Introduction
Oral squamous cell carcinoma (OSCC) is one of the
most common malignant epithelial tumors that arises in the oral
cavity (estimated 263,900 new cases in 2008 worldwide) (1). Although combination treatments
employing surgical resection and adjuvant treatments such as
radiotherapy and/or chemotherapy have improved, the disease-free
and overall survival rates in advanced OSCC have not improved
(2,3).
Periodontitis is a common chronic oral inflammatory
disease that causes destruction of periodontal tissues (4). Recent studies have demonstrated that
patients with periodontal disease have a 2–5-fold greater risk of
developing OSCC (4). One of the
major pathogens in periodontal disease is the gram-negative,
anaerobic bacterium Porphyromonas gingivalis. It can adhere
to both epithelial cells and gingival fibroblasts, and induce the
expression of pro-inflammatory cytokines involved in the
progression of periodontitis. Its primary virulence factor is
lipopolysaccharide (LPS) (5), which
has recently been revealed to play an important role in the
migration, invasion, lymphangiogenesis and metastasis of various
types of malignant tumor, including OSCC (6,7).
The induction of cyclooxygenase-2 (COX-2), an enzyme
that promotes cell proliferation and invasion, and suppresses
apoptosis (8), is considered to be
one of the key mechanisms by which LPS affects OSCC cells (9,10).
Upregulated COX-2 is frequently observed in head and neck squamous
cell carcinoma (HNSCC) (11), where
it is associated with a lower survival rate in patients with this
disease (12). Inhibition of COX-2
thus represents a potential approach for OSCC therapy. Celecoxib is
a selective COX-2 inhibitor that has recently been demonstrated to
possess antitumor effects in HNSCC (13,14);
however, its method of administration in OSCC has not yet been
established.
In the present study, to elucidate the association
between chronic periodontitis and OSCC progression, the effect of
P. gingivalis-derived LPS on OSCC cell proliferation was
examined both in vitro and in vivo. The antitumor
effects of celecoxib in an LPS-stimulated OSCC xenograft were
evaluated by assessing cell proliferation, apoptosis and the cell
cycle. Finally, the present study discusses the molecular
mechanisms underlying chronic periodontitis and OSCC, and the
therapeutic potential of celecoxib.
Materials and methods
Cell culture
The human OSCC cell line HSC-3 (Japanese Cancer
Research Resources Bank) was maintained in α-minimum essential
medium (α-MEM; Invitrogen; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Biowest), 100 IU/ml
penicillin and 100 mg/ml streptomycin (Invitrogen; Thermo Fisher
Scientific, Inc.) at 37°C in a 5% CO2 atmosphere.
Cell viability assay
The effect of celecoxib (Combi-Blocks, Inc.) or
P. gingivalis' LPS (InvivoGen, Inc.) on cell viability was
assessed using an MTS assay. Celecoxib was dissolved in dimethyl
sulfoxide (DMSO; Wako Pure Chemical Industries, Ltd.). The levels
of DMSO were minimized to avoid any potential DMSO-associated toxic
effects in the MTS assay. In all cases, the final DMSO
concentration was <0.1% in the cell cultures. HSC-3 cells were
plated at a density of 5,000 cells/well in 96-well plates and
incubated for 48 h at 37°C. Cell cultures were exposed to celecoxib
(0, 100 and 200 µM), LPS (10 µg/ml), or a combination of the two,
for 24 or 48 h. Following this, 20 µl of CellTiter 96 AQueous One
Solution Reagent (Promega Corporation) was added to each well and
incubated for 2 h at 37°C. The absorbance at 490 nm in each well
was then determined (SpectraMax M5; Molecular Devices, LLC).
Western blot analyses of COX-2 and
p53
HSC-3 cells were lysed in RIPA lysis buffer (Santa
Cruz Biotechnology, Inc.) containing 2 mM phenylmethylsulfonyl
fluoride, 1 mM sodium orthovanadate and 2% protease inhibitor
cocktail. The protein concentration was quantified by DC Protein
Assay (Bio-Rad Laboratories, Inc.). Protein samples (10 µg/lane)
were separated using 10–20% gradient gels and electro-transferred
to Immun-Blot PVDF Membranes (Bio-Rad Laboratories, Inc.).
Membranes were blocked using EzBlock Chemi (1:5; ATTO Corporation)
for 1 h at room temperature and then probed using the appropriate
primary antibodies, including anti-COX-2 (1:1,000; catalog no.
ab15191; Abcam), anti-p53 (1:1,000; catalog no. ab1101; Abcam) and
anti β-actin (1:1,000; catalog no. 8H10D10; Cell Signaling
Technology, Inc.) overnight at 4°C. After washing with
Tris-buffered saline with Tween 20 (TBS-T) three times, the
membranes were incubated with a horseradish peroxidase
(HRP)-conjugated AffiniPure goat anti-mouse IgG antibody (1:10,000;
catalog no. 115-035-072; Jackson ImmunoResearch Laboratories, Inc.)
for anti-p53 and anti β-actin or an HRP-conjugated AffiniPure goat
anti-rabbit IgG antibody (1:10,000; catalog no. 111-035-144;
Jackson ImmunoResearch Laboratories, Inc.) for anti-COX-2 for 1 h
at room temperature. Chemiluminescent signals were developed with
Western Lightning ECL Pro (PerkinElmer, Inc.) and detected using a
cooled charge-coupled device camera (LAS 4000 Mini; GE Healthcare
Life Sciences).
Nude mouse tumor model
A total of 32 5-week-old female nude BALB/c nu/nu
mice (21.4±1.2 g; CLEA Japan, Inc.) were maintained at 21–25°C and
40–70% humidity in a 12-h dark/light cycle, with continuous free
access to food and water. For stimulation, HSC-3 cells were exposed
to 10 µg/ml LPS for 48 h before implantation. HSC-3 and
LPS-stimulated HSC-3 cells (5×106 cells in 50 µl α-MEM)
were then mixed with an equal volume of Matrigel (BD Biosciences)
and injected into the flanks of mice during a short period of
anesthesia with 2% isoflurane (Abbott Pharmaceutical Co., 4 Ltd.).
