Introduction
Acute megakaryocytic leukemia (AMKL) is a rare type
of acute leukemia that was first reported in 1931 by Von Boros
(1). In subsequent years, patients
were rarely diagnosed with AMKL due to its low incidence and a lack
of accurate diagnostic criteria. In 1978, Breton-Gorius et
al (2) utilized immunoelectron
microscopy analysis with platelet peroxidase (PPO) to identify
megakaryocytes and increase the accuracy of diagnosing AMKL. AMKL
was then added to the French-American-British classification as
acute myeloid leukemia (AML) M7 in 1985, which identified the
diagnostic criteria as exhibiting >30% of blast cells of the
bone marrow nucleated cells, which were demonstrated as a lineage
derived from megakaryocytes by PPO staining (3). With the development and application of
flow cytometry, the diagnosis of AMKL became more accurate. In
2008, the World Health Organization (WHO) produced precise criteria
for diagnosing AMKL (4). In these
criteria, AMKL was diagnosed by the presence of ≥20% blasts of the
bone marrow nucleated cells, with >50% of the blasts being
megakaryoblasts in the bone marrow; or positive platelet-specific
antigens by bone marrow aspirate or biopsy, including factor VIII,
cycle of differentiation (CD)41, CD42 or CD61, as determined by
immunocytochemistry staining or immunophenotyping.
In the clinic, although there are now accurate
diagnostic criteria, the diagnosis of AMKL is challenging due to a
high incidence of myelofibrosis (MF), resulting in difficulty in
differentiating AMKL from acute MF (5). Clinical diagnostic experience with this
type of leukemia is also limited. When the percentage of blast
cells in the bone marrow is <20%, immunohistochemical staining
and immunophenotypic analysis are not performed; therefore, the
diagnosis of AMKL may lack accuracy when depending solely on the
information provided by cellular morphology (6). As a result, a comprehensive diagnosis is
important for this disease.
In the present study, the clinical characteristics,
experimental tests and survival times of 9 adult patients with
AMKL, who were recruited by the Sino-U.S. Shanghai Leukemia
Cooperative Group between June 2003 and December 2010, were
analyzed. Additionally, the diagnostic experience was summarized
and diagnostic recommendations for AMKL were provided.
Patients and methods
Patients
The Sino-U.S. Shanghai Leukemia Cooperative Group
(School of Public Health, Fudan University) diagnosed 623 patients
(Median age is 51 years old, and ranged from 18 to 88 years, the
percentage of males were 56.5%) with AML between June 2003 and
December 2010, and conducted follow-ups to determine their survival
time. Of these patients, 9 (1.4%) were diagnosed with AMKL. These
patients were diagnosed with AMKL according to the 2008 WHO
classification criteria (4) as
follows: i) The bone marrow aspirate exhibited a blast cell
infiltrate comprising ≥20% of all cells, and >50% of the blast
cells were identified as megakaryoblasts; ii) the expression of
CD41, CD42 and/or CD61 was positive, as demonstrated by flow
cytometry with monoclonal or polyclonal platelet-specific
antibodies; and iii) bone marrow aspiration was frequently
accompanied by MF, and in cases with fibrosis, a bone marrow biopsy
was required, and the cell of origin was required to be identified
as part of the megakaryocyte lineage. This was indicated by
positive immunocytochemical staining for platelet-specific
antigens, including factor VIII, CD41, CD42 and CD61. Patients were
be diagnosed with AMKL if they met at least one of the diagnostic
criteria. In the present study, all the patients with AMKL were
administered one, two or more intravenous (IV) courses of a
standard induction regimen that was comprised of 60
mg/m2 daunorubicin or 12 mg/m2 idarubicin per
day for 3 consecutive days, plus 200 mg/m2 cytarabine
that was continuously administered by IV infusion for 7 days.
Following one course of a standard induction, a number of patients
had no response and succumbed shortly after. Additionally, a number
of patients had complete remission or partial remission and
continued the regimen, receiving two or more courses of a standard
regimen.
Diagnostic process
i) If the percentage of blast cells was >20% in
the bone marrow of nucleated cells, and cell morphology was
demonstrated to be megakaryoblasts, as demonstrated using a bone
marrow smear, the diagnosis was AMKL. For this diagnosis, the
results of flow cytometry or immunocytochemical staining were
required to increase the accuracy of the diagnosis. ii) If the bone
marrow aspiration could not indicate a diagnosis of AMKL, detection
methods of flow cytometry and immunocytochemical staining were
vital, and the final diagnosis was frequently determined by
positive platelet-specific antigens. iii) If the bone marrow
aspiration diagnosis was not successful due to MF, a bone marrow
biopsy was the primary test method, and the final diagnosis was
determined by immunocytochemical staining for factor VIII, CD41,
CD42 or CD61.
Cell morphology, bone marrow
cellularity and cytochemical staining
Bone marrow aspirates and biopsies were obtained
from the posterior iliac crest. Bone marrow smears were conducted
by obtaining 0.2 ml marrow aspirate from each patient, in order to
perform cytomorphological classification and cytochemical staining,
including myeloperoxidase, periodic acid-Schiff, α naphthol-acetate
esterase, Sudan black B and non-specific esterase staining,
according to the International Committee for Standardization in
Hematology (7). Bone marrow
cellularity of the aspirate and biopsy were examined by routine
light microscopy (×100 magnification) and assessed with the
following grading: Definite hypercellularity, normal cellularity,
moderate hypocellularity and severe hypocellularity (8).
Flow cytometry immunophenotyping
An additional 3 ml bone marrow aspirate was used for
flow cytometry analysis. Mononuclear cells (MNCs) were isolated
from those samples using lymphocyte separation medium
(Ficoll®-paque Plus; GE Healthcare Life Sciences,
Chicago, IL, USA). The cells were suspended in PBS (GE HealthCare
Life Sciences) with 3% bovine serum albumin (A8020; Beijing
Solarbio Science & Technology Co., Ltd., Beijing, China) and
was adjusted to 1×106 MNCs/ml and an aliquot of 100 µl
was labeled with 5 or 20 µl fluorescein conjugated monoclonal
antibodies. The applied monoclonal antibodies were directed against
the following surface antigens: CD34 PE (550761, 1:5), CD45 PerCP
(564105, 1:20), human leukocyte antigen DR related antigen (HLA-DR
APC, 560896, 1:5), platelet-specific glycoproteins, CD41 FITC
(555469, 1:5) and CD61 PE (555754, 1:5), B-cell antigens, CD10 PE
(561002, 1:20) and CD19 FITC (560994, 1:5), T-cell antigens (CD2 PE
(555327, 1: 20), CD3 APC (555335, 1:5), CD4 APC-CY7 (561839, 1:20)
and CD7 FITC (555360, 1:5), myeloid antigens, CD13 APC (561698,
1:20), CD14 PE-CY7 (560919, 1:20), CD15 FITC (555401, 1:5), CD33 PE
(555450, 1:5), CD64 APC (561189, 1:20), CD117 PE (561682, 1:20) and
myeloperoxidase (556035, 1:20, BD Biosciences). All monoclonal
antibodies were obtained from BD Biosciences (San Jose, CA, USA).
