Introduction
Lung cancer is one of the most common malignant
tumors, exhibiting the highest mortality worldwide due to the
uncontrolled cell growth in tissues of the lung and high metastatic
ability (1). Non-small cell lung
cancer (NSCLC) accounts for 80–85% of lung cancer (2). EGF receptor tyrosine kinase inhibitors
(EGFR-TKI), such as gefitinib, have demonstrated notable
therapeutic efficacy in non-small cell lung cancer patients
(3,4).
However, nearly all patients develop drug resistance after a period
of EGFR-TKI treatment, which eventually limits the application of
EGFR-TKI (5). Therefore, developing
strategies to overcome drug resistance is significant for improving
prognosis and survival in the future.
A variety of molecular mechanisms are involved in
the acquisition of TKI resistance, including epithelial-mesenchymal
transition (EMT) (6). EMT is a
cellular process characterized by morphologic changes from an
epithelial phenotype to a mesenchymal phenotype, by which cells
lose epithelial cell-cell adhesions and gain the ability to move
through the extracellular matrix. EMT is often associated with
enhanced proliferation, invasion and metastasis in cancer cells
(7). During EMT, cells exhibit the
acquisition of mesenchymal markers, including Vimentin, type I
collagen, fibronectin and Snail, and the inhibition of cell
adhesion molecules, such as E-cadherin and catenin (8). Additionally, EMT is correlated with
chemotherapeutic sensitivity in several types of cancer and
sensitivity to EGFR-TKI in lung cancer cells, in particular
(9–11).
Napsin A, a member of the aspartic proteinase
family, is frequently expressed in normal kidney and lung tissue,
and is also present in lung adenocarcinoma (12–15).
Napsin A was reported to be negatively associated with the
malignancy degree of cancer cells (16–18). Cells
with low Napsin A expression or without Napsin A expression appear
to be prone to EMT (19). Hence, it
can be speculated that EMT might be suppressed by Napsin A
expression.
In the present study, Napsin A was overexpressed
into the lung cancer A549 cell line its effect was investigated on
their sensitivity to the EGFR-TKI, gefitinib. The results
demonstrated that Napsin A expression combined with gefitinib has a
synergistic inhibitory effect on cellular proliferation and a
promotive effect on cell apoptosis of gefitinib-resistant A549s. In
addition, Napsin A expression blocked the downregulation of
E-cadherin and the upregulation of Vimentin expression in
gefitinib-resistant cells. These data suggested that Napsin A
resensitized the resistant A549 cells to gefitinib by reversing EMT
and by inhibiting the activation of the integrin signaling pathway
via focal adhesion kinase (FAK).
Metarials and methods
Cell culture
The human NSCLC cell line A549 was purchased from
the Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences Institute (Shanghai, China). Cells were cultured in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) supplemented with 10% fetal bovine serum (FBS; Lonza
Group, Ltd., Basel, Switzerland), 5 mM L-glutamine, 5 mM
non-essential amino-acids, 100 U/ml penicillin and streptomycin
(Invitrogen; Thermo Fisher Scientific, Inc.), in a humidified 5%
CO2 incubator at 37°C. In preliminary experiments, A549
cells were detected to be sensitive to gefitinib and exhibited
E-cadherin and vimentin expression, which facilitates the analysis
of the EMT phenotype in response to Napsin A overexpression;
whereas vimentin expression in H322, H358 and H441 lung cancer
cells was negative or in trace amounts (data not shown). In
addition, Napsin A expression levels in parental A549 cells are
lower compared with other lung cancer cell lines (data not shown).
Therefore, the A549 cell line was selected for the present study,
with the hypothesis that exogenous overexpression of Napsin A in
A549 cells might result in more significant change in biological
function.
Development of the gefitinib-resistant
A549 cell line
Gefitinib-resistant A549 cells (A549-GFT) were
developed by exposing A549 cells to increasing concentration of
gefitinib [(also known as Iressa and ZD1839; Bioruler, Beijing
Bioway Biotechnology Co. Ltd., Beijing, China (http://a0002945.casmart.com.cn/)] ranging from
0.2 to 2 µmol/l in complete medium (20). Following exposure to increasing
concentration of gefitinib for at least 3 months, live cells that
developed resistance to 5 µmol/l gefitinib were collected, termed
A549-GFT and used in subsequent experiments.
Morphological observation of
gefitinib-resistant cells
Cells were cultured in 60 mm culture dishes for 24
h, and then treated with or without 2 µmol/l gefitinib for 48 h.
The medium was then removed and cells were washed once with
RPMI-1640 medium. Cell morphology was observed and photographed
using a vertical microscope (Olympus Corporation, Tokyo,
Japan).
