Introduction
Ewing's sarcoma (ES) commonly affects the growth of
metaphyseal bones. Although primary ES of the spine is rare
(1), it is commonly observed in the
sacrum. The highest incidence of ES is observed in patients in
their 20s, and mostly involves the long bones and the pelvis.
Spinal ES is divided into two groups: i) The sacral type, which
includes the sacrum and coccyx, the incidence of which is <5%
(2) and ii) the non-sacral type,
which includes the cervical, dorsal and lumbar vertebrae (3). The incidence of non-sacral ES is
>0.9%. In the majority of cases, vertebrae are affected
following the metastasis of ES, which originates elsewhere. It is
very rare to encounter primary vertebral ES if the sacrum is
excluded. Surgery combined with chemotherapy and radiotherapy to
control the progression of neurological deficits is associated with
a preferable outcome (3). The
treatment of ES is challenging, and there is currently no global
uniform treatment standard. The present report describes a case of
a middle aged patient with multiple ES in the spinal canal of
T12-L3 who was admitted to the First Affiliated Hospital of
Guangdong Pharmaceutical University (Guangzhou, China) in November
2016.
Case report
A 60-year-old male Chinese patient presented with
pain resembling electric shock in the waist and buttocks, which
occurred intermittently for 1 month, and incontinence for 1 week. A
neurophysical examination demonstrated weakness of the lower
extremities (power grade IV/V), decreased sensation below the ankle
joint (the right side was more affected compared with the left
side), two-sided knee tendon hyperreflexia and a positive Babinski
sign. Magnetic resonance imaging (MRI) is a medical imaging
technique used in radiology to form pictures of the anatomy and the
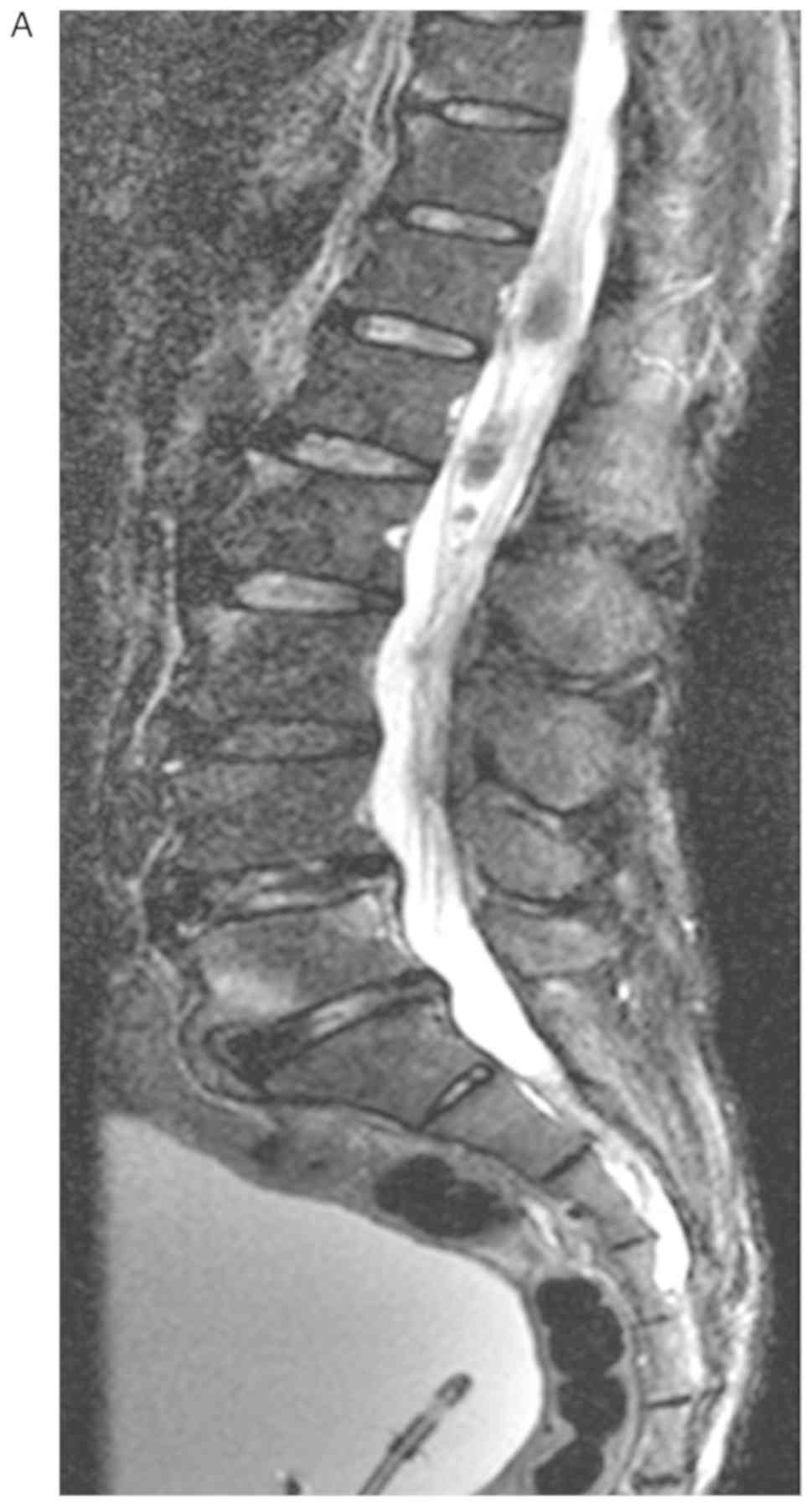
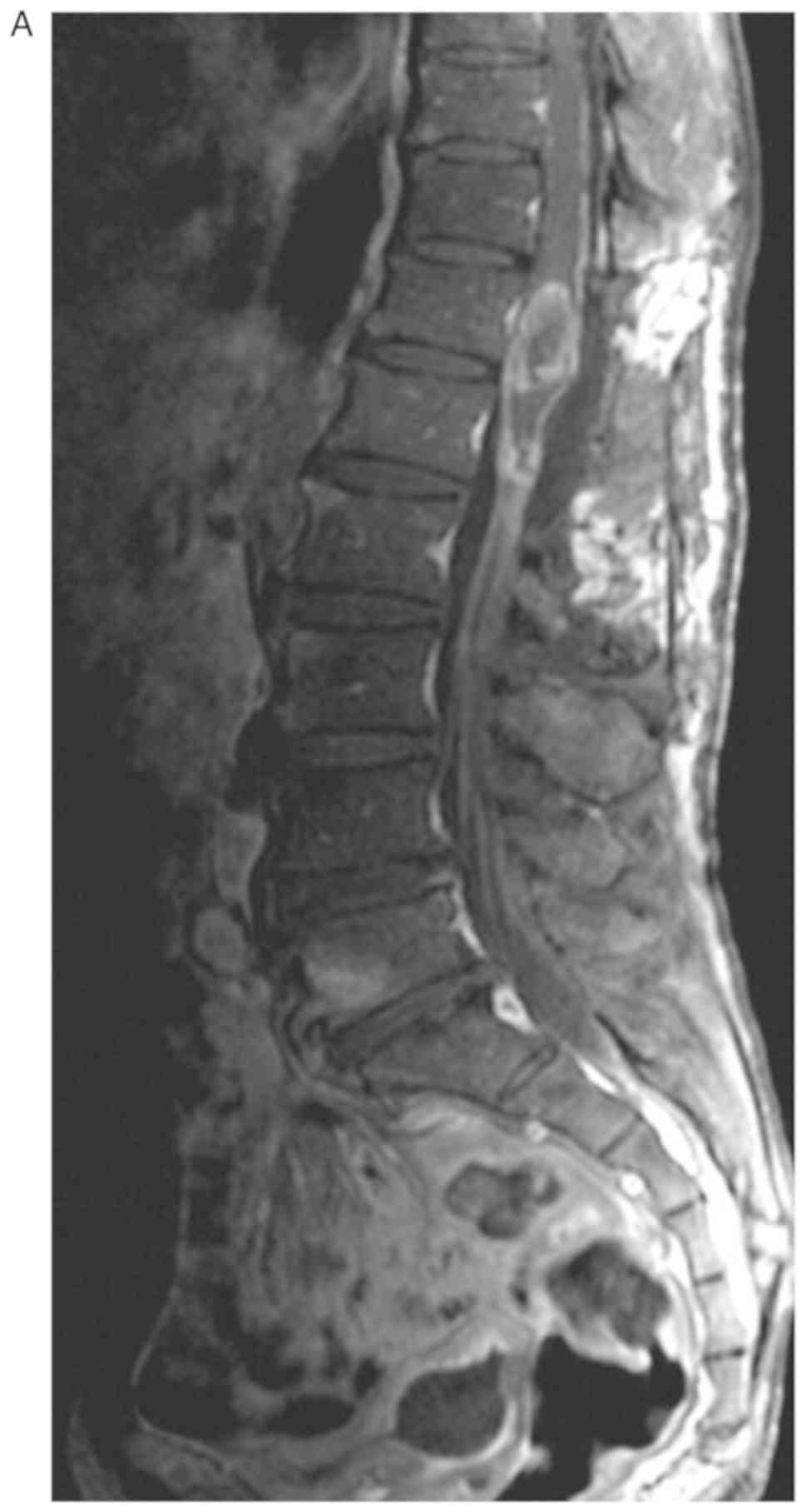
physiological processes of the body (4). MRI of the patient revealed multiple
inhomogeneous, oval-shaped nodules in the intradural and cauda
equina spaces of T12-L3. The largest nodule was ~23×11×10 mm, with
a high signal in T2-weighted, T2 spectral presaturation with
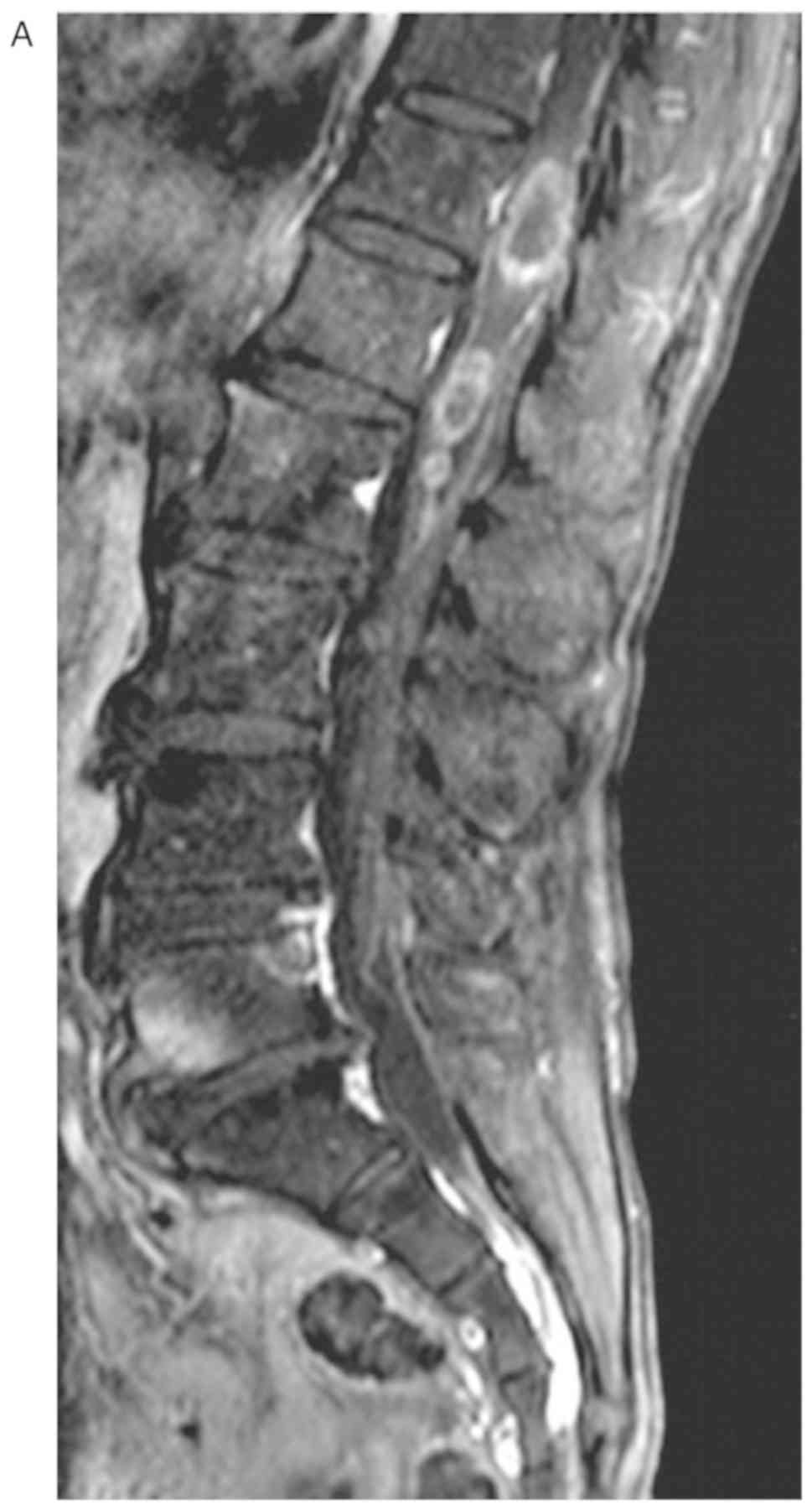
inversion recovery and T1-weighted imaging (Fig. 1), which was inhomogeneously
reinforced following gadolinium injection (Fig. 2). The oedema signal of T1 and T2 was
strip-like in the adjacent thoracic cord. The preoperative
diagnosis was spinal cord or cauda equina occupying lesions,
considering multiple metastases of the cauda equina in the spinal
canal of T12-L3. The patient underwent a T12-L3 laminectomy.