Subsequently, the cell suspension was incubated with these
antibodies in the dark for 15 min at 4°C, then washed in PBS with
5% EDTA and centrifuged (200 × g) for 5 min at 4°C three times.
Following this, the cells were suspended in 500 µl PBS for further
use. The immunophenotyping was performed with the BD FACS Canton
flow cytometer using FCS Express 3.0 software (De Novo Software,
Glendale, CA, USA). Gating was set with CD45 and 90° light-scatter
parameters in order to exclude erythrocytes, platelets and
subcellular debris. A total of 1×104 cells was acquired
and the percentage of cells expressing the marker was
calculated.
Immunohistochemical staining
The samples from the bone marrow biopsy were fixed
in B5 stationary liquid (10% formaldehyde) (mercuric chloride 6.0
g, anhydrous sodium acetate 1.25 g, distilled water 90 ml and
formaldehyde 10 ml) for 2 h at room temperature, followed by
processing in 70% absolute ethyl alcohol, decalcification in Decal
(EDTA bisodic salt) for 2.5 h at room temperature and ordinary
processing with paraffin embedding. The paraffin-embedded samples
were placed on slides (Dako ChernMate Capillary Gap slides; Dako;
Agilent Technologies, Inc., Santa Clara, CA, USA) and stored in a
warm incubator at 56°C. After 2 h, the samples were rinsed with
water and washed with dimethylbenzene, and then, washed by graded
concentration of absolute ethyl alcohol (100, 95, 90, 80 and 70%).
Then, the samples were sealed in 3% H2O2 for
10 min at room temperature and washed in distilled water, three
times at room temperature. Subsequently, the samples underwent
optimized antigen retrieval procedures, the 0.01 M sodium citrate
buffer (pH 6.0) (C1010; Beijing Solarbio Science & Technology
Co., Ltd.) was heated in microwave oven to 95°C, and kept the
samples in boiled sodium citrate buffer for 15 min. After that, it
was cooled at room temperature. Then the samples were washed in
distilled water for 3 min, two times at room temperature, and in
PBS for 5 min, three times at room temperature, and were sealed
with 5% bovine serum albumin (Gibco; Thermo Fisher Scientific,
Inc., MA, USA) for 20 min at room temperature. The applied
monoclonal antibodies (Dako; Agilent Technologies, Inc.) were
against factor VIII (ab236284, 1:200, Abcam, Cambridge, MA, USA),
CD42 (ab134087, 1:250, Abcam) and CD61 (ab7166, 1:250, Abcam), and
were incubated with the samples overnight at 4°C. Subsequently, the
sections were washed in Tris-buffered saline (TBS) three times for
10 min each at room temperature, and then incubated with
biotinylated rabbit anti-goat secondary antibody (1:250; Vector
Laboratories, Inc., Burlingame, CA, USA) for 60 min at room
temperature, followed by washing with TBS three times for 10 min
each. Following this, the sections were processed with
avidin-biotin complex (TA-015-BB, Thermo Fisher Scientific Inc.)
reagent for 30 min at room temperature, incubated in
3,3′diaminobenzidine solution (710008, Sera Care Inc, MA, USA) for
30 min in the dark and counterstained with hematoxylin (0701;
Beijing Solarbio Science & Technology Co., Ltd.). Subsequently,
the sections were dehydrated in an ascending graded series of
absolute ethyl alcohols (70, 80, 90, 95 and 100%), cleared in
xylene and cover-slipped with neutral balsam. These samples were
observed in a light microscope (Nikon 801; ×60 and ×100) and the
imaging Software NIS-Elements F 3.00, SP7 (Build 547) (both Nikon
Corporation, Tokyo, Japan). The stain intensity was classified as
-, +, ++, +++. -, No staining is observed. + positive staining is
observed in less than 25% cells. ++ positive staining in 25–49%
cells. +++ extensive positive staining in more than 50% cells.
Gomori silver impregnation
staining
The Gomori silver impregnation staining kit (G1800;
Beijing Solarbio Science & Technology Co., Ltd.) were used. The
stationary process, paraffin embedding and dehydration procedure
were the same as described previously. The oxidizing agent Gomori
was added to the samples for 5 min at room temperature, washed with
running water for 30 sec. Subsequently, the samples were bleached
by oxalic acid solution for 2 min, then washed by running water for
2 min, by distilled water for 1 min, all the process was performed
at room temperature. After that, Samples were stained with ammonium
ferric sulfate solution (5%) for 5 min, washed by tap water for 1
min, by distilled water for 2 min. Silver ammonia solution was used
to stain samples for 3 min, then distilled water was used to wash
the solution for 1 min. Reducing agent of Gomori was added to the
samples for 1 min and washed by running water for 10 min at room
temperature. Subsequently, the sections were dehydrated in an
ascending graded series of absolute ethyl alcohols (70, 80, 90, 95
and 100%), cleared in xylene and cover-slipped with a neutral
balsam.
Evaluation of MF
MF was graded according to the European Consensus
Grading system (9) as follows: MF-0,
scattered linear reticulin with no intersections; MF-1, loose
network of reticulin with numerous intersections, particularly in
perivascular areas; MF-2, diffuse and dense increases in reticulin
with extensive intersections; and occasionally with only focal
bundles of collagen and/or focal osteosclerosis MF-3, diffuse and
dense increases in reticulin with extensive intersections, with
coarse bundles of collagen, often associated with significant
osteosclerosis.