Tumor-bearing mice were then randomly divided into four groups (n=8
in each group): i) A control group; ii) an LPS-treated group; iii)
a celecoxib-treated group; and iv) an LPS + celecoxib-treated
group. In all cases, tumors were allowed to grow to ~60
mm3 prior to the treatment. The celecoxib-treated group
and LPS + celecoxib-treated group were fed powder feed containing
1,500 ppm celecoxib, as previously described (15); the control and LPS-treated groups
were fed common powder feed alone. Mice were assessed twice a week
for 28 days, and tumor growth was determined by estimating tumor
volume, using the formula 0.5 × length × width2, as
previously described (16). Each
cage contained four mice; the dietary intake of powdered feed per
day in the cage was measured to calculate an estimate of the
dietary intake for each mouse. The limitation associated with this
method of drug administration was that dietary intake was not an
actual dose, but estimated dose. One mouse was euthanized when
bleeding from the ulceration of the tumor was observed. On day 28
post-administration, the mice were euthanized using carbon dioxide
gas (20%/min gradual displacement) and monitored for 5 min to
confirm cardiac arrest, and the tumors were removed along with the
surrounding tissue and overlying skin (2 mm from the tumor) for
subsequent histological examination as previously described
(16). The specimens were fixed
immediately with 4% paraformaldehyde for 24 h at room temperature
and embedded in paraffin. All animal experiments were approved by
the Animal Ethics Committee of the University of Fukui (no. 29105)
and performed in accordance with the Guide for the Care and Use of
Laboratory Animals (published by the National Institutes of Health)
(17).
Immunohistochemistry
The paraffin-embedded tissues were sliced into 4
µm-thick sections. Ki-67 and p21 were stained using indirect
immunoperoxidase staining (ImmPRESS Reagent kit; Vector
Laboratories, Ltd.). Tissue sections were deparaffinized in Clear
Plus (Falma, Co., Ltd.), dehydrated in 100% ethanol for 15 min at
room temperature and autoclaved in 1 mM EDTA 2Na and 10 mM Tris
buffer (pH 9.0) at 95°C for 30 min for antigen retrieval.
Endogenous peroxide activity was eliminated by treatment with 0.3%
H2O2 in methanol for 30 min at room
temperature. The ImmPRESS reagent with 2.5% normal horse serum
(undiluted; Vector Laboratories, Ltd.) was used to block
non-specific immunoreactions for 10 min at room temperature. After
incubation with a primary monoclonal rabbit anti-human antibody
against either Ki-67 (1:100; catalog no. ab16667) or p21 (1:100;
catalog no. ab109520) (both from Abcam) for 2 h at room
temperature, the sections were incubated with ImmPRESS polymer
anti-rabbit IgG reagent (undiluted; cat. no. MP-7800; Vector
Laboratories, Ltd.) for 30 min at room temperature.
Immunoreactivity was visualized using 3,3′-diaminobenzidine
(Dojindo Molecular Technologies, Inc.) for 5 min at room
temperature. The sections were also counterstained with hematoxylin
for 5 min at room temperature. The sections were rinsed in PBS
between all steps. Ki-67-stained or p21-stained tissue sections
were observed under an Olympus AX80 light microscope (Olympus
Corporation) at ×400 magnification, and the number of Ki-67- or
p21-positive cells were counted in each individual microscopic
field. At least five microscopic fields per section were used for
the subsequent analysis.
Cell death assay
The terminal deoxynucleotidyl-transferase- mediated
dUTP nick end labeling (TUNEL) method was used to evaluate
apoptosis using the in situ Apoptosis Detection kit (Takara
Bio, Inc.). Briefly, the sections were deparaffinized for 15 min,
dehydrated in 100% ethanol for 15 min and permeabilized using 10
µg/ml proteinase K (Invitrogen; Thermo Fisher Scientific, Inc.) for
10 min at room temperature. Endogenous peroxide activity was
blocked with 3% H2O2 for 5 min at room
temperature. The sections were incubated with 50 µl labeling
reaction mixture (consisting of TdT Enzyme 5 µl + Labeling Safe
Buffer 45 µl) for 90 min at 37°C and reacted with 70 µl anti-FITC
horseradish peroxidase (undiluted; cat no. MK503; Takara Bio, Inc.)
for 30 min at 37°C. Immunoreactivity was visualized using
3,3′-diaminobenzidine (Dojindo Molecular Technologies, Inc.). The
sections were counterstained with hematoxylin for 5 min at room
temperature. The apoptosis index was determined by calculating the
ratio of the number of TUNEL-positive cells to the total number of
tumor cells (avoiding necrotic tumor areas) from a minimum of five
randomly selected microscopic fields in each individual section
using an Olympus AX80 light microscope (Olympus Corporation) at
×400 magnification as previously described (16).
Statistical analysis
All experiments were independently repeated at least
three times. Numerical values are expressed as the mean ± standard
deviation. Differences between experimental groups were analyzed
using one-way analysis of variance followed by Dunnett's test (cell
viability at 24 and 48 h after celecoxib treatment), Turkey-Kramer
multiple comparison test (cell viability, tumor volumes, mouse body
weights, the expression levels of Ki-67 and p21 and apoptosis index
in response to celecoxib with/without LPS treatment) and unpaired
Student's t-test (cell viability 24 and 48 h after LPS treatment
and the COX-2/β-actin ratio). P<0.05 was considered to indicate
a statistically significant difference.