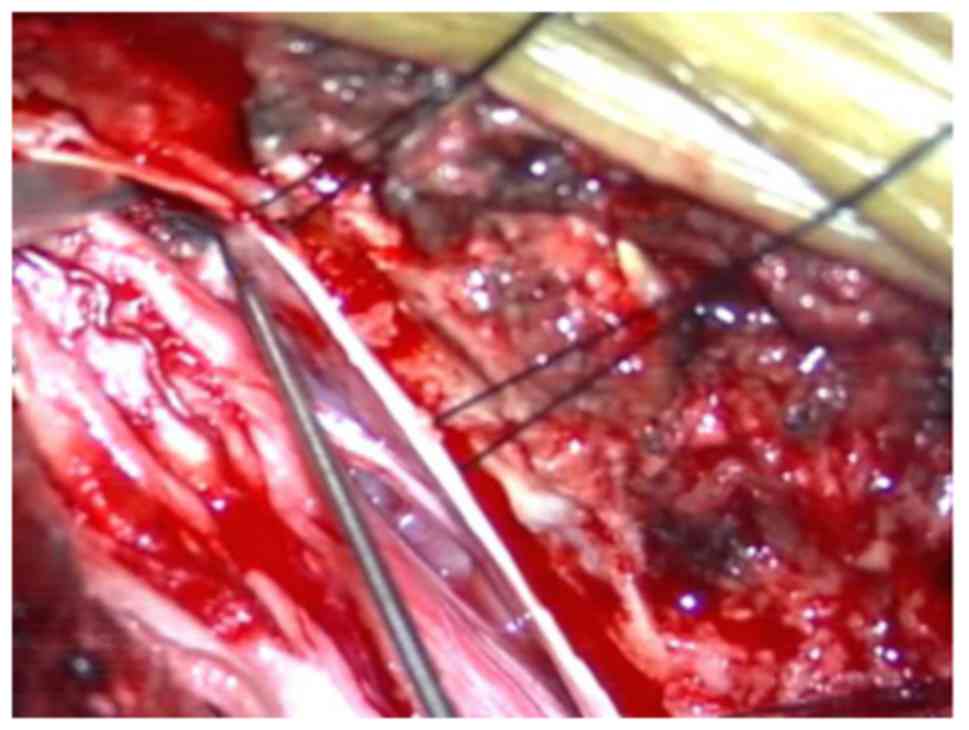
Opening of the dura mater revealed an expansion of the conus
medullaris and two non-enveloped, soft, greyish-white occupied
lesions of the cauda equina (Fig.
3). The occupied lesions were partly abscised using
microsurgical tools. Sarcoma was diagnosed with frozen section
analysis while the surgery was being performed. The nerve tracts
were saved, and the remaining occupied lesions were completely
removed (Fig. 4). Opening of the
white expansion of the conus medullaris revealed soft,
greyish-brown tissue (Fig. 5). The
tissue was removed using microsurgical tools, and the
histopathological results were consistent with ES. Postoperative
MRI did not detect the existence of a remaining tumour. Soft tissue
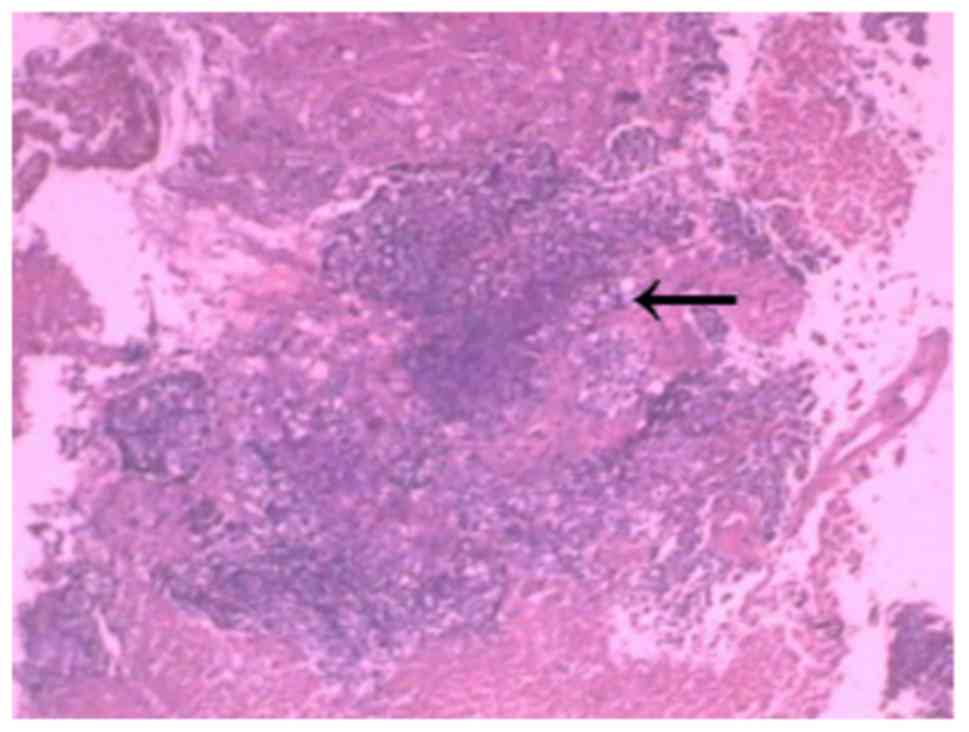
swelling was presented after surgery (Fig. 6). Immunohistochemistry of the tumour
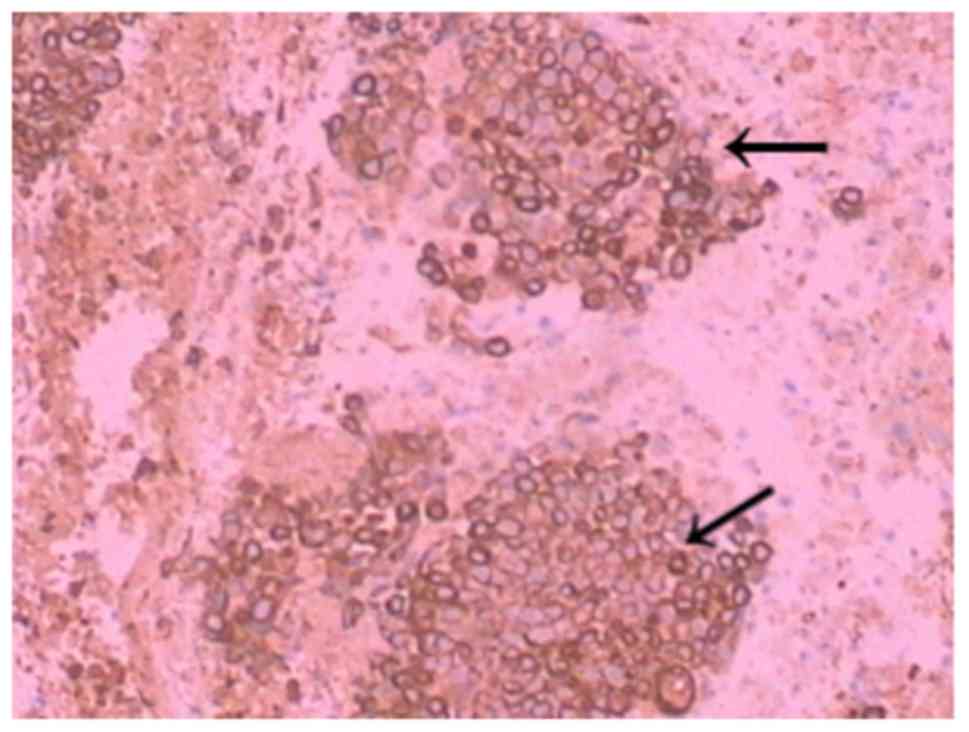
revealed hypercellular areas, tissue with monomorphic small blue
circular cells lacking cytoplasm with focal clearing of the
cytoplasm arranged in sheets and compact nest patterns (Fig. 7). The tumour cells were not arranged