Detection of the t(9;22) mutation
Blood samples were obtained from the posterior
superior iliac spine by bone marrow aspiration and collected in a
heparinized tube. Blood samples (1 ml) were cultured in a culture
dish containing 6 ml RPMI-1640 (Hyclone; GE Healthcare Life
Sciences, Logan, UT, USA) supplemented with 10% fetal calf serum
(Gibco; Thermo Fisher Scientific, Inc.), 1% antibiotics and 100 µl
10 mg/ml phytohemagglutinin for each patient. After 24 h at 37°C,
50 µl colchicine (0.1 mg/ml) was added in culture dish and
incubated at 37°C for 60 min. Subsequently, the samples were
centrifuged at 200 × g for 10 min at room temperature, the
supernatant was removed and then 5 ml 0.075 M KCl was added.
Following this, the samples were incubated for 20 min at 37°C,
followed by centrifugation at 200 × g for 10 min at room
temperature. The supernatant was discarded and cells were
resuspended in 1 ml cold fixative (methanol: Acetic acid, 3:1),
incubated for 0.5 h at room temperature, and washed three times at
200 × g for 5 min each at room temperature. Finally, cells were
suspended in 3 drops of the fixative. Slides were prepared by
dropping a suspension of the pellet onto clean glass slides and
dried at 37°C for 10 min. Giemsa staining was performed for 30 min
at room temperature, then it was examined under a light microscope
(×100 magnification), followed by G-banding for analysis.
Primarily, 20–30 good-quality (The mitotic phase was complete and
independent and the chromosome of intact cells was observed)
metaphases were screened. Chromosome abnormalities were described
according to the standard of The International System for Human
Cytogenetic Nomenclature (10).
Detection of the Janus kinase
(JAK2)V617F mutation
Total DNA was isolated from bone marrow MNCs using
the QIAamp DNA Blood Mini kit (Qiagen GmbH, Hilden, Germany),
according to the manufacturer's protocols. DNA quality was assessed
using a Bio-Rad Experion electrophoretogram instrument (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The purity of the DNA was
determined by measuring the optical density of the 260/280 ratio
using a NanoDrop spectrophotometer. DNA samples were then stored at
−20°C for further use. All DNA samples were amplified in the C1000
Touch™ Thermal Cycler (Bio-Rad Laboratories, Inc.) using the Takara
Premix Taq™ HS polymerase chain reaction (PCR) kit, UNG plus
(Takara Bio, Inc., Otsu, Japan). In this system, a 50-µl reaction
mixture contained the following components: 0.25 µl Takara TaqHS
DNA polymerase, 5 µl 10X PCR buffer for UNG plus, 4 µl dU plus dNTP
mixture, 0.5 µl UNG, 1 µl each 20 pmol/µl primer, 2 µl genomic DNA
(200 ng) and 34.25 µl diethyl pyrocarbonate-treated water. The
mixture was amplified at 94°C for 4 min and subsequently subjected
to 36 cycles of 94°C for 45 sec, 60°C for 45 sec and 72°C for 45
sec, and then extended at 72°C for 8 min. Patients and controls
were genotyped by a DNA tetra-primer Amplification Refractory
Mutation Screening (ARMS) assay (11), a method that uses 2 primer pairs to
amplify the normal and mutant sequences plus a positive control
band specifically in a single reaction. Primers included mismatches
to maximize discrimination of the two alleles (in lowercase) and
mutant/wild-type-specific bases (underlined). The PCR primers were
as follows: Forward outer (FO), 5′-TCCTCAGAACGTTGATGGCAG-3′;
reverse outer (RO), 5′-ATTGCTTTCCTTTTTCACAAGAT-3′; forward
wild-type-specific (Fwt), 5′-GCATTTGGTTTTAAATTATGGAGTATaTG-3′, and
reverse-mutant-specific (Rmt), 5′-GTTTTACTTACTCTCGTCTCCACAaAA-3′
(The subsequent amplicons were electrophoresed on a 1.5% agarose
gel and visualized following staining with ethidium bromide. The
samples were visualized with a gel imaging system (Tanon 2500R;
Tanon Science & Technology Co., Ltd. Shanghai, China).
Results
Clinical characteristics and survival
time
Among all the patients with AMKL, the white blood
cell (WBC) count of 3 patients was normal, while 4 patients had a
low WBC count and 2 patients had an increased WBC count. A total of
8 patients exhibited different degrees of anemia, 6 patients
exhibited elevated lactate dehydrogenase levels, 6 patients
exhibited hyperpyrexia and 6 patients exhibited bleeding from the
skin or the mucosa. Peripheral blasts were observed in 6 patients,
with a range of 2–30% (Table I).
 | Table I.Characteristics of patients with acute
megakaryocytic leukemia. |
Table I.
Characteristics of patients with acute
megakaryocytic leukemia.
| Patient no. | Sex | Age, years | WBC count
(×109/l) | RBC count
(×1012/l) | Plts (×109/l) | Hb, g/l | LDH, IU/l | Peripheral blasts,
% | Fever | Bleeding | Splenomegaly | Hepatomegaly | Lymphadenopathy | Sternal
tenderness | Induction
regimen | Survival time,
months |
|---|
| 1 | M | 53 | 2.58 | 1.36 | 6.68 | 48.3 | 773 | 5 | + | + | – | – | + | – | DA | 3 |
| 2 | M | 58 | 2.53 | 1.68 | 42.4 | 50.3 | 138 | 15 | + | + | – | – | – | + | DA | 2 |
| 3 | F | 68 | 5.02 | 3.77 | 324 | 96.6 | 1392 | 30 | + | + | 6 cm below the
rib | – | – | – | DA | 24 |
| 4 | M | 66 | 13.7 | 1.7 | 460 | 73.3 | 1451 | 0 | – | + | Splenectomy for
splenomegaly, 1 year ago at diagnosis | – | – | – | Hu, DA | 20 |
| 5 | F | 66 | 6.39 | 2.47 | 57.4 | 81.3 | 619 | 0 | – | + | 4 cm below the
rib | – | – | – | IA | 10 |
| 6 | M | 54 | 35.0 | 1.88 | 173 | 55.1 | 987 | 10 | – | – | Splenectomy for
trauma, 10 years ago at diagnosis | 5 cm below the
rib | – | – | Hu, IA | 13 |
| 7 | M | 58 | 2.04 | 3.84 | 181 | 121 | 180 | 2 | + | – | – | – | + | – | DA | 6 |
| 8 | M | 53 | 3.62 | 2.0 | 118 | 56.3 | 476 | 2 | + | – | – | – | – | – | DA | 3 |
| 9 | F | 60 | 6.71 | 3.21 | 72.4 | 77.1 | 190 | 0 | + | + | – | – | – | – | IA | 1.1 |
The group consisted of 3 patients who exhibited
marked splenomegaly. Additionally, patient no. 4 underwent
splenectomy at diagnosis. A total of 2 patients exhibited
superficial lymphadenopathy, 1 patient exhibited hepatomegaly
(patient no. 6 underwent splenectomy due to trauma at diagnosis)
and 1 patient exhibited sternal tenderness (Table I).