Western blot analysis
Cells were treated with different doses of gefitinib
as indicated. The cells were then washed twice with PBS and total
protein was extracted using M-PER Mammalian Protein Extraction
Reagent (Pierce; Thermo Fisher Scientific, Inc.) according to the
manufacturer's manual. The proteins (50 µg) were separated by 12%
SDS-PAGE (Beijing Solarbio Science & Technology Co., Ltd.) and
transferred to polyvinylidene fluoride membranes (Millipore; Merck
KGaA, Darmstadt, Germany). The membranes were blocked with 5%
non-fat dried milk in TBST for 1 h, and incubated with the
following specific primary antibodies overnight at 4°C: Mouse
monoclonal anti-E-cadherin (cat. no. sc-21791; 1:2,000), mouse
monoclonal anti-Vimentin (cat. no. sc-6260; 1:2,000), anti-GAPDH
(cat. no. sc-365062; 1:3,000; all Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), rabbit polyclonal anti-FAK (cat. no. 3285;
1:1,000), rabbit monoclonal anti-phospho-FAK (Tyr397; cat. no.
8556; 1:1,000; both Cell Signaling Technology, Inc., Danvers, MA,
USA) and rabbit monoclonal anti-Napsin A (cat. no. ab133249;
1:10,000; Abcam, Cambridge, UK). Membrane were then incubated with
horseradish peroxidase-conjugated secondary antibodies goat
anti-mouse (cat. no. sc-2005; 1:2,000) and goat anti-rabbit IgG
(cat. no. sc-2004; 1:2,000; both Santa Cruz Biotechnology, Inc.)
for 2 h at room temperature. Development was performed using
enhanced chemiluminescence detecting reagent (Amersham; GE
Healthcare, Chicago, IL, USA). The protein blots were quantified by
densitometry using QuantityOne software (Bio-Rad Laboratories,
Inc., Hercules, CA, USA), and the amounts were expressed relative
to the internal reference GAPDH.
Overexpression of Napsin A
The Napsin A-overexpressing plasmid, PLJM1-Napsin A,
was constructed by cloning the full-length human Napsin A cDNA into
the PLJM1 lentivirus vector (Addgene, Cambridge, CA, USA). cDNA was
amplified using reverse transcription-polymerase chain reaction
(RT-PCR). The PLJM1-Napsin A or the empty vehicle plasmid was
cotransfected with pVSVG and pGag-pol plasmids (Clontech
Laboratories, Inc., Mountainview, CA, USA) into HEK293T cells using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) to
produce virus, according to the manufacturer's manual. The
lentivirus supernatant was harvested 48 h post-transfection and
used to infect A549 cells in order to obtain Napsin A stable
overexpressing cells. Puromycin (Sigma-Aldrich; Merck KGaA) was
used to screen positive clones.
Proliferation assay
Cell proliferation was evaluated by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma Aldrich; Merck KGaA) assay. A total of 2,000 cells were
seeded into each well of a 96-well plate in 100 µl of medium and
incubated with or without various concentrations of gefitinib for
different time periods at 37°C in a 5% CO2 incubator.
Then cells were incubated with 20 µl of 5 mg/ml MTT for 4 h, and
then cells were lysed for 10 min by addition of 200 µl dimethyl
sulfoxide. Absorbance was measured at 490 nm using a Rainbow
microplate reader (Tecan Group Ltd., Mannedorf, Switzerland). Cell
proliferation was expressed as a % of the untreated control.
Apoptosis assay
Cells were grown to 80% confluence and treated with
5 µmol/l gefitinib for 48 h. Apoptosis was analyzed using an
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI)
assay (BD Biosciences, Franklin Lakes, NJ, USA) following the
manufacturer's instructions. The amount of phosphatidylserine on
the outer surface of the plasma membrane (a biochemical alteration
unique to membranes of apoptotic cells) and the amount of PI (a dye
that easily enters dead cells or cells in the late stages of
apoptosis and binds DNA, but does not bind the plasma membrane of
viable cells) were detected by flow cytometry using a FACSCalibur
(BD Biosciences, San Jose, CA, USA). Data were analyzed using
CellQuest software (BD Biosciences). Cells with phosphatidylserine
on their surface were considered to be apoptotic.
Statistical analysis
All experiments were repeated at least in
triplicate. Statistical analysis was performed with SPSS 11.0
(SPSS, Inc., Chicago, IL). Values are presented as the mean ±
standard error of the mean. One-way analysis of variance was used
to assess differences between groups. The Duncan's new multiple
range test was subsequently employed for pairwise comparisons
followed by the Bonferroni correction. P<0.05 was considered to
indicate a statistically significant difference.