Results
Effect of celecoxib on HSC-3 cells in
vitro
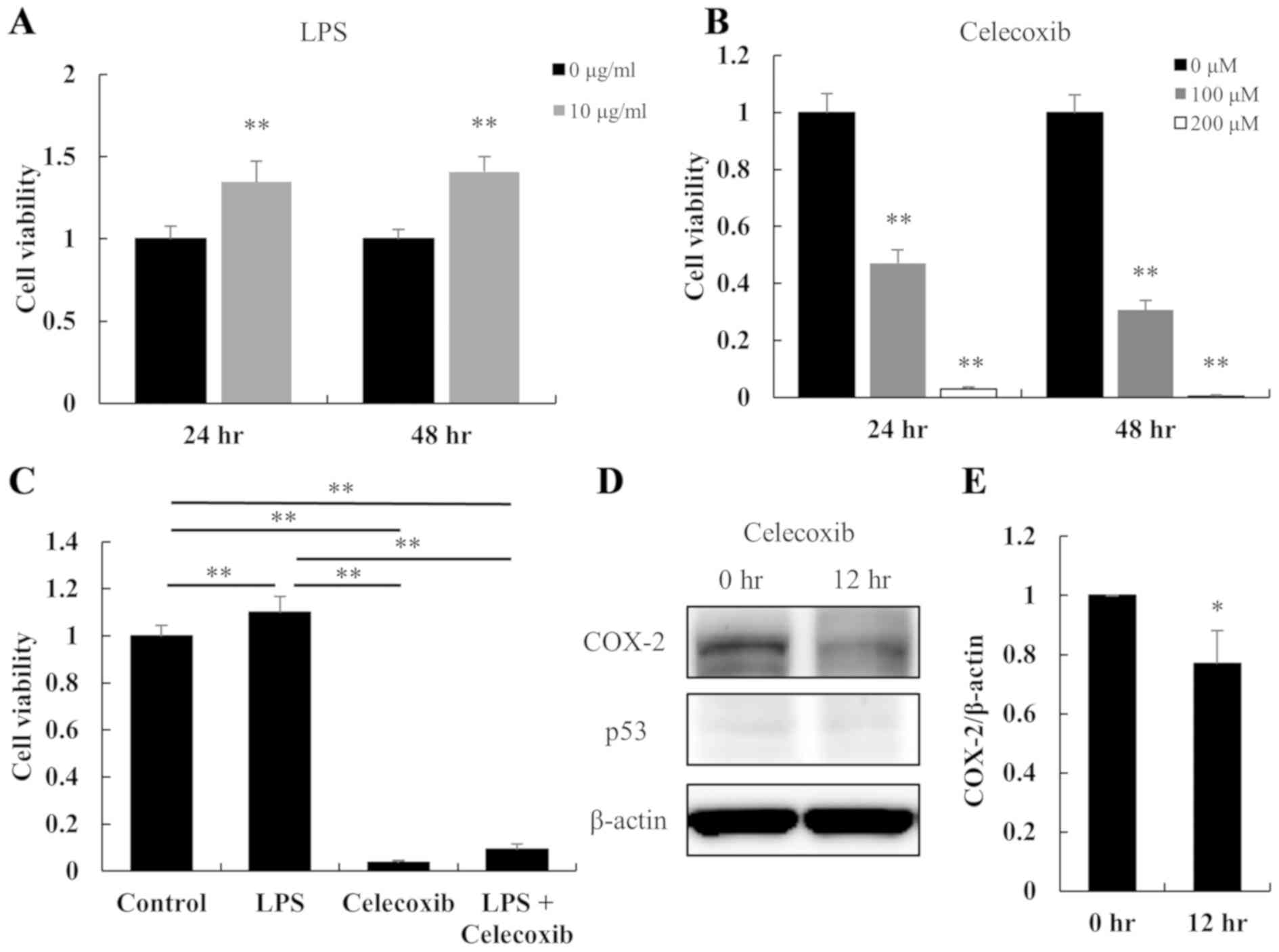
LPS treatment for 24 and 48 h increased the
viability of HSC-3 cells (P<0.01; Fig. 1A); whereas, celecoxib decreased cell
viability in a dose- and time-dependent manner (P<0.01; Fig. 1B), indicating that the cells were
sensitive to celecoxib. The proliferation of LPS-treated HSC-3
cells was significantly inhibited by treatment with celecoxib (100
µM for 48 h) (P<0.01; Fig. 1C).
The protein expression levels of COX-2 and p53 with/without
celecoxib treatment were also examined in HSC-3 cells via western
blotting. Compared with untreated cells, treatment of HSC-3 cells
with 100 µM celecoxib downregulated the protein expression levels
of COX-2 after 12 h, but there was little change in p53 expression
levels (Fig. 1D). The COX-2/β-actin
ratios in the HSC-3 cells were significantly decreased by the
celecoxib treatment (Fig. 1E).