in well-formed rosettes. The round-to-oval nuclei had finely
distributed chromatin and small nucleoli. Mitotic figures were
infrequent. Additionally, foci of necrosis, haemorrhage and oedema
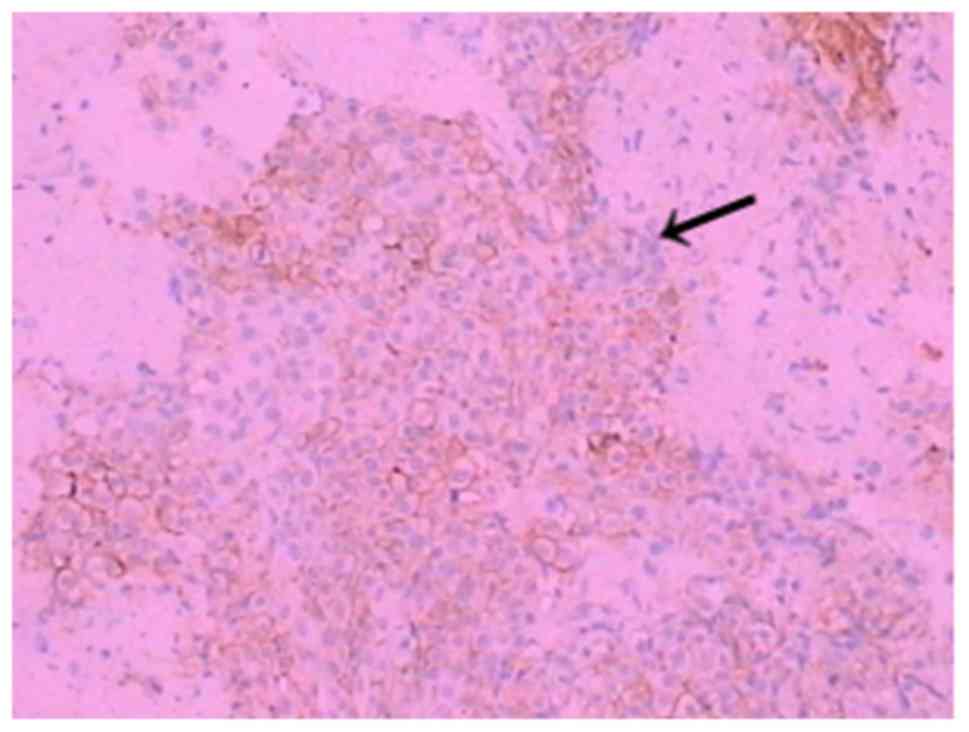
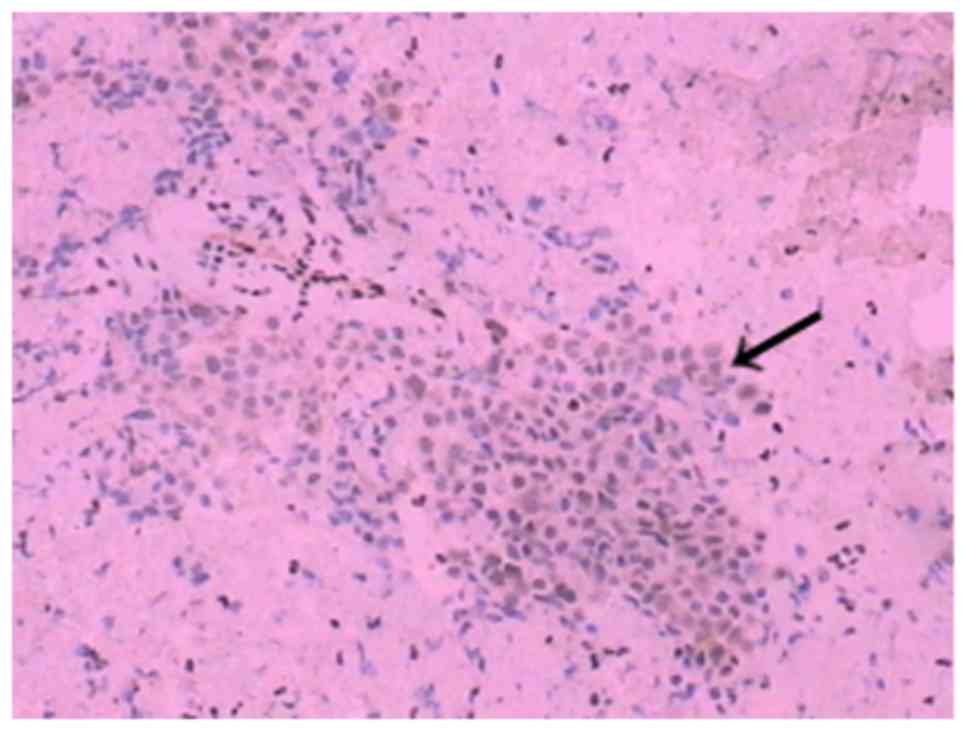
within the tumour were observed. Immunohistochemistry was performed
on tissue sections to investigate the presence of certain proteins
in tumour cells. The antibodies (OriGene Technologies, Inc.) used
in the immunohistochemistry experiment were monoclonal antibodies
secreted by a B lymphocyte clone. The immunohistochemical analysis
revealed positivity for cluster of differentiation 99 (CD99; clone
PCB1) (Fig. 8), foetal-liver
infusion 1 (FLI-1; clone MRQ-1) (Fig.
9), cytokeratin (CK; clone AE1/AE3) (Fig. 10), negativity for CD34, CD20, CD3,
SOX-10, glial fibrillary acidic protein, progesterone receptor,
α-fetoprotein, CD117, placental alkaline phosphatase, synapsin,
glycoprotein hormone α chain, prostate-specific antigen, napsin A,
transcription termination factor 1, interleukin-12 subunit β and