All patients were treated with cytarabine and
daunorubicin/idarubicin. A total of 4 patients (patient nos. 3–6)
achieved complete remission, 1 patient (patient no. 7) achieved
partial remission and the remaining 4 patients exhibited no
response. The median overall survival time of all patients was 6
months (range, 1.1–24.0 months).
Bone marrow smear and bone marrow
cellularity
On the bone marrow smears, irregular forms of
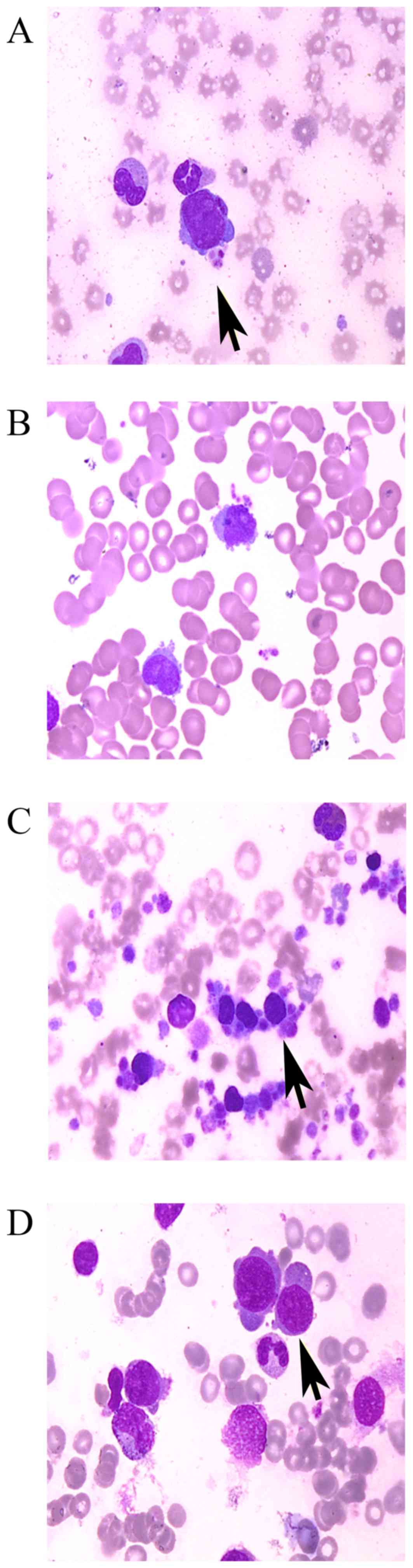
megakaryoblasts, pseudopodia or flocculence were frequently
observed around the edge of the cytoplasm, which was occasionally
accompanied by platelet aggregation (Fig.
1). Patient nos. 3 and 6 exhibited a notable increase in
megakaryoblasts, as demonstrated by bone marrow aspiration, and the
percentage of megakaryoblasts was 24 and 25%, respectively. For the
other patients, it was not possible to accurately identify the
cells as megakaryoblasts by observing cell morphology, therefore,
the percentage of megakaryoblasts was reported as 0%. The bone
marrow cellularity of these 2 patients (patient nos. 3 and 6) was
moderately hypocellular or hypercellular, respectively (Table II). The percentage of bone marrow
blasts in 2 patients (patient nos. 4 and 9) was <5%, while it
was >30% in 4 patients and it ranged from 20–30% in 3 patients
(Table II).
| Table II.Bone marrow smear and cytochemical
staining. |
Table II.
Bone marrow smear and cytochemical
staining.
| Patient no. | Bone marrow
cellularity | Marrow blasts,
% | Megakaryoblasts,
% | No. of
megakaryocytes | POX | PAS | NAE | SBB | NCE |
|---|
| 1 | Hypercellular | 30.0 | 0 | 12 | – | +++ | +++ | N/A | N/A |
| 2 | Hypercellular | 60.5 | 0 | 6 | – | +++ | ++~+++ | – | – |
| 3 | Moderately
hypocellular | 28.0 | 24 | >500 | – | +++ | ++ | – | – |
| 4 | Severely
hypocellular | 2.0 | 0 | 38 | ++ | N/A | N/A | ++ | ++ |
| 5 | Hypercellular | 31.0 | 0 | 89 | – | – | +++ | N/A | N/A |
| 6 | Hypercellular | 29.0 | 25 | >300 | N/A | N/A | N/A | N/A | N/A |
| 7 | Hypercellular | 59.0 | 0 | 21 | – | +++ | ++ | – | – |
| 8 | Moderately
hypercellular | 22.0 | 0 | 14 | – | ++++ | ++ | – | – |
| 9 | Hypercellular | 3.5 | 0 | 143 | N/A | N/A | N/A | N/A | N/A |
Bone marrow biopsy and
immunohistochemical staining
The bone marrow cellularity was also evaluated with
a bone marrow biopsy. A total of 6 patients exhibited moderate or
severe hypocellular bone marrow cellularity. The bone marrow
cellularity of patient nos. 1, 2, 6 and 8 was severely, moderately,
severely and moderately hypocellular, respectively, as demonstrated
by bone marrow biopsy (Table III);
however, the cellularity was hypercellular or moderate
hypercellular, as indicated by bone marrow aspiration (Table II). The cellularity of the other
patients was consistent between the results of the bone marrow
aspiration and bone marrow biopsy. Gomori silver staining
demonstrated that 7 patients had moderate or marked MF, and the
remaining 2 patients were negative for the condition. Additionally,
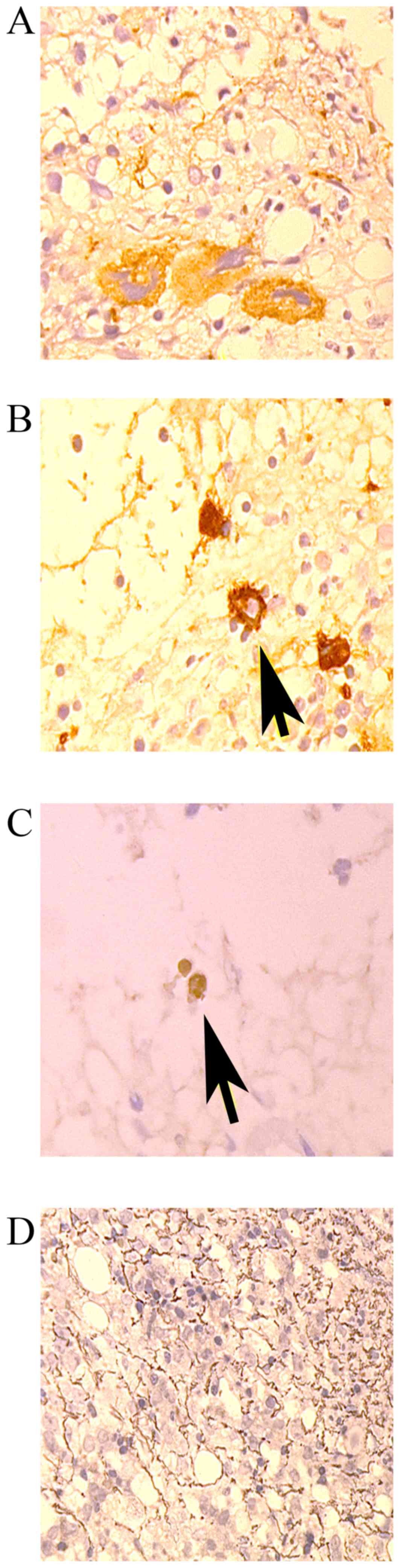
7 patients exhibited the expression of platelet-specific antigens,
including factor VIII, CD61, CD41 or CD42, as demonstrated by
immunohistochemical staining (Fig. 2;
Table III).