Effect of celecoxib on OSCC tumor
growth in vivo
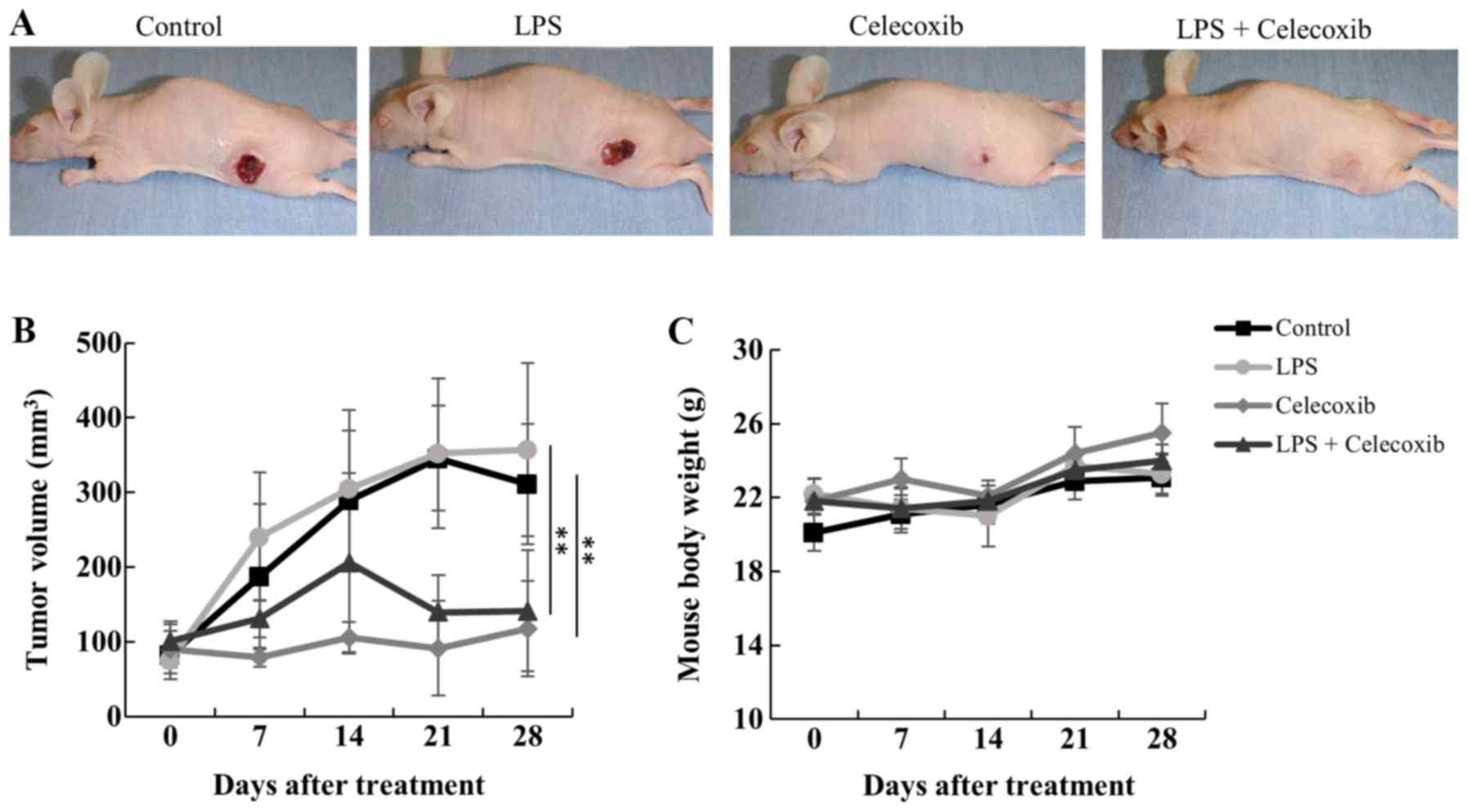
The antitumor effects of celecoxib were examined in
control and LPS-treated OSCC tumors in nude mice. After 28 days of
treatment, tumor volumes were significantly decreased in both
celecoxib-treated and LPS + celecoxib-treated mice compared with
control and LPS-treated mice (P<0.01; Fig. 2A and B). There were no significant
differences body weight in the four treatment groups (Fig. 2C). Dietary intake throughout the
experimental period (3.7–4.6 g/day) is presented in Table I.
 | Table I.Estimated amount of dietary intake of
powdered feed with/without celecoxib. |
Table I.
Estimated amount of dietary intake of
powdered feed with/without celecoxib.
|
| Estimated quantity
of dietary intake per day for each mouse, g/day |
|---|
|
|
|
|---|
| Group | 0–7 days | 8–14 days | 15–21 days | 22–28 days |
|---|
| Control | 3.8 | 4.1 | 4.5 | 4.1 |
| LPS | 4.0 | 4.1 | 4.6 | 4.6 |
| Celecoxib | 4.2 | 4.5 | 4.3 | 4.5 |
| LPS +
celecoxib | 3.8 | 3.7 | 4.3 | 4.1 |