Results
Development of gefitinib-resistant
A549 lung cancer cell line
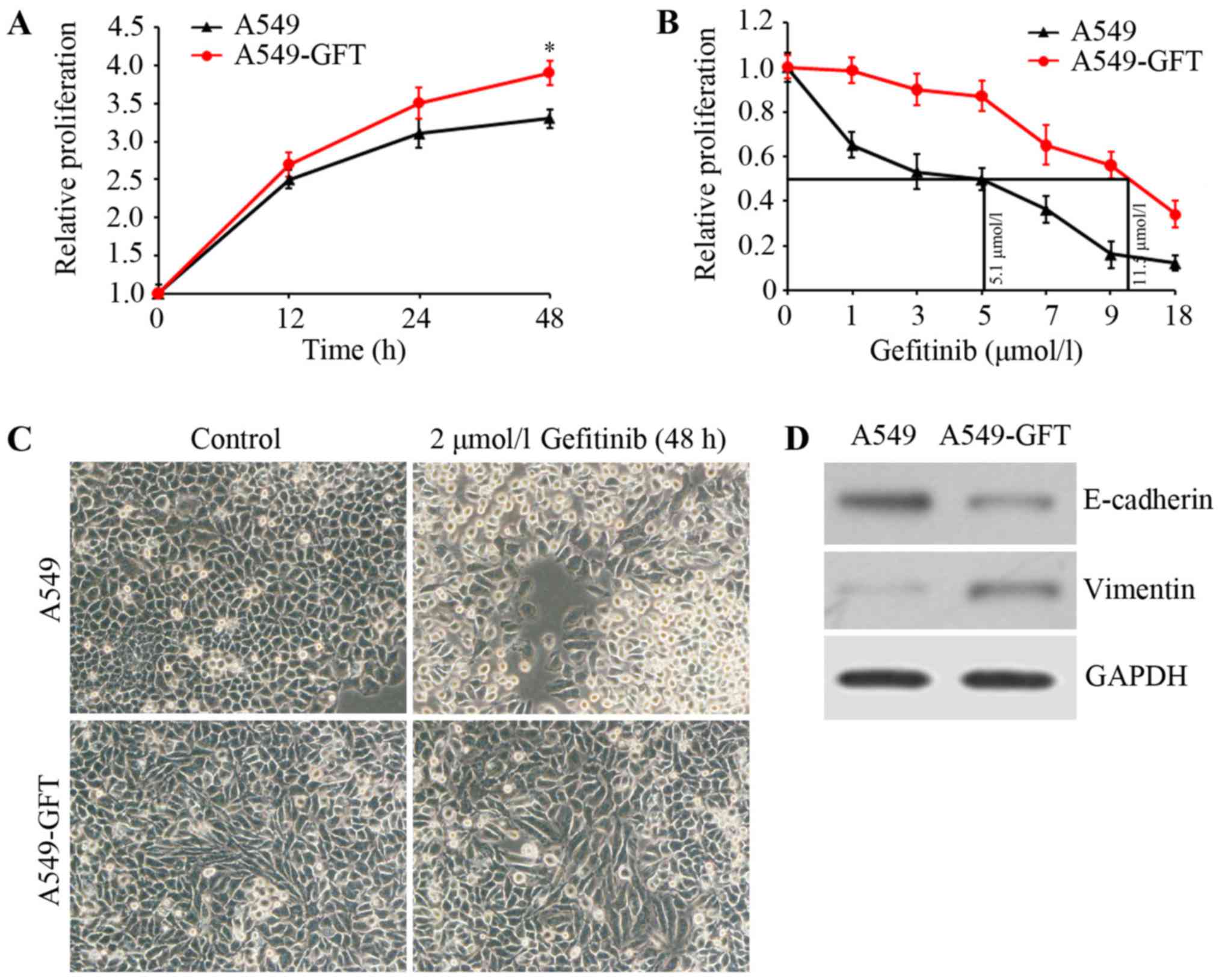
To investigate the mechanism underlying gefitinib
resistance in lung cancer, a gefitinib-resistant lung cancer cell
model (A549-GFT) was first constructed by exposing the A549 lung
cancer cell line to increasing concentrations of gefitinib. Cell
growth was assessed using MTT assay and growth curves of A549-GFT
and A549 cells were calculated. The data demonstrated that A549-GFT
cells grew faster compared with A549 cells at the same time
interval (Fig. 1A). Additionally, the
IC50 of gefitinib was determined by exposing A549-GFT
and A549 cells to different concentrations of gefitinib for 48 h.
The IC50 values of the two cells were respectively
calculated to be 11.5 and 5.1 µmol/l (Fig. 1B). Further analysis of the
gefitinib-resistant cells revealed that A549-GFT cells exhibited an
EMT phenotype, characterized by spindle-like morphology, decreased
E-cadherin expression and increased Vimentin expression (Fig. 1C and D). Finally, the two cell lines
were treated with 2 µmol/l gefitinib for 48 h and their morphology
was inspected by microscopy. As expected, A549 cells appeared round
and with obvious blebbing, which are features of apoptosis
(Fig. 1C). However, no significant
changes in A549-GFT morphology were observed (Fig. 1C) following gefitinib treatment,
confirming that the cells were gefitinib-resistant.