| Table III.Bone marrow biopsy and
immunohistochemical stain. |
Table III.
Bone marrow biopsy and
immunohistochemical stain.
| Patient no. | Bone marrow
cellularity | MF | Immunohistochemical
stain |
|---|
| 1 | Severely
hypocellular | 0 | FVIII+/CD61+ |
| 2 | Moderately
hypocellular | 0 |
FVIII+++/CD42+/CD61+ |
| 3 | Moderately
hypocellular | 2 | FVIII+/CD61+ |
| 4 | Severely
hypocellular | 3 | N/A |
| 5 | Hypercellular | 3 |
FVIII+/CD42+/CD61+ |
| 6 | Severely
hypocellular | 2 | N/A |
| 7 | Hypercellular | 3 |
FVIII+/CD42+/CD61+ |
| 8 | Moderately
hypocellular | 2 |
FVIII+/CD41+/CD42+/CD61+ |
| 9 | Hypercellular | 3 |
FVIII+/CD61+/CD42- |
Immunophenotype
Immunophenotypic analysis was performed on all
patients. The blast cells of 6 patients primarily expressed
myeloid-specific antigens, including CD13 and CD33. A total of 3
patients primarily expressed platelet-specific antigens, including
CD41 and CD61. The blast cell percentages of patient no. 4 and
patient no. 9 were <20% by bone marrow smear analysis; however,
the percentage of CD41- and CD61-positive cells was >90%, as
demonstrated by flow cytometry. In 5 patients, the percentage of
CD34-positive cells was >20% (Table
IV).
| Table IV.Immunophenotype of patients with
acute megakaryocytic leukemia. |
Table IV.
Immunophenotype of patients with
acute megakaryocytic leukemia.
| Patient no. | CD2, % | CD3, % | CD4, % | CD7, % | CD10, % | CD13, % | CD14, % | CD19, % | CD33, % | CD34, % | CD41, % | CD61, % | CD64, % | MPO, % | CD117, % | HLA-DR, % |
|---|
| 1 | 2 | 6 | 39 | 44 | 2 | 63 | 19 | 2 | 88 | 0 | 17 | 15 | 24 | 28 | 12 | 46 |
| 2 | 0 | 0 | 0 | 0 | 57 | 44 | 0 | 0 | 54 | 0 | N/A | N/A | 0 | 84 | 17 | 15 |
| 3 | 2 | 0 | 2 | 3 | 2 | 10 | 5 | 0 | 62 | 62 | N/A | N/A | 22 | 29 | 9 | 60 |
| 4 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 2 | 22 | 99 | 99 | 0 | 0 | 0 | 5 |
| 5 | 0 | 0 | 30 | 78 | 2 | 89 | 2 | 0 | 95 | 88 | N/A | N/A | 10 | 4 | 0 | 9 |
| 6 | 0 | 2 | 7 | 0 | 0 | 3 | 0 | 0 | 17 | 24 | 92 | 92 | 0 | 5 | 8 | 10 |
| 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 89 | 0 | N/A | N/A | 0 | 2 | 13 | 52 |
| 8 | 0 | 0 | 0 | 2 | 39 | 32 | 0 | 1 | 80 | 0 | N/A | N/A | 64 | 81 | 12 | 16 |
| 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 40 | 98 | 98 | 0 | 6 | 3 | 3 |
Karyotypic analysis
A total of 6 patients exhibited an abnormal
karyotype. Additionally, in 5 patients (5/6 patients) complex
karyotypes were observed. The karyotypes of patient nos. 3 and 5
contained the translocation t(9;22)(q34;11.2) (Table V).
| Table V.Karyotype of patients with acute
megakaryocytic leukemia. |
Table V.