Effect of celecoxib on OSCC cells in
vivo
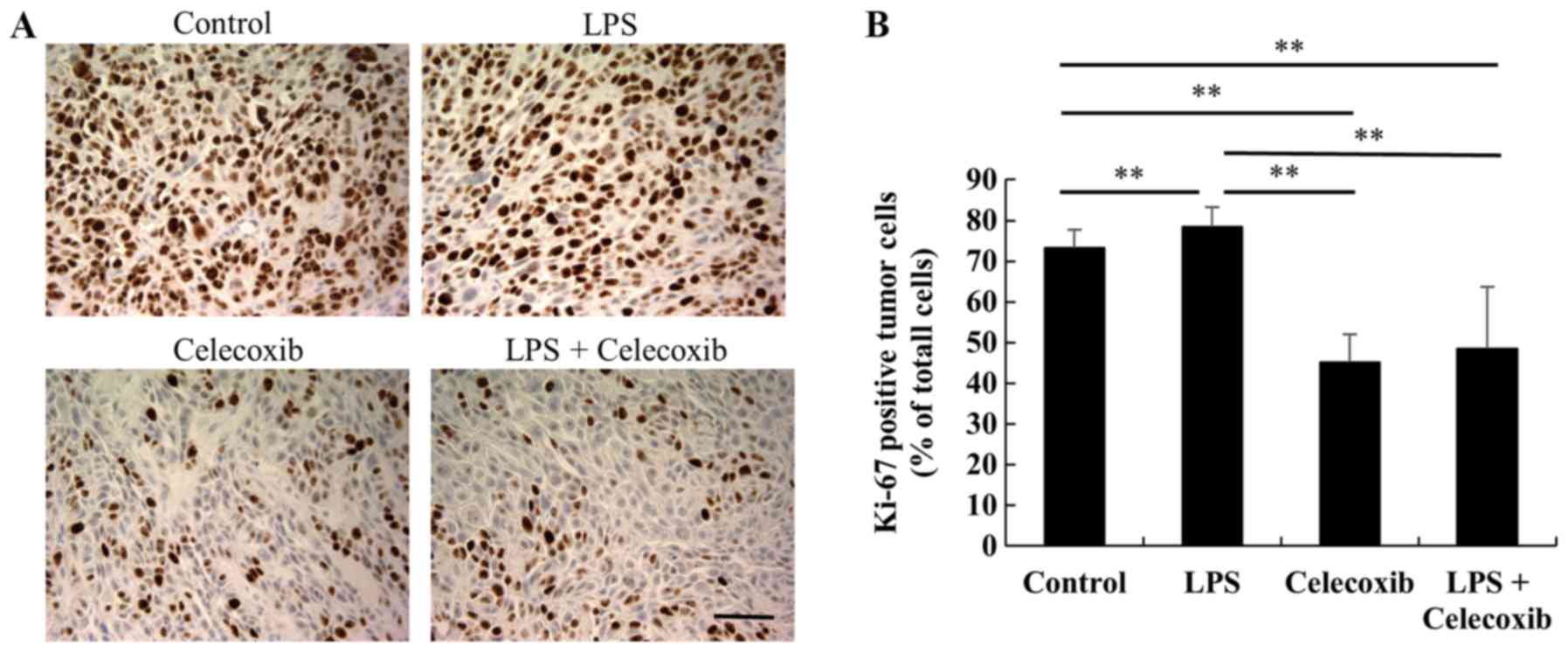
Ki-67 expression levels were significantly decreased
in celecoxib-treated and LPS + celecoxib-treated groups when
compared with the control and LPS-treated groups (P<0.01) and
increased in the LPS-treated group compared with the control group
(P<0.01), indicating that cell proliferation was decreased
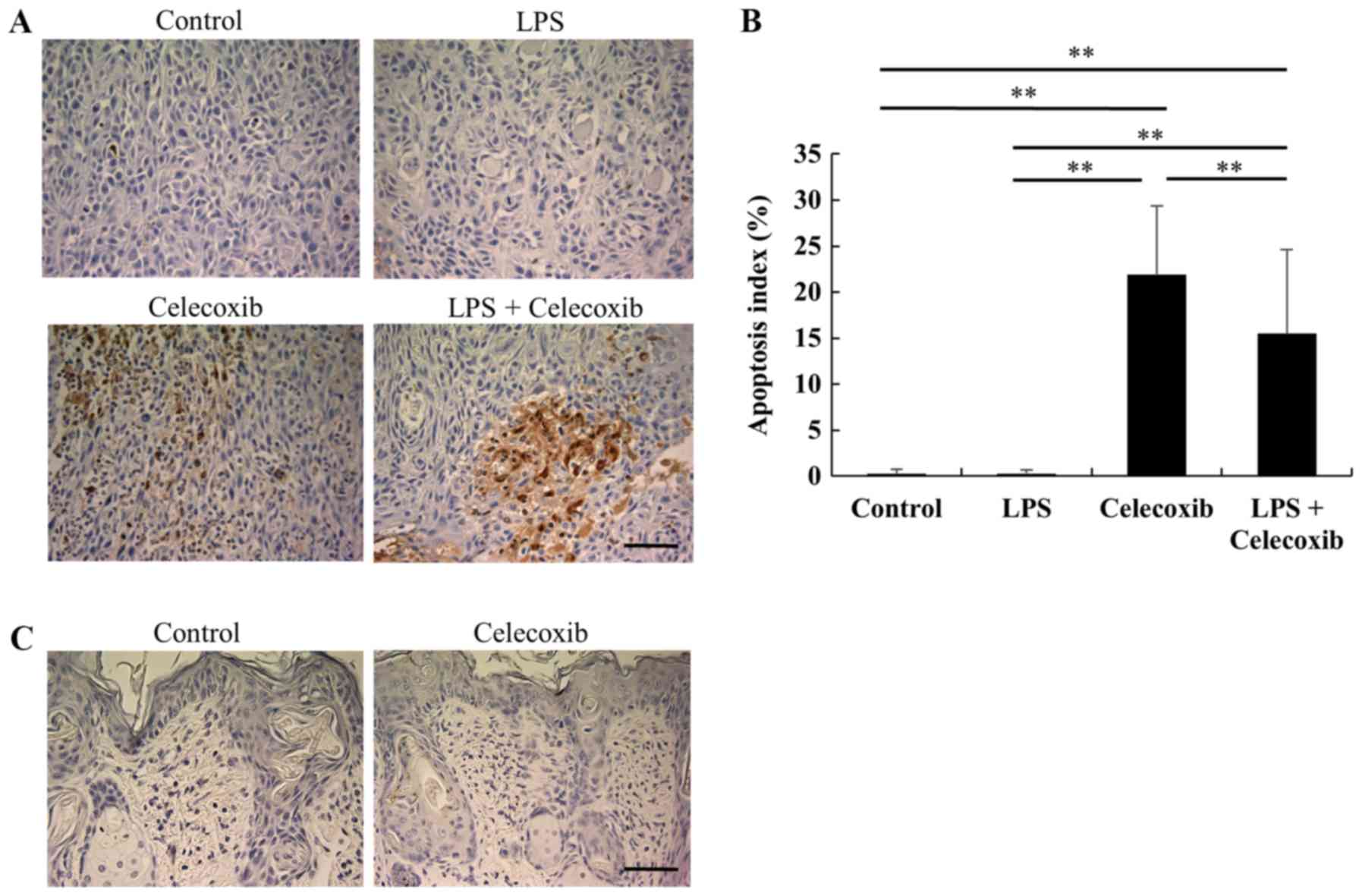
(Fig. 3A and B). Furthermore, TUNEL
staining revealed that the apoptotic indices in the
celecoxib-treated and LPS + celecoxib-treated groups were
significantly higher than in the control and LPS-treated groups
(P<0.01), suggesting that celecoxib induced apoptosis in the
control and LPS-treated OSCC xenografts (Fig. 4A and B). The apoptotic indices in the
LPS + celecoxib-treated group were significantly lower compared
with those in the celecoxib-treated group (P<0.01; Fig. 4A and B). Apoptosis was not induced in
non-tumorous skin and mucosa tissues surrounding the tumors
following celecoxib treatment (Fig.