tumour protein P63, and 80% of the cells were positive for Ki-67,
which supported the diagnosis. Based on the patient's positive
immunohistochemistry results, an MRI at the corresponding position
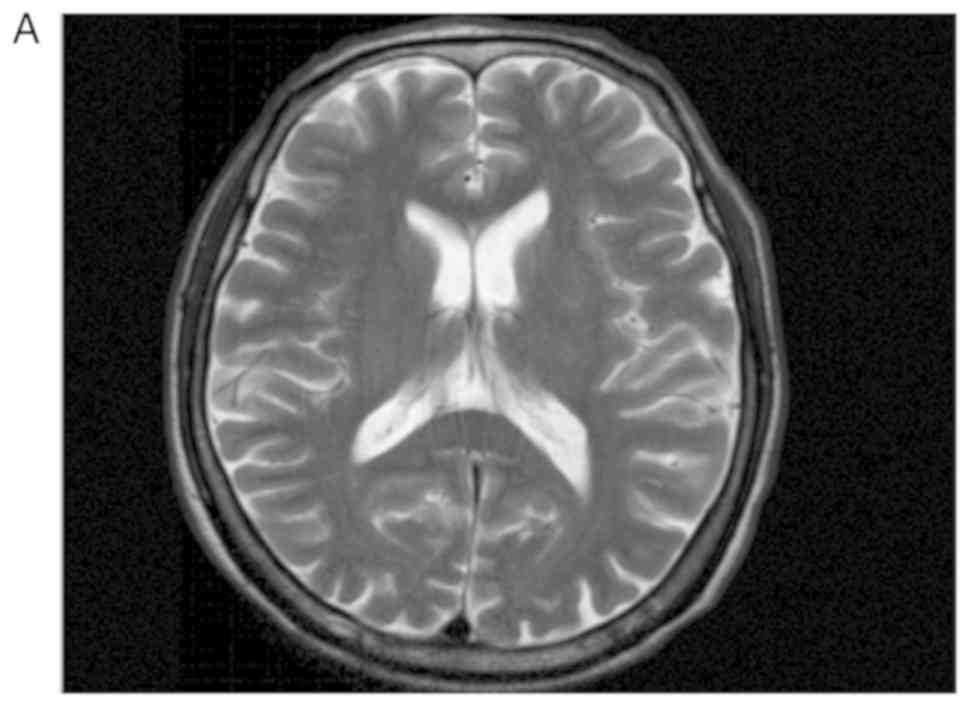
was performed to identify metastatic tumors and primary lesions. No
suspicious lesions were identified in the patient's brain, lungs or
prostate (Fig. 11). Fluorescence
in situ hybridization (FISH) is a molecular cytogenetic
technique that uses fluorescent probes that bind to the parts of a
nucleic acid sequence with a high degree of sequence
complementarity; fluorescence microscopy can be used to detect the
fluorescent probe bound to the chromosomes (5,6). Genetic
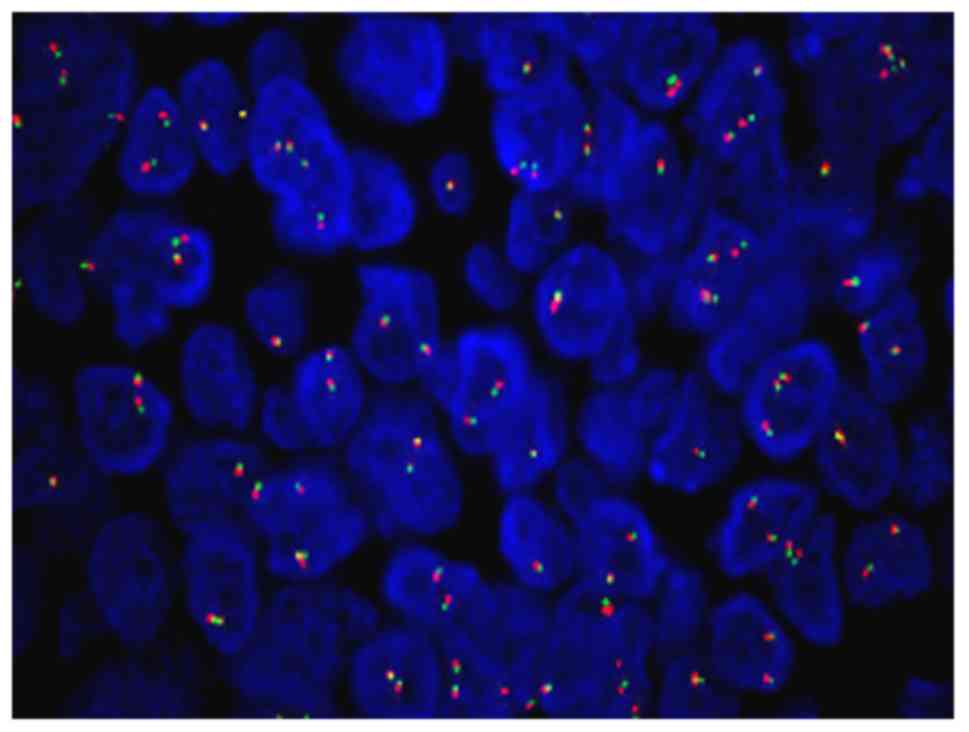
FISH analysis of the Ewing's sarcoma breakpoint region 1 (EWSR1)
gene in 200 interphase cells from the patient demonstrated no
specific cytogenetic abnormalities. Each signal pattern exhibited
the following: 2F, 32.0%; 1F, 10%; 3F, 45.0%; 4F, 4.0%; 1G1R1F,
2.5%; 1G1R2F, 2.0% and 5F, 4.5% (F, G and R stand for fusion, green
and red, respectively; Fig. 12).