Napsin A overexpression resensitizes
resistant A549 cells to gefitinib
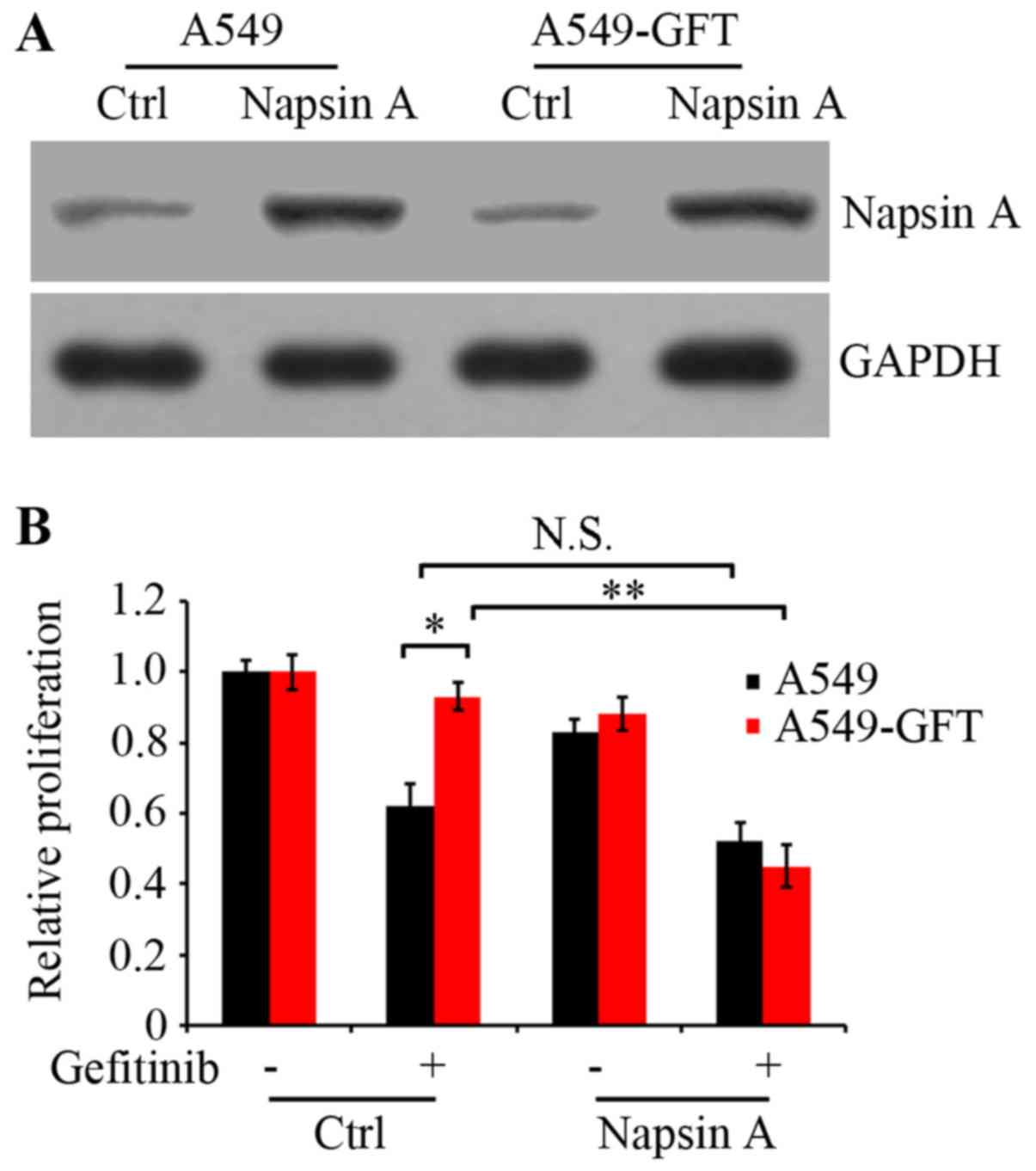
To examine the effect of Napsin A on gefitinib
resistance in lung cancer cells, the PLJM1-Napsin A expression
vector was constructed and used to increase the expression of
Napsin A. Western blot analysis confirmed that the Napsin A
expression levels in the PLJM1-Napsin A-transduced A549 and
A549-GFT cells were markedly elevated compared with the control
cells (Fig. 2A). Next, A549 and
A549-GFT cells were transiently transduced with empty control
vector or PLJM1-Napsin A expression vector, and cultured in the
presence or absence of 2 µmol/l gefitinib for 48 h. Cell
proliferation was then analyzed by MTT assay. The results
demonstrated that cell proliferation of Napsin A-overexpressing
A549-GFT cells was significantly suppressed by gefitinib treatment
compared with gefitinib-treated control A549-GFT cells (P=0.006;
Fig. 2B). The growth of control
A549-GFT cells was not nearly affected by gefitinib treatment and
exhibited significantly increased survival compared with the
parental A549 cells (P=0.019; Fig.