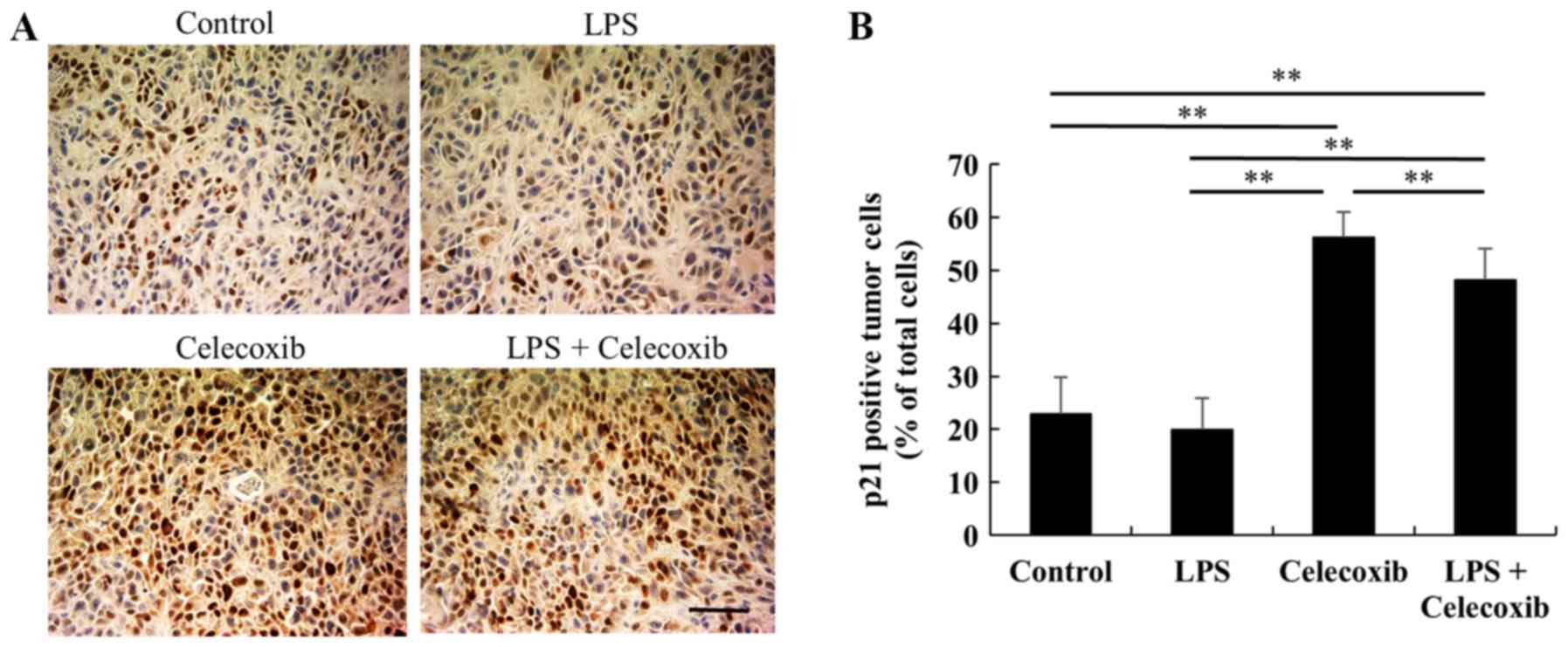
4C). Furthermore, there were no significant changes in p21
expression levels between the control and LPS-treated groups
(Fig. 5A and B), whereas the
celecoxib-treated and LPS + celecoxib-treated groups had
significantly increased levels of p21 compared with the control and
LPS-treated groups (P<0.01), indicating that celecoxib
upregulates p21 expression in OSCC xenografts (Fig. 5A and B). The levels of p21 in the LPS
+ celecoxib-treated groups were significantly decreased compared in
the celecoxib-treated groups (Fig. 5A
and B).
Discussion
In the present study, P. gingivalis-derived
LPS was used to investigate the molecular mechanisms underlying
periodontitis in the Toll-like receptor (TLR) 4-expressing OSCC
cell line HSC-3 (7). LPS binds
directly to the TLR4/myeloid differentiation factor 2 receptor
complex (18) activating the myeloid
differentiation factor 88 signaling pathway, which in turn
activates mitogen-activated protein kinase (MAPK) and the
transcription factor nuclear factor-κB, which play an important
role in cell proliferation (19).
Previous reports have demonstrated that LPS stimulation of TLR4
promotes breast cancer growth in nude mice (6), and, consistently, LPS caused a
significant increase in OSCC cell proliferation in vitro in
the present study.
To assess whether COX-2 plays a role in LPS-induced
proliferation of OSCC cells, the antitumor effect of the COX-2
inhibitor celecoxib was investigated. Celecoxib significantly
decreased the proliferation of OSCC cells and inhibited tumor
growth in a xenograft model. The expression of Ki-67 was also
decreased, and apoptosis was significantly increased in the
celecoxib-treated group. This coincides with previous reports that
celecoxib significantly decreases the viability of OSCC cells by
inhibiting phosphorylated protein kinase B, cyclin D1 or the
epithelial-to-mesenchymal transition, as well as by inhibiting
tumor growth in OSCC xenografts (14,20).
In the present study, celecoxib treatment
downregulated the protein expression levels of COX-2 in OSCC cells.
This finding is supported by previous reports that celecoxib
attenuates COX-2 expression and inhibits the growth of OSCC cells
(21,22). On the other hand, celecoxib has been
indicated to inhibit the cell survival of both COX-2-expressing and
non-expressing colon carcinoma cells (23), indicating that the effects of
celecoxib are independent of COX-2, and there could be other
targets/effects that have yet to be defined.
The concentrations of celecoxib used in the present
study were much higher than those used in oral administration for
humans (24), so the results
obtained from the present study may not be directly extrapolatable
to humans. However, the antitumor effects of celecoxib have
previously been established in epidemiological studies and in
clinical trials (24), indicating
that celecoxib may be effective even at lower concentrations in
humans. Furthermore, no significant weight loss was observed in
celecoxib-treated mice in the present study, suggesting that
celecoxib did not exhibit toxicity or cause adverse effects.