FISH analysis shows fusion cell <10%, indicating that the
specimen was considered FISH-negative. Adjuvant chemotherapy was
suggested; the patient accepted radical surgery followed by
combination chemotherapy, but the disease continued to progress.
Following chemotherapy treatment, the patient suffered from
depression and refused any further treatment; the patient was lost
to follow-up as they did not return.
Discussion
ES is a developmental tumour characterized by
balanced chromosomal translocations and the formation of new fusion
genes. ES is an aggressive tumour with high occurrence of
metastasis in children and young teenagers, and is caused by
chromosomal fusion in EWSR1 genes (7). ES can affect any bone, but mostly
affects the lower extremities (45%), followed by the pelvis (20%),
upper extremities (13%), axial skeleton and ribs (13%) or face (2%)
(7). The femur is affected the most
frequently, especially in the midshaft. However, ES is rarely
observed in the spinal canal. Typically, the tumour consists of
small circular cells with regular circular nuclei containing finely
dispersed chromatin and inconspicuous nucleoli, as well as a narrow
rim of a clear or pale cytoplasm, which can be observed by light
microscopy (2). Ultrastructural
examination demonstrates that the tumour includes primitive cells
with a smooth nuclear surface, scanty organelles and cytoplasmic
glycogen in pools or aggregates (8).
Tumours with similar histology also arise in soft tissues, such as
peripheral primitive neuroectodermal tumours, neuroepitheliomas and
Askin tumours (8). Pain is the most
common symptom in patients with ES. Usually, the disease is
concealed, and the pain may exist for a long time before the
patient seeks medical treatment. Initial pain may be mild and
intermittent and may respond to nonsurgical treatment (8). The average delay between symptom onset
and diagnosis is 34 weeks. According to a previous report, the
average time is 15 weeks between symptom onset and the first visit,
and 19 weeks between the initial visit and a correct diagnosis
(8). If the patient continues to
experience symptoms, a radiographic examination is important as it
can identify the primary lesion, when first diagnosed and during
follow-up. In addition to pain, patients may experience fever,
erythema and swelling (9).
Laboratory tests may reveal an increase in the white blood cell
count, the red blood cell rate (erythrocyte sedimentation rate) and
C-reactive protein, which may be misdiagnosed as osteomyelitis
(9). The patient described in the
present case report was a 60-year-old male patient who admitted to
hospital following a surgical pathology diagnosis. Clinical
literature attributes this type of pathology to rare diseases
(10), and a limited number of
reports of ES in the spine have been published (10,11). ES
is rare not only in middle-aged patients, but also in the spinal
canal. In the present case, no tumours were identified in the
spinal cord or vertebrae; they were distributed in the spinal
canal, and the patient's EWSR1 FISH test was negative. No previous
reports describing similar symptoms, diagnosis and complications
were found in published literature. The diagnosis is further
complicated by the similarity of the biopsy results of ES to those
of pus (11), and the tissue may be
sent to the microbiology department instead of the pathology
department; the majority of biopsy specimens should be sent for
bacterial culture and pathology. A number of common
immunohistochemical markers expressed in ES, such as CD99, FLI-1
and CK, provide valuable support for ES diagnosis. CD99 is a 32 kDa
glycoprotein encoded by the MIC2 gene, and is used to diagnose ES
with high sensitivity (11,12); the sensitivity is 95%, although the
specificity is low (12–14). According to a previous report by
Vural et al (14), FLI-1
expression was detected in 7/8 (87.5%) ES cases, and CD99
expression was detected in 10/11 (90%) ES cases. CD99 is the most
sensitive immunohistochemical marker for ES. However, the
expression of these markers has also been described in
T-lymphoblastic lymphoma, rhabdomyosarcoma, synovial sarcoma and
small cell anaplastic osteosarcoma (15–18).
Therefore, ES may be misdiagnosed if based solely on the expression
of CD99. FLI-1, as well as CK, is sensitive but less specific for
ES compared with CD99 (11,13). Previous studies have demonstrated
(19–25) that both markers are expressed in
various other round cell tumours (19). The immunohistochemistry results of at
least CD99 and FLI-1 are primarily markers of high specificity in
the diagnosis of ES (20–22).