Karyotype of patients with acute
megakaryocytic leukemia.
| Patient no. | Karyotype |
|---|
| 1 |
73,XYY,+2,der(5)t(5;19)(q10;q10),+der(5)t(5;19),-6,-7,+8,+8,-9,-11,-13,+15,+15,+15,-16,18,+19,+mar1×2[6]/74,idem,+add(5)(q10)[3]/73,idem,-6,+8,+11,der(8;17)(q10;q10)[3]/72,idem,
der(8;19)(q10;p10),+13,-15,
del(20)(q11.2q12)[2]/74,idem,t(8;8)(q13;p13),+13[3]/46,XY,
der(4)t(4;9)(p16;q10),-9,
der(20)(t9;20)(p10;q13.3),+mar2[2]/46,XY[5] |
| 2 | 46,XY |
| 3 |
46,XY,t(9;22)(q34;q11.2)[30]/92,
idemx2[2]/184,idemx4[2] |
| 4 |
46,XYdel(13)(q12q14)[20] |
| 5 |
63,XXX,-4,-5,-7,-9,t(9;22)(q34;q11.2),+10,der(12;21)(p10;p10),-17,-18,-20,+21,der(22)t(9;22)[13]/63,sl,
der(12;21),+add(12)(q11.1)[2]/65,sdl1,+12,-add(12),+21,+der(22)t(9;22)[5] |
| 6 | 46,XY |
| 7 | 46,XY |
| 8 |
43,X,-Y,del(5)(q11.1),-13,add(14)(p11.1),add(15)(p11.1),add(16)(p13.3),-17,-18,-19,+mar1,+mar2[5]/46,XY[15] |
| 9 |
44,XX,i(5)(p10),-7,-12,der(21)t(?;21)(?;q22)t(?;12)(?;q13)[10]/43,idem,-3,+add(12)(p11.1),add(14)(q32),-15,+21,-der(21)[10] |
JAK2V617F mutation
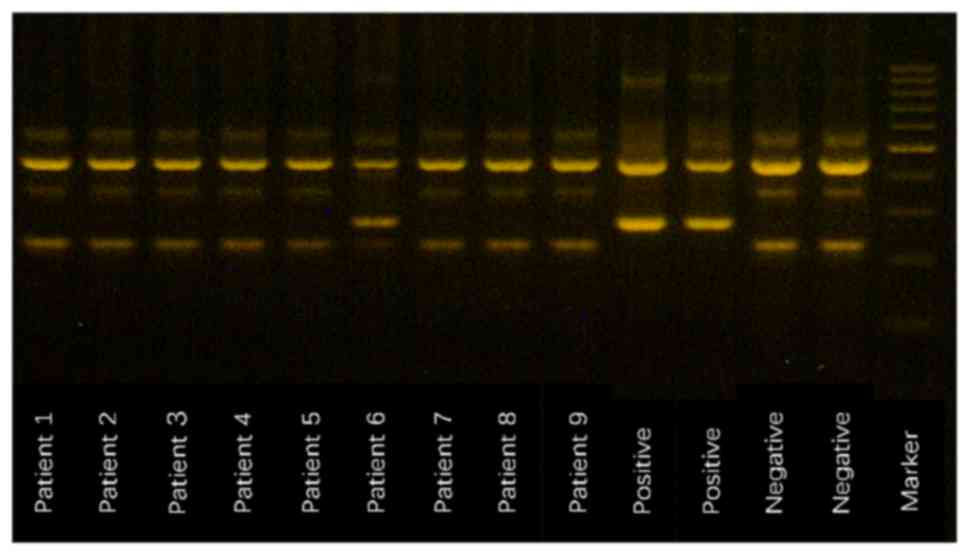
Primers FO and RO flanked the JAK2 exon 12 and
generated a control 463-bp band in all patients. Primers Fwt and RO
produced a 229-bp wild-type-specific product, and primers FO and
Rmt generated a 279-bp mutant-specific product. Patient no. 6 was
positive for the JAK2V617F mutation (Fig.
3).
Discussion
AMKL is a rare subtype of AML comprising only ~1% of
all AML cases (12,13). In China, a limited number of patients
had been diagnosed with AMKL due to its low incidence. In the
present study, only 1.4% of patients with AML were diagnosed with
AMKL. In the clinic, AMKL may not be considered due to its low
incidence. When clinicians diagnose AMKL, bone the marrow biopsy
frequently exhibits proliferation of abnormal megakaryoblasts and
extensive fibrosis (3).
Immunohistochemical staining and flow cytometry frequently express
platelet-specific antigens, including factor VIII, CD41, CD42 or
CD61; however, no specific cytogenetic abnormalities are detected
in patients with AMKL, including abnormalities of chromosomes 3 and
8, and translocations, such as t(8;17), t(1;5) and t(10;17), which
have previously been reported in adult patients with AML-M7
(14–17).
In the clinic, patients may be diagnosed with AMKL
upon finding >20% of blasts and >50% megakaryoblasts among
all blasts, as determined by bone marrow aspiration; however, the
diagnosis of AMKL is frequently challenging due to a high incidence
of MF, resulting in the failure of the bone marrow aspiration and
inconsistency between aspiration and biopsy. In the present study,
the bone marrow cellularity of patient nos. 1, 2, 6 and 8 was
hypocellular to different degrees upon bone marrow biopsy; however,
the cellularity was hypercellular upon bone marrow aspiration.
Primarily, in other types of AML, if the bone marrow aspiration
demonstrates hypocellular bone marrow cellularity, the result of
the biopsy is frequently hypercellular; however, in AMKL, due to
extensive MF, following bone marrow aspiration, the samples
selected by biopsy are frequently insufficient, therefore the
opposite result occurs.
In the present study, 7 patients presented with
moderate or extensive MF, which frequently results in ‘dry taps’
and diagnosis inaccuracy in the morphological examination of bone
marrow; thus, in these 7 patients with MF, it was difficult to
obtain the exact number of blast cells. Therefore, >20% blasts
and >50% megakaryoblasts among the blasts were not essential
diagnostic criteria for AMKL, and it was necessary to conduct a
comprehensive diagnosis that depended on the results of bone marrow
biopsy, flow cytometry and immunohistochemical staining.
Furthermore, the diagnosis could not be confirmed under conditions
where the percentage of blast cells was <20% (18). In the present study, the percentage of
blast cells in patient no. 4 and patient no. 9 was <5%, and this
outcome may be associated with severe MF, which is demonstrated
with strong positive Gomori staining or dilution of bone marrow
blood; however, flow cytometry demonstrated the percentage of blast
cells was >20%, and the expression levels of CD41 or CD61 were
significantly increased (both >90%). Immunohistochemical
staining also demonstrated that the expression of factor VIII was
notably positive in patient no. 9, therefore leading to a diagnosis
of AMKL.