Nonetheless, other studies have suggested that adverse effects may
be decreased and controlled using the oral administration of
antitumor drugs (25,26), and that celecoxib may be eligible for
such application as well. Furthermore, a recent study has revealed
that, in HNSCC, a combined metronomic oral celecoxib treatment is
advantageous when compared with intravenous single-agent therapy
(24); thus, this approach may also
improve future results using celecoxib therapy.
In the LPS-stimulated OSCC xenografts, celecoxib
upregulated the expression of p21 which induces cell cycle arrest
and inhibits tumor development (27). Celecoxib has been demonstrated to
inhibit the G0/G1-to-S-phase transition by
increasing the expression of p21, thereby decreasing tumor growth
in colon cancer (23). In the
present study, the expression levels of p53 exhibited little change
in the OSCC cells exposed to celecoxib. Since it is known that p21
expression is upregulated by both p53-dependent and independent
pathways (27), celecoxib may
exhibit its antitumor effect in LPS-stimulated OSCC xenografts by
upregulating p21 expression and halting the cell cycle through a
p53-independent pathway.
In the present study, HSC-3 cells were stimulated
with LPS for 48 h before implantation. This protocol has been
supported by reports that describe the pre-treatment of carcinoma
cells with LPS prior to implantation in nude mouse xenograft models
(6,28). Stimulation of breast carcinoma cells
with LPS for 48 h before implantation increased tumor volumes and
weights in a xenograft model of breast carcinoma (6). Similarly, stimulation of cluster of
differentiation 133+ hepatoma cells with LPS for 1 week
before implantation caused an increase in the growth of tumors in
nude mice compared with those without LPS (28). Although the duration of LPS
stimulations prior to implantation varied between these two
reports, both clearly indicate that pre-treatment with LPS has
growth-promoting effects in vivo. In the present study,
LPS-induced tumor growth was not observed in vivo, although
LPS treatment significantly increased the proliferation of OSCC
cells in vitro. The limitation of the method of LPS
administration used in the present study is that this was a
one-time LPS treatment in vivo. In a murine breast cancer
model, LPS was intraperitoneally injected for 3 consecutive days
and it was observed that this promoted lung metastasis (29). Therefore, intraperitoneal injection
of LPS following implantation may improve the experimental model
used in the present study.
Understanding the mortality and clinical
abnormalities caused by celecoxib is important for chemotherapy.
One study reported the adverse effects of celecoxib in patients
with locoregionally advanced nasopharyngeal carcinoma (30). According to this report,
administration of celecoxib concurrently with nasopharyngeal
radiotherapy caused toxicities such as mucositis, weight loss,
dermatitis and otitis, although no episodes of toxic mortality
occurred with the treatment (30).
Furthermore, a regimen of celecoxib combined with radiation was
well tolerated in patients with brain metastases, and
celecoxib-associated toxicity was limited to a mild skin reaction
(31). These reports suggest that
celecoxib can be safely administrated to patients with carcinomas
such as lung or breast carcinoma or melanoma, including those with
metastases.
A limitation of the present study is that it
utilizes a single cell line, HSC-3, for both the in vitro
and in vivo experiments. In a previous study, an
HSC-3-bearing nude mouse model was successfully established and
utilized (16). For the in
vitro experiments, previous studies support the data from the
present study that celecoxib inhibits the growth of OSCC cells
(14,20).
The clinical implications of the present study are
important. First, P. gingivalis-derived LPS can stimulate
tumor growth by interacting with OSCC cells. Thus, it poses a risk
for developing OSCC (4). Secondly,
celecoxib could be used for the effective prevention and treatment
of LPS-stimulated OSCC. Further experiments are required for the
future clinical development of celecoxib as an OSCC treatment.
In conclusion, the data from the present study
revealed that P. gingivalis-derived LPS can stimulate the
growth of OSCC tumors. Celecoxib suppressed the proliferation of
LPS-stimulated OSCC cells and inhibited tumor growth by increasing
p21 expression levels and inducing apoptosis in OSCC xenografts.
These results suggest that celecoxib administration could provide
an effective prevention and treatment strategy for LPS-stimulated
OSCC.
Acknowledgements
Not applicable.
Funding
The present study was funded in part by the
Grant-in-Aid for Young Scientists (grant no. 18K17196) and the
Grant-in-Aid for Scientific Research (grant no. 18K09719) from the
Japan Society for the Promotion of Science.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HisY, HitY and KS designed the experiments. HisY,
HitY, SM, SY, KO, HI and KS performed the experiments. HisY, HitY,
MO, TR, TK and MK analyzed the data. HisY, HitY and KS wrote the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the Animal
Ethics Committee of the University of Fukui (approval no.
29105).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Neville BW and Day TA: Oral cancer and
precancerous lesions. CA Cancer J Clin. 52:195–215. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marta GN, Riera R, Bossi P, Zhong LP,
Licitra L, Macedo CR, de Castro Junior G, Carvalho AL, William WN
Jr and Kowalski LP: Induction chemotherapy prior to surgery with or
without postoperative radiotherapy for oral cavity cancer patients:
Systematic review and meta-analysis. Eur J Cancer. 51:2596–2603.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Javed F and Warnakulasuriya S: Is there a
relationship between periodontal disease and oral cancer? A
systematic review of currently available evidence. Crit Rev Oncol
Hematol. 97:197–205. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jain S and Darveau RP: Contribution of
Porphyromonas gingivalis lipopolysaccharide to periodontitis.
Periodontol 2000. 54:53–70. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang H, Wang B, Wang T, Xu L, He C, Wen H,
Yan J, Su H and Zhu X: Toll-like receptor 4 prompts human breast
cancer cells invasiveness via lipopolysaccharide stimulation and is
overexpressed in patients with lymph node metastasis. PLoS One.