EWSR1 is the most commonly translocated gene in
sarcoma and is associated with a variety of mesenchymal lesions,
such as ES, proliferative small circle cell tumour, clear cell
sarcoma, vascular-like fibres, histiocytoma, extramedullary
mucinous chondrosarcoma and mucinous liposarcoma (23–25). The
detection of genetic FISH showed rupture abnormalities of the EWSR1
gene can assist in the differential diagnosis of ES and peripheral
primitive neuroectodermal tumours; however, positivity does not
necessarily indicate ES, and not all cases of ES are
EWSR1-positive, which suggests that EWSR1 is not specific to ES
(26,27). EWSR1 rearrangement can be visualized
by FISH; as soft tissue ES is diagnostically challenging, FISH
analysis is a useful confirmatory diagnostic tool (28). However, as in the majority of
instances in which a split-apart approach is used, the results of
molecular genetics must be evaluated in the context of morphology.
The patient described in the present report was subjected to FISH
analysis to detect a fusion partner, but no rearrangements were
identified. According to the presence of the EWSR1 gene
rearrangement in only one of the three carcinomas (Ewing's sarcoma
and hidradenoma of the skin and mucoepidermoid carcinoma of the
salivary glands) studied by Möller et al (28), it may be concluded that the patient
described in the present study was negative for EWSR1 gene
rearrangement, suggesting that other mechanisms may be involved in
the pathogenesis of this tumour type. This case was sent to
Professor Anjia Han at the Department of Pathology of the First
Affiliated Hospital of Sun Yat-sen University (Guangzhou, China)
and other professors, who considered the lesion to be a malignant
tumour, but did not exclude ES or metastatic cancer. Therefore,
this case may be described as an EWSR1-negative ES in the primary
spinal canal. Surgical treatment of tumours in the spine, pelvis or
even the spinal canal is controversial (29). In addition to surgery and
chemotherapy, several novel molecular targets for ES treatment have
recently been identified and investigated in preclinical and
clinical settings; treatments targeting the function of receptor
tyrosine kinases, fusion protein EWSR1 and mTOR have demonstrated
promising results (30). There has
also been an increasing interest in the immune responses of
patients with ES; immunotherapies using T cells, NK cells, cancer
vaccines and monoclonal antibodies have been considered for ES,
especially for recurrent cases (30). In the patient described in the
present report, due to the sudden occurrence of spinal cone
constriction and following multidisciplinary consultation, as well
as consultation with the patient and his family, it was decided to
immediately perform surgery to remove the tumour mass and relieve
the spinal cord compression. The median survival time of patients
with all stages of ES is 26.14 months, and the median survival of
patients in the late metastatic stage is 5.6 months (31); although no randomized studies of ES
occurrence in the spinal canal have been published,
ifosfamide-based chemotherapy may have a positive impact on overall
survival (32). A multidrug
chemotherapy regimen for bone ES treatment can be administered;
however, different responses to chemotherapy regimens have been
observed. The most effective drugs include vincristine, actinomycin
D, high-dose cyclophosphamide, doxorubicin, etoposide and
ifosfamide (32). In the present
case, radical surgery was performed, followed by the administration
of combination chemotherapy, but the disease continued to progress.
Thus, the best solution for each patient may be established
following extensive discussions with the patient and his/her
family.
In conclusion, the present report described for the
first time a rare case of a Chinese patient with localized ES
occurring in the spinal canal, as confirmed by genetic and
ultrastructure analyses. This case and previous reports have
revealed that surgery combined with chemotherapy and radiotherapy
may contribute to significant improvements in survival; however,
for each patient, the best treatment plan can be established
through discussions with the patient and his/her family.
Acknowledgements
The authors would like to thank Professor Anjia Han
from the Department of Pathology of the First Affiliated Hospital
of Sun Yat-sen University (Guangzhou, China).
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
DZ conceived the study. DY acquired the data and
performed the literature search. DY and JZ designed the study and
analysed the data. DY and DZ prepared and edited the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethical Committee of
the First Affiliated Hospital of Guangdong Pharmaceutics University
(Guangzhou, China). The study conformed to the relevant regulatory
standards.
Patient consent for publication
Written informed consent was obtained from the
patient for this study. All patient identifiers have been
removed.
Competing interests
The authors state that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ES
|
Ewing's sarcoma
|
|
CD
|
cluster of differentiation
|
|
FLI-1
|
foetal-liver infusion 1
|
|
CK
|
cytokeratin
|
|
MRI
|
magnetic resonance imaging
|
References
|
1
|
Zucman J, Delattre O, Desmaze C,
Plougastel B, Joubert I, Melot T, Peter M, De Jong P, Rouleau G,
Aurias A, et al: Cloning and characterization of the Ewing's
sarcoma and peripheral neuroepithelioma t(11;22) translocation
breakpoints. Genes Chromosomes Cancer. 5:271–277. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Haresh KP, Chinikkatti SK, Prabhakar R,
Rishi A, Rath GK, Sharma DN and Julka PK: A rare ease of intradural
extramedullary Ewing's sarcoma with skip metastasis in the spine.
Spinal Cord. 46:582–584. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gardner LJ, Ayala AG, Monforte HL and
Dunphy CH: Ewing sarcoma/peripheral primitive neuroectodermal tumor
adult abdominal tumors with an Ewing sarcoma gene rearrangement
demonstrated by fluorescence in situ hybridization in paraffin
sections. Appl Immunohistochem Mol Morphol. 12:160–165. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ramos-Vara JA and Miller MA: When tissue
antigens and antibodies get along: Revisiting the technical aspects
of immunohistochemistry-the red, brown, and blue technique. Vet
Pathol. 51:42–87. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pernthaler A, Pernthaler J and Amann R:
Fluorescence in situ hybridization and catalyzed reporter
deposition for the identification of marine bacteria. Appl Environ
Microbiol. 68:3094–3101. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wagner M, Horn M and Daims H: Fluorescence
in situ hybridisation for the identification and characterisation
of prokaryotes. Curr Opin Microbiol. 6:302–309. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grier HE: The Ewing family of tumors.
Ewing's sarcoma and primitive neuroectodermal tumors. Pediatr Clin
North Am. 44:991–1104. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burchill SA: Ewing's sarcoma: Diagnostic,
prognostic, and therapeutic implications of molecular
abnormalities. J Clin Pathol. 56:96–102. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iwamoto Y: Diagnosis and treatment of
Ewing's sarcoma. Jpn J Clin Oncol. 37:79–89. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ozturk E, Mutlu H, Sonmez G, Vardar Aker
F, Cinar Basekim C and Kizilkaya E: Spinal epidural extraskeletal
Ewing sarcoma. J Neuroradiol. 34:63–67. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hung YP, Fletcher CD and Hornick JL:
Evaluation of NKX2-2 expression in round cell sarcomas and other
tumors with EWSR1 rearrangement: Imperfect specificity for Ewing
sarcoma. Mod Pathol. 29:370–380. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ahmed AA, Nava VE, Pham T, Taubenberger
JK, Lichy JH, Sorbara L, Raffeld M, Mackall CL and Tsokos M: Ewing
sarcoma family of tumors in unusual sites: Confirmation by rt-PCR.