Due to the rare incidence of AMKL and cost-saving
considerations, conventional antibody combinations for surface
antigens in diagnosing AML do not include CD41 and CD61, which may
result in a missed diagnosis of AMKL. The 9 patients in the present
study were not tested for the antigens of CD41 and CD61 at first,
which made the diagnosis challenging depending on the results of
cell morphology only. Finally, only 4 patients (patient nos. 1, 4,
6 and 9) underwent detection of expression levels of CD41 and CD61
by flow cytometry, and these patients were diagnosed with AMKL. The
remaining 5 patients did not have data available to determine the
expression levels of CD41 and CD61 antigens as they were not
requested by the clinicians. In these 5 cases, the final diagnosis
of AMKL depended on bone marrow biopsy and immunohistochemical
staining. We suggest that when immunophenotypic analysis fails to
diagnose AMKL, bone marrow biopsies should be analyzed by
immunocytochemical staining for platelet-specific antigens,
including factor VIII, CD41, CD42 or CD61. Compared with other
types of leukemia, flow cytometry, bone marrow biopsy and
immunocytochemical staining produce more notable results when
patients are diagnosed with AMKL; thus, a comprehensive diagnosis
in AMKL should be emphasized. We recommend that CD41 and CD61
analysis should be contained in the diagnosis of AMKL, if
available.
Furthermore, clinical presentations may indicate the
cause of AMKL and assist clinicians with diagnosing this type of
disease accurately. In AMKL, 20–30% of patients frequently exhibit
splenomegaly and hepatomegaly, without lymphadenopathy (12,13). In
the present 9 patients, 3 exhibited splenomegaly, 1 exhibited
hepatomegaly, possibly due to compensatory hepatomegaly (this
patient underwent splenectomy for trauma 10 years ago prior to
diagnosis), and 2 patients exhibited superficial lymphadenopathy.
Patient no. 4 underwent splenectomy 1 year ago prior to diagnosis
for unexplained splenomegaly and intolerable symptoms, and the
diagnosis has now been confirmed as AMKL. It was considered that
this patient may have had a history of myeloproliferative neoplasm
(MPN). The patient was diagnosed with AMKL; however they may have
had a history of MPN, meaning that AMKL may have transformed from
MPN, therefore, they were diagnosed with secondary AMKL.
Notably, chromosomal karyotypic analysis may also
indicate the cause of AMKL. Patient nos. 3 and 5 exhibited
translocation of chromosomes 9 and 22, t(9;22), which primarily
occurs in chronic myeloid leukemia (CML) (19). In patients with AMKL, the t(9;22)
chromosomal translocation may be exhibited in the acute stages of
CML (20). Patient nos. 3 and 5 had
no prior history of CML, and it is possible that CML was not
previously diagnosed; thus, the diagnosis may be the blast crisis
of CML. In the clinic, primary AMKL with Philadelphia chromosome is
rare; therefore, this type of disease is frequently considered as
secondary AMKL (21). Secondary AMKL
is frequently reported from the transformation of CML, essential
thrombocytosis (ET) and primary MF (PV) (20,22,23). In
the present study, 2 patients exhibited the Philadelphia chromosome
(patient nos. 3 and 5), and patient no. 4 had a history of
splenomegaly, which may be associated with MPNs. Furthermore, it
was considered that the proportion of secondary AMKL was notably
high. Additionally, all patients also underwent the detection of
the JAK2 gene mutation, for which patient no. 6 was positive. This
patient underwent a splenectomy due to trauma 10 years ago at
diagnosis, and whether or not the spleen was enlarged at the time
was unclear. However, JAK2V617F is frequently exhibited in ET and
PV (11,24,25), but
not in AMKL, according to at least one report (26). Thus, it was speculated that the
transformed AMKL of this patient was secondary to MPN.
In previous reports, patients with AMKL have short
survival times and a poor prognosis, with median survival times
that range from 5–10 months (12,13,15,27).
In the 9 patients in the present study, the median survival time
was 6 months, which was consistent with the median survival time
reported from other groups outside of mainland China (12,13,15,27).
Thus, it was considered that the survival time may be associated
with the high proportion of secondary AMKL, bone marrow fibrosis
and a complex karyotype. If patients with AMKL receive a regimen of
cytarabine combined with anthracycline, ~50% patients could achieve
complete remission (15). Compared
with traditional chemotherapy, allogeneic hematopoietic stem cell
transplantation is a better choice in patients with complete
remission, and benefits could be observed from this mode of therapy
(28); however, the recurrence rate
is high and the survival time is notably short (29). Novel treatment regimens require
investigation for this type of disease in the future.
Acknowledgements
The authors would like to thank the Sino-U.S.
Shanghai Leukemia Cooperative Group for their efforts.
Funding
The present study was supported by a grant awarded
by the Key Construction Building Subject of a Three-Year Action
Plan of the Fourth Round of a Public Health Project, Shanghai,
China (grant no. 15GWZK0801).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JG and XW designed the experiments; GZ performed and
analyzed most of the experiments; WW performed some experiments; GZ
wrote the manuscript. All authors read and approved the final
manuscript.
Ethical approval and consent to
participate
All procedures performed in studies involving human
participants were approved by the Ethics Committee of Huashan
Hospital, Fudan University and in accordance with the 1964 Helsinki
declaration and its later amendments or comparable ethical
standards. Informed consent was obtained from all individual
participants included in the study.
Patient consent for publication
Informed consent was obtained from all individual
participant included in the study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Von Boros J and Korenyi A: Uber einen fall
von akuter megakaryocyblasten-leukamie, zugleich einige bemerkungen
zum Problem der akuten leukemie. Z Klin Med. 118:679–718. 1931.(In
German).
|
|
2
|
Breton-Gorius J, Reyes F, Duhamel G,
Najman A and Gorin NC: Megakaryoblastic acute leukemia:
Identification by the ultrastructural demonstration of platelet
peroxidase. Blood. 51:45–60. 1978.PubMed/NCBI
|
|
3
|
Bennett JM, Catovsky D, Daniel MT,
Flandrin G, Galton DA, Gralnick HR and Sultan C: Criteria for the
diagnosis of acute leukemia of megakaryocyte lineage (M7). A report
of the French-American-British Cooperative Group. Ann Intern Med.
