. The results of the current study indicate that
ADRB2 antagonists may therefore represent a promising therapeutic
strategy for HCC.
ADRB2 is primarily distributed at the cell membrane,
although its expression in different cells and tissues varies
(16). ADRB2 is expressed in
pancreatic cancer BxPC-3, MIA PaCa-2 and Panc-1 cell lines
(17,18); breast cancer IBH-4, IBH-6 and
MDA-MB-231 cell lines (19); and
human astrocytoma U118 and 1321N1 cell lines (20). The present study revealed the presence
of high ADRB2 expression in HCC SMMC-7721, Huh7 and Hep3B cell
lines, but no expression in the normal liver L02 cell line.
Kassahun et al (14) reported
that the ADRB2 density was higher in HCC liver membranes than the
density in the nonadjacent nontumor liver membranes. In addition,
ADRB2 protein expression was increased 1.5-fold when compared with
nonmalignant controls (14), which
was consistent with the results of the present study in HCC cell
lines, indicating that ADRB2 was upregulated in human HCC. However,
Kassahun et al (14) did not
investigate the mechanisms responsible for this change in the
growth of HCC and the nature of this alteration further. The
changes in ADRB2 expression observed in HCC tissues and cell lines
in the current study may reflect cellular defects following tumor
growth and progression.
The association between β-adrenergic antagonism and
cancer has been well established. Zhang et al (21) reported that ADRB2 inhibition induces
G1/S phase arrest and apoptosis in pancreatic cancer
cells via the Ras/Akt/nuclear factor-κB (NF-κB) pathway. They also
observed that ADRB2 antagonists suppress pancreatic cancer cell
invasion by inhibiting cAMP response element binding protein, NF-κB
and activator protein-1 (18). In
addition, Liao et al (22)
reported that the β-adrenoreceptor antagonist propranolol enhanced
the sensitivity of gastric cancer cells to radiation by inhibiting
β-adrenergic receptors and the downstream NF-κB/vascular
endothelial growth factor/epidermal growth factor
receptor/cyclooxygenase-2 pathway. In the present study, ADRB2
antagonism induced G2/M phase arrest and apoptosis in
HCC cells via inhibiting Bcl-2, cyclin B1 and CDK1. However, Pérez
Piñero et al (19) reported
that the β-adrenoreceptor agonists isoprenaline and salbutamol
inhibited breast cancer cell proliferation and tumor growth, an
effect that could be reversed by treatment with the
β-adrenoreceptor antagonist propranolol, indicating that the role
of ADRB2 signaling is complex and the actions of the agonists or
antagonists can elicit a wide range of effects. ADRB2 agonists can
stimulate activation of Src tyrosine kinase and Ras, and activation
of the Raf/MEK/ERK and PI3 K/Akt pathways, promoting tumor growth
and disease progression (17,23). Taken together, alternation of ADRB2
signaling, by treatment with either agonists or antagonists, could
markedly change the cellular processes, potentially leading to
tumor growth inhibition.
In summary, the results of the present study
demonstrated the growth inhibition effects of ADRB2 antagonism in
HCC cells. This inhibitory role was likely mediated by induction of
apoptosis and G2/M phase cell cycle arrest. However, the
molecular mechanisms by which altered levels of ADBR2 regulate and
orchestrate cellular processes require further investigation to
achieve an improved understanding of the role of ADBR2 in cancer
cells. Overall, these results provide a possible therapeutic
approach for the treatment of human HCC by antagonism of ADBR2.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
DD performed all the experiments, analyzed and
interpreted the data, JZ contributed to the cell culture and qPCR.
JY design this study, analyzed the data and wrote the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to publish
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Artinyan A, Mailey B, Sanchez-Luege N,
Khalili J, Sun CL, Bhatia S, Wagman LD, Nissen N, Colquhoun SD and
Kim J: Race, ethnicity, and socioeconomic status influence the
survival of patients with hepatocellular carcinoma in the United
States. Cancer. 116:1367–1377. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Antoni MH, Lutgendorf SK, Cole SW, Dhabhar
FS, Sephton SE, McDonald PG, Stefanek M and Sood AK: The influence
of bio-behavioural factors on tumour biology: Pathways and
mechanisms. Nat Rev Cancer. 6:240–248. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thaker PH, Han LY, Kamat AA, Arevalo JM,
Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori
M, et al: Chronic stress promotes tumor growth and angiogenesis in
a mouse model of ovarian carcinoma. Nat Med. 12:939–944. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cherezov V, Rosenbaum DM, Hanson MA,
Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI,
Kobilka BK and Stevens RC: High-resolution crystal structure of an
engineered human beta2-adrenergic G protein-coupled receptor.