2B). However, under identical experimental conditions, there
was no significant difference in the proliferation between Napsin
A-overexpressing and control A549 cells (Fig. 2B).
Napsin A overexpression promotes
gefitinib-induced apoptosis in A549-GFT cells
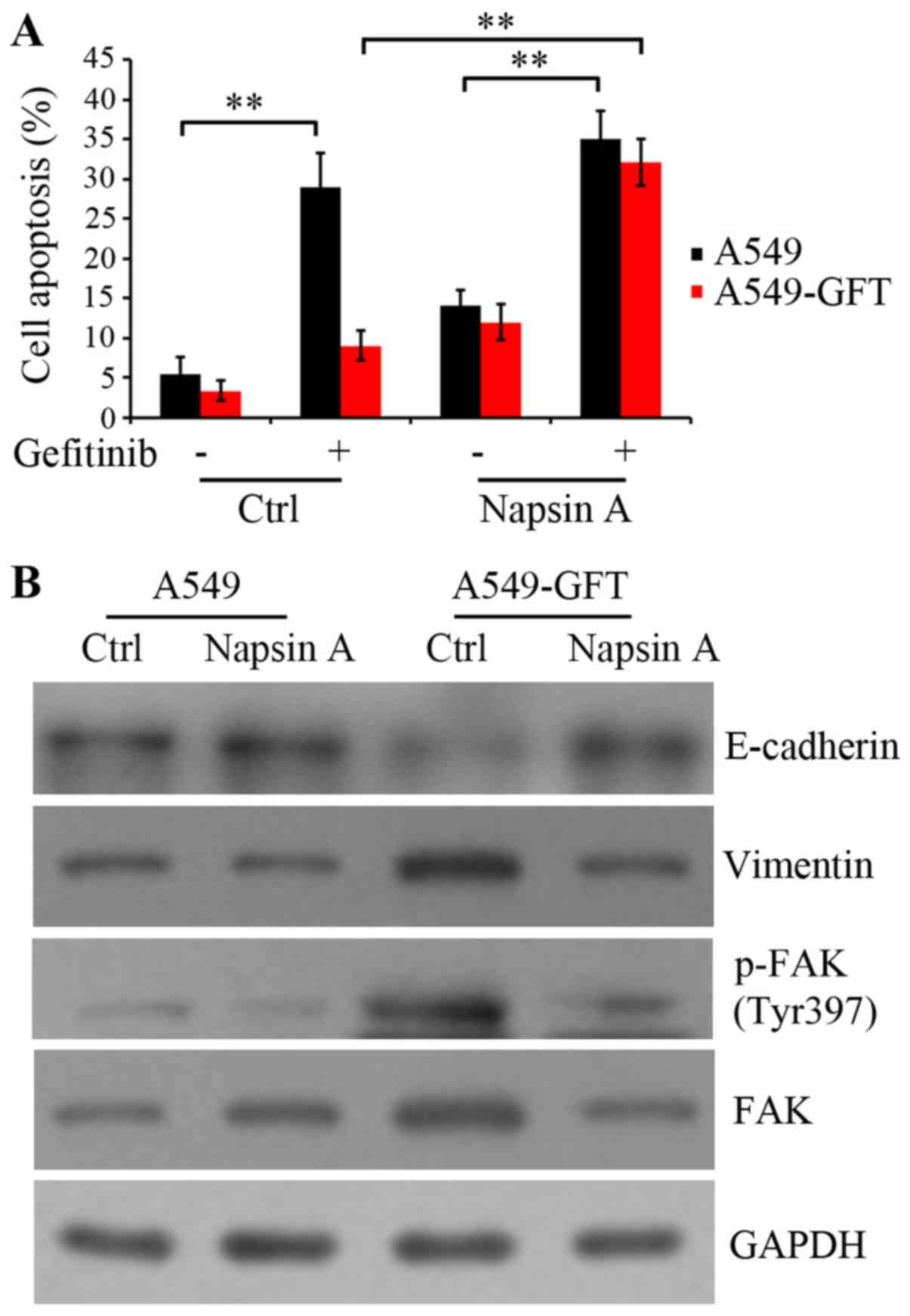
To clarify the mechanism by which Napsin A
overexpression resensitized the resistant A549-GFT cells to
gefitinib, apoptosis was evaluated by the Annexin V-FITC/PI assay.