103:460–462. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Swerdlow SH, Campo E, Harris NL, Jaffe ES,
Pileri SA, Stein H, Thiele J and Vardiman JW: WHO Classification of
Tumours of Haematopoietic and Lymphoid Tissues. 4th edition. Lyon:
pp. 136–137. 2008
|
|
5
|
den Ottolander GJ, te Velde J, Brederoo P,
Geraedts JP, Slee PH, Willemze R, Zwaan FE, Haak HL, Muller HP and
Bieger R: Megakaryoblastic leukaemia (acute myelofibrosis): A
report of three cases. Br J Haematol. 42:9–20. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Strauchen JA: Criteria for the diagnosis
of acute megakaryocytic leukemia. Blood. 97:18982001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shibata A, Bennett JM, Castoldi GL,
Catovsky D, Flandrin G, Jaffe ES, Katayama I, Nanba K, Schmalzl F,
Yam LT, et al: Recommended methods for cytological procedures in
haematology. International committee for standardization in
haematology (ICSH). Clin Lab Haematol. 7:55–74. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gruppo RA, Lampkin BC and Granger S: Bone
marrow cellularity determination: Comparison of the biopsy,
aspirate, and buffy coat. Blood. 49:29–31. 1977.PubMed/NCBI
|
|
9
|
Thiele J, Kvasnicka HM, Facchetti F,
Franco V, van der Walt J and Orazi A: European consensus on grading
bone marrow fibrosis and assessment of cellularity. Haematologica.
90:1128–1132. 2005.PubMed/NCBI
|
|
10
|
Gonzalez GJ and Meza-Espinoza JP: Use of
the International System for Human Cytogenetic Nomenclature (ISCN).
Blood. 108:3952–3953. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jones AV, Kreil S, Zoi K, Waghorn K,
Curtis C, Zhang L, Score J, Seear R, Chase AJ, Grand FH, et al:
Widespread occurrence of the JAK2 V617F mutation in chronic
myeloproliferative disorders. Blood. 106:2162–2168. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pagano L, Pulsoni A, Vignetti M, Mele L,
Fianchi L, Petti MC, Mirto S, Falcucci P, Fazi P, Broccia G, et al:
Acute megakaryoblastic leukemia: Experience of GIMEMA trials.
Leukemia. 16:1622–1626. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tallman MS, Neuberg D, Bennett JM,
Francois CJ, Paietta E, Wiernik PH, Dewald G, Cassileth PA, Oken MM
and Rowe JM: Acute megakaryocytic leukemia: The Eastern Cooperative
Oncology Group experience. Blood. 96:2405–2411. 2000.PubMed/NCBI
|
|
14
|
Duchayne E, Fenneteau O, Pages MP, Sainty
D, Arnoulet C, Dastugue N, Garand R and Flandrin G: GroupeFrançais
d'Hématologie Cellulaire; Groupe Français de Cytogénétique
Hématologique: Acute megakaryoblastic leukaemia: A national
clinical and biological study of 53 adult and childhood cases by
the Groupe Francais d'Hematologie Cellulaire (GFHC). Leuk Lymphoma.
44:49–58. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oki Y, Kantarjian HM, Zhou X, Cortes J,
Faderl S, Verstovsek S, O'Brien S, Koller C, Beran M, Bekele BN, et
al: Adult acute megakaryocytic leukemia: An analysis of 37 patients
treated at M.D. Anderson Cancer Center. Blood. 107:880–884. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ahmad F, Dalvi R, Das BR and Mandava S:
Novel t(8;17)(q23; q24.2) and t(9;22) (p24.1;q12.2) in acute
megakaryoblastic leukemia AML-M7 subtype in an adult patient.
Cancer Genet Cytogenet. 193:112–115. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang E and Stoecker M: ‘Cannibalistic’
phagocytosis in acute megakaryoblastic leukemia (AML M7) with
t(10;17) (p15; q22). Leuk Lymphoma. 51:1944–1947. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hahn AW, Li B, Prouet P, Giri S, Pathak R
and Martin MG: Acute megakaryocytic leukemia: What have we learned.
Blood Rev. 30:49–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Druker BJ: Translation of the Philadelphia
chromosome into therapy for CML. Blood. 112:4808–4817. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Karkuzhali P, Shanthi V and Usha T: A case
of chronic myeloid leukaemia presenting as megakaryocytic blast
crisis (AML M7). Ecancermedicalscience. 7:3752013.PubMed/NCBI
|
|
21
|
Akahoshi M, Oshimi K, Mizoguchi H, Okada
M, Enomoto Y and Watanabe Y: Myeloproliferative disorders
terminating in acute megakaryoblastic leukemia with chromosome 3q26
abnormality. Cancer. 60:2654–2661. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Radaelli F, Mazza R, Curioni E, Ciani A,
Pomati M and Maiolo AT: Acute megakaryocytic leukemia in essential
thrombocythemia: An unusual evolution. Eur J Haematol. 69:108–111.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mesa RA, Li CY, Kettering RP, Schroeder
GS, Knudson RA and Tefferi A: Leukemic transformation in
myelofibrosis with myeloid metaplasia: A single-institution
experience with 91 cases. Blood. 105:973–977. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vianelli N, Baravelli S and Gugliotta L:
Acute megakaryoblastic transformation of essential thrombocythemia.
Haematologica. 81:288–289. 1996.PubMed/NCBI
|
|
25
|
Patiño-Sarcinelli F, Knecht H, Pechet L,
Pihan G, Savas L and Snyder LM: Leukemia with megakaryocytic
differentiation following essential thrombocythemia and
myelofibrosis. Case report and review of the literature. Acta
Haematol. 95:122–128. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Swaminathan S, Madkaikar M, Ghosh K,
Vundinti BR, Kerketta L and Gupta M: Novel immunophenotypic and
morphologic presentation in acute myeloid leukemia (AML) with JAK2
V617F mutation. Eur J Haematol. 84:180–182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Giri S, Pathak R, Prouet P, Li B and
Martin MG: Acute megakaryocytic leukemia is associated with worse
outcomes than other types of acute myeloid leukemia. Blood.
124:3833–3834. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Garderet L, Labopin M, Gorin NC, Polge E,
Baruchel A, Meloni G, Ortega J, Vossen J, Bunjes D, Leverger G, et
al: Hematopoietic stem cell transplantation for de novo acute
megakaryocytic leukemia in first complete remission: A
retrospective study of the European Group for Blood and Marrow
Transplantation (EBMT). Blood. 105:405–419. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ishiyama K, Yamaguchi T, Eto T, Ohashi K,
Uchida N, Kanamori H, Fukuda T, Miyamura K, Inoue Y, Taguchi J, et
al: Acute megakaryoblastic leukemia, unlike acute erythroid
leukemia, predicts an unfavorable outcome after allogeneic HSCT.
Leuk Res. 47:47–53. 2016. View Article : Google Scholar : PubMed/NCBI
|