Science. 318:1258–1265. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hara MR, Kovacs JJ, Whalen EJ, Rajagopal
S, Strachan RT, Grant W, Towers AJ, Williams B, Lam CM, Xiao K, et
al: A stress response pathway regulates DNA damage through
β2-adrenoreceptors and β-arrestin-1. Nature. 477:349–353. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Quốc Lu'o'ng KV and Nguyễn LT: The roles
of beta-adrenergic receptors in tumorigenesis and the possible use
of beta-adrenergic blockers for cancer treatment: Possible genetic
and cell-signaling mechanisms. Cancer Manag Res. 4:431–445.
2012.PubMed/NCBI
|
|
9
|
Cole SW and Sood AK: Molecular pathways:
Beta-adrenergic signaling in cancer. Clin Cancer Res. 18:1201–1206.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barron TI, Connolly RM, Sharp L, Bennett K
and Visvanathan K: Beta blockers and breast cancer mortality: A
population-based study. J Clin Oncol. 29:2635–2644. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Melhem-Bertrandt A, Chavez-Macgregor M,
Lei X, Brown EN, Lee RT, Meric-Bernstam F, Sood AK, Conzen SD,
Hortobagyi GN and Gonzalez-Angulo AM: Beta-blocker use is
associated with improved relapse-free survival in patients with
triple-negative breast cancer. J Clin Oncol. 29:2645–2652. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shan T, Ma Q, Zhang D, Guo K, Liu H, Wang
F and Wu E: β2-adrenoceptor blocker synergizes with gemcitabine to
inhibit the proliferation of pancreatic cancer cells via apoptosis
induction. Eur J Pharmacol. 665:1–7. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kassahun WT, Guenl B, Ungemach FR, Jonas S
and Abraham G: Expression and functional coupling of liver
β2-adrenoceptors in the human hepatocellular carcinoma.
Pharmacology. 89:313–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Katritch V, Cherezov V and Stevens RC:
Structure-function of the G protein-coupled receptor superfamily.
Annu Rev Pharmacol Toxicol. 53:531–556. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pérez-Sayáns M, Somoza-Martin JM,
Barros-Angueira F, Diz PG, Gándara Rey JM and García-García A:
Beta-adrenergic receptors in cancer: Therapeutic implications.
Oncol Res. 19:45–54. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weddle DL, Tithoff P, Williams M and
Schuller HM: Beta-adrenergic growth regulation of human cancer cell
lines derived from pancreatic ductal carcinomas. Carcinogenesis.
22:473–479. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang D, Ma QY, Hu HT and Zhang M:
β2-adrenergic antagonists suppress pancreatic cancer cell invasion
by inhibiting CREB, NFκB and AP-1. Cancer Biol Ther. 10:19–29.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pérez Piñero C, Bruzzone A, Sarappa MG,
Castillo LF and Lüthy IA: Involvement of α2- and β2-adrenoceptors
on breast cancer cell proliferation and tumour growth regulation.
Br J Pharmacol. 166:721–736. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Toll L, Jimenez L, Waleh N, Jozwiak K, Woo
AY, Xiao RP, Bernier M and Wainer IW: {Beta}2-adrenergic receptor
agonists inhibit the proliferation of 1321N1 astrocytoma cells. J
Pharmacol Exp Ther. 336:524–532. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang D, Ma Q, Wang Z, Zhang M, Guo K,
Wang F and Wu E: β2-adrenoceptor blockage induces G1/S phase arrest
and apoptosis in pancreatic cancer cells via Ras/Akt/NFκB pathway.
Mol Cancer. 10:1462011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liao X, Che X, Zhao W, Zhang D, Bi T and
Wang G: The β-adrenoceptor antagonist, propranolol, induces human
gastric cancer cell apoptosis and cell cycle arrest via inhibiting
nuclear factor κB signaling. Oncol Rep. 24:1669–1676.
2010.PubMed/NCBI
|
|
23
|
Schuller HM: Mechanisms of smoking-related
lung and pancreatic adenocarcinoma development. Nat Rev Cancer.
2:455–463. 2002. View
Article : Google Scholar : PubMed/NCBI
|