Pediatr Dev Pathol. 9:488–495. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Llombart-Bosch A, Machado I, Navarro S,
Bertoni F, Bacchini P, Alberghini M, Karzeladze A, Savelov N,
Petrov S, Alvarado-Cabrero I, et al: Histological heterogeneity of
Ewing's sarcoma/PNET: An immunohistochemical analysis of 415
genetically confirmed cases with clinical support. Virchows Arch.
455:397–411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vural C, Uluoğlu O, Akyürek N, Oğuz A and
Karadeniz C: The evaluation of CD99 immunoreactivity and EWS/FLI1
translocation by fluorescence in situ hybridization in central
PNETs and Ewing's sarcoma family of tumors. Pathol Oncol Res.
17:619–625. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ambros IM, Ambros PF, Strehl S, Kovar H,
Gadner H and Salzer-kuntschik M: MIC2 is a specific marker for
Ewing's-sarcoma and peripheral primitive neuroectodermal tumors.
Evidence for a common histogenesis of Ewing's sarcoma and
peripheral primitive neuroectodermal tumors from MIC2 expression
and specific chromosome aberration. Cancer. 67:1886–1893. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gerald WL, Ladanyi M, de Alava E,
Cuatrecasas M, Kushner BH, LaQuaglia MP and Rosai J: Clinical,
pathologic, and molecular spectrum of tumors associated with
t(11;22)(p13;q12): Desmoplastic small round-cell tumor and its
variants. J Clin Oncol. 16:3028–3036. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Devaney K, Vinh TN and Sweet DE:
Small-cell osteosarcoma of bone: An immunohistochemical study with
differential diagnostic considerations. Hum Pathol. 24:1211–1225.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Riopel M, Dickman PS, Link MP and Perlman
EJ: MIC2 analysis in pediatric lymphomas and leukemias. Hum Pathol.
25:396–399. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mhawech-Fauceglia P, Herrmann FR, Bshara
W, Odunsi K, Terracciano L, Sauter G, Cheney RT, Groth J,
Penetrante R and Mhawech-Fauceglia P: Friend leukaemia
integration-1 expression in malignant and benign tumours: A
multiple tumour tissue microarray analysis using polyclonal
antibody. J Clin Pathol. 60:694–700. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Saxena R, Sait S and Mhawech-Fauceglia P:
Ewing sarcoma/primitive neuroectodermal tumor of the kidney: A case
report. Diagnosed by immunohistochemistry and molecular analysis.
Ann Diagn Pathol. 10:363–366. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhong J, Chen N, Chen X, Gong J, Nie L, Xu
M and Zhou Q: Peripheral primitive neuroectodermal tumor of the
kidney in a 51-year-old female following breast cancer: A case
report and review of the literature. Oncol Lett. 9:108–112. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pinto A, Dickman P and Parham D:
Pathobiologic markers of the Ewing sarcoma family of tumors: State
of the art and prediction of behaviour. Sarcoma. 2011:8561902011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mazur MA, Gururangan S, Bridge JA,
Cummings TJ, Mukundan S, Fuchs H, Larrier N and Halperin EC:
Intracranial Ewing sarcoma. Pediatr Blood Cancer. 45:850–856. 1999.
View Article : Google Scholar
|
|
24
|
Llombart-Bosch A, Machado I, Navarro S,
Bertoni F, Bacchini P, Alberghini M, Karzeladze A, Savelov N,
Petrov S, Alvarado-Cabrero I, et al: Histological heterogeneity of
Ewing's sarcoma/PNET: An immunohistochemical analysis of 415
genetically confirmed cases with clinical support. Virchows Arch.
455:397–411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Weidner N and Tjoe J: Immunohistochemical
profile of monoclonal antibody O13: Antibody that recognizes
glycoprotein p30/32MIC2 and is useful in diagnosing Ewing's sarcoma
and peripheral neuroepithelioma. Am J Surg Pathol. 18:486–494.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lessnick SL and Ladanyi M: Molecular
pathogenesis of Ewing sarcoma: New therapeutic and transcriptional
targets. Annu Rev Pathol. 7:145–159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Romeo S and Dei Tos AP: Soft tissue tumors
associated with EWSR1 translocation. Virchows Arch. 456:219–234.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Möller E, Stenman G, Mandahl N, Hamberg H,
Mölne L, van den Oord JJ, Brosjö O, Mertens F and Panagopoulos I:
POU5F1, encoding a key regulator of stem cell pluripotency, is
fused to EWSR1 in hidradenoma of the skin and mucoepidermoid
carcinoma of the salivary glands. J Pathol. 215:78–86. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sankar S, Bell R, Stephens B, Zhuo R,
Sharma S, Bearss DJ and Lessnick SL: Mechanism and relevance of
EWS/FLI-mediated transcriptional repression in Ewing sarcoma.
Oncogene. 32:5089–5100. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Naing A, LoRusso P, Fu S, Hong DS,
Anderson P, Benjamin RS, Ludwig J, Chen HX, Doyle LA and Kurzrock
R: Insulin growth factor-receptor (IGF-1R) antibody cixutumumab
combined with the mTOR inhibitor temsirolimus in patients with
refractory Ewing's sarcoma family tumors. Clin Cancer Res.
18:2625–2631. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ladenstein R, Pötschger U, Le Deley MC,
Whelan J, Paulussen M, Oberlin O, van den Berg H, Dirksen U, Hjorth
L, Michon J, et al: Primary disseminated multifocal Ewing sarcoma:
Results of the Euro-EWING 99 trial. J Clin Oncol. 28:3284–3291.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Grier HE, Krailo MD, Tarbell NJ, Link MP,
Fryer CJ, Pritchard DJ, Gebhardt MC, Dickman PS, Perlman EJ, Meyers
PA, et al: Addition of ifosfamide and etoposide to standard
chemotherapy for Ewing's sarcoma and primitive neuroectodermal
tumor of bone. N Engl J Med. 348:694–701. 2003. View Article : Google Scholar : PubMed/NCBI
|