A549-GFT and A549 cells were transduced with empty control and
Napsin A-expressing vector, and exposed to 2 µmol/l gefitinib for
48 h. The data indicated that gefitinib treatment induced the
apoptosis of both Napsin A-overexpressing and control A549 cells
compared to the corresponding untreated control cells (P=0.006 and
P=0.009, respectively; Fig. 3A).
Notably, Napsin A overexpression significantly stimulated the
gefitinib-induced apoptosis in A549-GFT cells compared with the
A549-GFT cells transduced with the empty vector (P=0.005; Fig. 3A). However, gefitinib treatment did
not significantly induce apoptosis in empty vector-transduced
A549-GFT cells compared with empty vector-transduced parental A549
cells (Fig. 3A). These results
suggest that Napsin A may be correlated with the resistance of lung
cancer A549 cells to gefitinib and Napsin A expression may increase
the sensitivity of resistant cells to gefitinib by enhancing
gefitinib-induced apoptosis.
Additionally, the protein expression levels of
E-cadherin, Vimentin and FAK were detected, because they serve a
critical role in integrin-mediated signaling pathway and are
correlated with cell transformation (21–23).
Autophosphorylation of FAK at residue Tyr397 results in the
activation of the integrin signaling pathway. The results
demonstrated that Napsin A overexpression rescued the expression of
E-cadherin and blocked the upregulation of Vimentin in A549-GFT
cells (Fig. 3B). Furthermore, Napsin
A overexpression resulted in an inhibition of FAK expression, as
well as its phosphorylation at Tyr397 in resistant A549-GFT cells
(Fig. 3B). These findings suggested
that Napsin A reversed the EMT and enhanced the sensitivity of
A549-GFT cells to gefitinib, potentially through the inhibition of
FAK-mediated integrin signaling.
Discussion
Lung cancer, a common malignancy with very high
mortality, seriously threatens human health (1). The majority of lung cancers are
diagnosed as NSCLC (2). EGFR-TKIs,
such as gefitinib, have been used as the standard therapy in NSCLCs
with EGFR-activating mutations (3,4). However,
nearly all patients eventually succumb to recurrence due to
developing drug resistance; therefore the therapeutic efficacy of
EGFR-TKIs is significantly limited. Therefore, exploring novel
therapeutic strategies to overcome the acquisition of EGFR-TKI
resistance is necessary. The present study demonstrated that Napsin
A was able to attenuate the drug resistance of lung cancer A549
cells to gefitinib and Napsin A in combination with gefitinib
significantly inhibited cancer cell growth in vitro.
In the present study, gefitinib-resistant lung
cancer A549-GFT cells were firstly generated by simulating the
condition of drug resistance development in vivo, using an
increasing concentration of gefitinib over a long period of time.
Results from a cell proliferation assay revealed that the
IC50 of gefitinib in the drug-resistant A549-GFT cells
at 48 h was markedly increased compared with the parental A549
cells. In addition, gefitinib treatment at 2 µmol/l concentration
did not significantly affect the proliferation of A549-GFT cells,
while under identical conditions, the proliferation of the parental
A549 cells was reduced by ~80%. These findings confirmed that
A549-GFT cells were resistant to gefitinib and could be used for
subsequent experiments. Notably, gefitinib resistance was
demonstrated to cause changes in A549 cell morphology, from
cobblestone-like epithelial cells into slender spindle-shaped
cells, suggesting that the resistant cells had undergone. EMT is
characterized by the loss of cell adhesion molecules, such as
E-cadherin, and elevation of mesenchymal markers, such as Vimentin
(8). The present results demonstrated
that gefitinib-resistant A549-GFT cells displayed downregulation of
E-cadherin expression and upregulation of Vimentin expression
compared with parental A549 cells. These data supported that the
development of resistance to gefitinib in A549 cells induced EMT. A
previous study has indicated that EMT is involved in the
acquisition of TKI resistance (6).
Hence, it was speculated that EMT might be the mechanism underlying
the acquisition of gefitinib resistance in A549 cells.
Napsin A has been reported to inhibit EMT in A549
cells (24). In the present study,
Napsin A-overexpressing A549 and gefitinib-resistant A549-GFT cell
lines were constructed. It was demonstrated that Napsin A
overexpression remarkably suppressed EMT in gefitinib-resistant
cells, resulting in the restoration of E-cadherin expression and
the downregulation of Vimentin expression. Furthermore, Napsin A
overexpression significantly enhanced the sensitivity of resistant
A549-GFT cells to gefitinib by inhibiting cell proliferation and
inducing cell apoptosis. In addition, the results demonstrated that
FAK was inhibited following Napsin A overexpression in resistant
A549-GFT cells. FAK has an important role in integrin signaling
(21,25–27). FAK
gets activated by integrin and subsequently regulates several
signaling pathways, including signal transducer and activator of
transcription 1 (STAT1), mitogen-activated protein kinase (MAPK)
and phosphoinositide 3-kinase (PI3K) signaling (21–23),
resulting in the initiation of cell proliferation and
transformation. Integrin aggregation by ligand binding results in
the oligomerization of FAK, which is mediated by talin.
Autophosphorylation of FAK at residue Tyr397 results in the binding
of the SH2 domain of Src and Fyn, which then phosphorylate a number
of FAK-associated proteins, including paxilin, tensin and the
docking protein p130CAS (Crk-Associated Substrate). In addition,
phosphorylation of Tyr397 leads to the recruitment of other
SH2-containing proteins, including PI3K, phospholipase-Cγ and the
adapter protein growth factor receptor-bound protein 7 (GRB7)
(5), this resulting in the activation
of the integrin signaling pathway (28). The carboxyl terminal of the Napsin A
contains an Arg-Gly-Asp (RGD) sequence, which recognizes and binds
integrins on the cell surface (29).
Integrins mediate cell adhesion and signal transduction between
cells and the extracellular matrix (ECM) (30), and they are an important regulator of
cell proliferation, apoptosis, migration, and metastasis (31,32). In
the present study, it may be possible that Napsin A repressed the
interaction between integrins and ECM through RGD sequence-mediated
interaction with integrin, thus hindering integrin signaling
pathway and inhibiting cell proliferation and transformation by
downregulating FAK expression and phosphorylation in
gefitinib-resistant A549-GFT cells. Further investigation needs to
be performed in order to test this hypothesis.
In conclusion, the present data demonstrated that
Napsin A overexpression combined with the EGFR-TKI gefitinib may be
an effective method to improve the sensitivity of lung cancer cells
to gefitinib. Napsin A could be delivered to tumor tissues through
viral vectors or liposomes in order to elevate its expression level
as a potential clinical gene treatment strategy for patients with
gefitinib-resistant lung tumors. Future studies will employ
additional gefitinib-resistant lung cancer cell lines in order to
confirm that this effect is not specific to A549 cells, and to
further investigate the association between gefitinib resistance
and Napsin A expression in lung cancer.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ, XL and ZW were major contributors in the
conception and design of the research and revision of the
manuscript for important intellectual content. Acquisition of data
was performed by JY. YZ and TX were the major contributors in the
analysis and interpretation of data and statistical analysis.
Drafting the manuscript was performed by ZW.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
PDQ Adult Treatment Editorial Board:
Non-small cell lung cancer treatment (PDQ®): Patient Version, . PDQ
cancer information summaries [Internet]. Nat Cancer Inst (US);
Bethesda: https://www.ncbi.nlm.nih.gov/books/NBK65917/April
13–2017
|
|
2
|
Pao W and Girard N: New driver mutations
in non-small-cell lung cancer. Lancet Oncol. 12:175–180. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Soria JC, Mok TS, Cappuzzo F and Jänne PA:
EGFR-mutated oncogene-addicted non-small cell lung cancer: Current
trends and future prospects. Cancer Treat Rev. 38:416–430. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nguyen KS and Neal JW: First-line
treatment of EGFR-mutant non-small-cell lung cancer: The role of
erlotinib and other tyrosine kinase inhibitors. Biologics.
6:337–345. 2012.PubMed/NCBI
|
|
5
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu PF, Zhu YP, Yang CH, Wang YF and Wang
GH: The Mechanism and countermeasures on the secondary resistance
of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor
(EGFR-TKI). Anti-Tumor Pharm. 5:42015.
|
|
7
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Robert G, Gaggioli C, Bailet O, Chavey C,
Abbe P, Aberdam E, Sabatié E, Cano A, de Herreros Garcia A,
Ballotti R and Tartare-Deckert S: SPARC represses E-cadherin and
induces mesenchymal transition during melanoma development. Cancer
Res. 66:7516–7523. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Voulgari A and Pintzas A:
Epithelial-mesenchymal transition in cancer metastasis: Mechanisms,
markers and strategies to overcome drug resistance in the clinic.
Biochim Biophys Acta. 1796:75–90. 2009.PubMed/NCBI
|
|
10
|
Neel DS and Bivona TG: Secrets of drug
resistance in NSCLC exposed by new molecular definition of EMT.
Clin Cancer Res. 19:3–5. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Uramoto H, Iwata T, Onitsuka T, Shimokawa
H, Hanagiri T and Oyama T: Epithelial-mesenchymal transition in
EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res.
30:2513–2517. 2010.PubMed/NCBI
|
|
12
|
Tatnell PJ, Powell DJ, Hill J, Smith TS,
Tew DG and Kay J: Napsins: New human aspartic proteinases.
Distinction between two closely related genes. FEBS Lett.
441:43–48. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chuman Y, Bergman A, Ueno T, Saito S,
Sakaguchi K, Alaiya AA, Franzén B, Bergman T, Arnott D, Auer G, et
al: Napsin A, a member of the aspartic protease family, is
abundantly expressed in normal lung and kidney tissue and is
expressed in lung adenocarcinomas. FEBS Lett. 462:129–134. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schauer-Vukasinovic V, Bur D, Kling D,
Grüninger F and Giller T: Human napsin A: Expression,
immunochemical detection, and tissue localization. FEBS Lett.
462:135–139. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hirano T, Auer G, Maeda M, Hagiwara Y,
Okada S, Ohira T, Okuzawa K, Fujioka K, Franzén B, Hibi N, et al:
Human tissue distribution of TA02, which is homologous with a new
type of aspartic proteinase, napsin A. Jpn J Cancer Res.
91:1015–1021. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hirano T, Gong Y, Yoshida K, Kato Y,
Yashima K, Maeda M, Nakagawa A, Fujioka K, Ohira T, Ikeda N, et al:
Usefulness of TA02 (napsin A) to distinguish primary lung
adenocarcinoma from metastatic lung adenocarcinoma. Lung Cancer.
41:155–162. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ueno T, Linder S and Elmberger G: Aspartic
proteinase napsin is a useful marker for diagnosis of primary lung
adenocarcinoma. Br J Cancer. 88:1229–1233. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hirano T, Auer G, Maeda M, Hagiwara Y,
Okada S, Ohira T, Okuzawa K, Fujioka K, Franzén B, Hibi N, et al:
Human tissue distribution of TA02, which is homologous with a new
type of aspartic proteinase, napsin A. Jpn J Cancer Res.
91:1015–1021. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ueno T, Elmberger G, Weaver TE, Toi M and
Linder S: The aspartic protease napsin A suppresses tumor growth
independent of its catalytic activity. Lab Invest. 88:256–263.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Meena AS, Sharma A, Kumari R, Mohammad N,
Singh SV and Bhat MK: Inherent and acquired resistance to
paclitaxel in hepatocellular carcinoma: Molecular events involved.
PLoS One. 8:e615242013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hauck CR, Hsia DA and Schlaepfer DD: The
focal adhesion kinase-a regulator of cell migration and invasion.
IUBMB Life. 53:115–119. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie B, Zhao J, Kitagawa M, Durbin J, Madri
JA, Guan JL and Fu XY: Focal adhesion kinase activates Stat1 in
integrin-mediated cell migration and adhesion. J Biol Chem.
276:19512–19523. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bhowmick NA, Zent R, Ghiassi M, McDonnell
M and Moses HL: Integrin beta 1 signaling is necessary for
transforming growth factor-beta activation of p38MAPK and
epithelial plasticity. J Biol Chem. 276:46707–46713. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zheng JX, Guan SH, Xu Q, Liu JZ and Song
P: Inhibition of epithelial-mesenchymal transition in A549 cell by
transfected Napsin A. Chin Med J (Engl). 125:2734–2740.
2012.PubMed/NCBI
|
|
25
|
Hauck CR, Sieg DJ, Hsia DA, Loftus JC,
Gaarde WA, Monia BP and Schlaepfer DD: Inhibition of focal adhesion
kinase expression or activity disrupts epidermal growth
factor-stimulated signaling promoting the migration of invasive
human carcinoma cells. Cancer Res. 61:7079–7090. 2001.PubMed/NCBI
|
|
26
|
Sieg DJ, Hauck CR, Ilic D, Klingbeil CK,
Schaefer E, Damsky CH and Schlaepfer DD: FAK integrates
growth-factor and integrin signals to promote cell migration. Nat
Cell Biol. 2:249–256. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sieg DJ, Hauck CR and Schlaepfer DD:
Required role of focal adhesion kinase (FAK) for
integrin-stimulated cell migration. J Cell Sci. 112:2677–2691.
1999.PubMed/NCBI
|
|
28
|
Parsons JT, Martin KH, Slack JK, Taylor JM
and Weed SA: Focal adhesion kinase: A regulator of focal adhesion
dynamics and cell movement. Oncogene. 19:5606–5613. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ruoslahti E: RGD and other recognition
sequences for integrins. Annu Rev Cell Dev Biol. 12:697–715. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Juliano RL: Signal transduction by cell
adhesion receptors and the cytoskeleton: Functions of integrins,
cadherins, selectins, and immunoglobulin-superfamily members. Annu
Rev Pharmacol Toxicol. 42:283–323. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hynes RO: Integrins: Bidirectional,
allosteric signaling machines. Cell. 110:673–687. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Yang J, Dai C, Wu C and Liu Y: Role
for integrin-linked kinase in mediating tubular epithelial to
mesenchymal transition and renal interstitial fibrogenesis. J Clin
Invest. 112:503–516. 2003. View Article : Google Scholar : PubMed/NCBI
|