9:e1099802014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He Z, Deng R, Huang X, Ni Y, Yang X, Wang
Z and Hu Q: Lipopolysaccharide enhances OSCC migration by promoting
epithelial-mesenchymal transition. J Oral Pathol Med. 44:685–692.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu CH, Chang SH, Narko K, Trifan OC, Wu
MT, Smith E, Haudenschild C, Lane TF and Hla T: Overexpression of
cyclooxygenase-2 is sufficient to induce tumorigenesis in
transgenic mice. J Biol Chem. 276:18563–18569. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kojima M, Morisaki T, Izuhara K, Uchiyama
A, Matsunari Y, Katano M and Tanaka M: Lipopolysaccharide increases
cyclo-oxygenase-2 expression in a colon carcinoma cell line through
nuclear factor-kappa B activation. Oncogene. 19:1225–1231. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gallo O, Franchi A, Magnelli L, Sardi I,
Vannacci A, Boddi V, Chiarugi V and Masini E: Cyclooxygenase-2
pathway correlates with VEGF expression in head and neck cancer.
Implications for tumor angiogenesis and metastasis. Neoplasia.
3:53–61. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chan G, Boyle JO, Yang EK, Zhang F, Sacks
PG, Shah JP, Edelstein D, Soslow RA, Koki AT, Woerner BM, et al:
Cyclooxygenase-2 expression is up-regulated in squamous cell
carcinoma of the head and neck. Cancer Res. 59:991–994.
1999.PubMed/NCBI
|
|
12
|
Gallo O, Masini E, Bianchi B, Bruschini L,
Paglierani M and Franchi A: Prognostic significance of
cyclooxygenase-2 pathway and angiogenesis in head and neck squamous
cell carcinoma. Hum Pathol. 33:708–714. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shin DM, Zhang H, Saba NF, Chen AY,
Nannapaneni S, Amin AR, Müller S, Lewis M, Sica G, Kono S, et al:
Chemoprevention of head and neck cancer by simultaneous blocking of
epidermal growth factor receptor and cyclooxygenase-2 signaling
pathways: Preclinical and clinical studies. Clin Cancer Res.
19:1244–1256. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chiang SL, Velmurugan BK, Chung C, Lin SH,
Wang ZH, Hua CH, Tsai MH, Kuo TM, Yeh KT, Chang PY, et al:
Preventive effect of celecoxib use against cancer progression and
occurrence of oral squamous cell carcinoma. Sci Rep. 7:62352017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu XF, Xie CG, Wang XP, Liu J, Yu YC, Hu
HL and Guo CY: Selective inhibition of cyclooxygenase-2 suppresses
the growth of pancreatic cancer cells in vitro and in vivo. Tohoku
J Exp Med. 215:149–157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoshida H, Yoshimura H, Matsuda S, Ryoke
T, Kiyoshima T, Kobayashi M and Sano K: Effects of peritumoral
bevacizumab injection against oral squamous cell carcinoma in a
nude mouse xenograft model: A preliminary study. Oncol Lett.
15:8627–8634. 2018.PubMed/NCBI
|
|
17
|
National Research Council, . Guide for the
Care and Use of Laboratory Animals8th. The National Academies
Press; Washington, DC: 2011
|
|
18
|
Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh
SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ and Lee JO: Crystal
structure of the TLR4-MD-2 complex with bound endotoxin antagonist
Eritoran. Cell. 130:906–917. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu YC, Yeh WC and Ohashi PS: LPS/TLR4
signal transduction pathway. Cytokine. 42:145–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abrahão AC, Giudice FS, Sperandio FF and
Pinto Junior Ddos S: Effects of celecoxib treatment over the AKT
pathway in head and neck squamous cell carcinoma. J Oral Pathol
Med. 42:793–798. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kwak YE, Jeon NK, Kim J and Lee EJ: The
cyclooxygenase-2 selective inhibitor celecoxib suppresses
proliferation and invasiveness in the human oral squamous
carcinoma. Ann N Y Acad Sci. 1095:99–112. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li WZ, Wang XY, Li ZG, Zhang JH and Ding
YQ: Celecoxib enhances the inhibitory effect of cisplatin on
Tca8113 cells in human tongue squamous cell carcinoma in vivo and
in vitro. J Oral Pathol Med. 39:579–584. 2010.PubMed/NCBI
|
|
23
|
Grösch S, Tegeder I, Niederberger E,
Bräutigam L and Geisslinger G: COX-2 independent induction of cell
cycle arrest and apoptosis in colon cancer cells by the selective
COX-2 inhibitor celecoxib. FASEB J. 15:2742–2744. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Patil VM, Noronha V, Joshi A, Muddu VK,
Dhumal S, Bhosale B, Arya S, Juvekar S, Banavali S, D'Cruz A, et
al: A prospective randomized phase II study comparing metronomic
chemotherapy with chemotherapy (single agent cisplatin), in
patients with metastatic, relapsed or inoperable squamous cell
carcinoma of head and neck. Oral Oncol. 51:279–286. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Argiris A, Li Y, Murphy BA, Langer CJ and
Forastiere AA: Outcome of elderly patients with recurrent or
metastatic head and neck cancer treated with cisplatin-based
chemotherapy. J Clin Oncol. 22:262–268. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kawano S, Zheng Y, Oobu K, Matsubara R,
Goto Y, Chikui T, Yoshitake T, Kiyoshima T, Jinno T, Maruse Y, et
al: Clinicopathological evaluation of pre-operative
chemoradiotherapy with S-1 as a treatment for locally advanced oral
squamous cell carcinoma. Oncol Lett. 11:3369–3376. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer
Ther. 1:639–649. 2002.PubMed/NCBI
|
|
28
|
Lai FB, Liu WT, Jing YY, Yu GF, Han ZP,
Yang X, Zeng JX, Zhang HJ, Shi RY, Li XY, et al: Lipopolysaccharide
supports maintaining the stemness of CD133(+) hepatoma cells
through activation of the NF-κB/HIF-1α pathway. Cancer Lett.
378:131–141. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li S, Xu X, Jiang M, Bi Y, Xu J and Han M:
Lipopolysaccharide induces inflammation and facilitates lung
metastasis in a breast cancer model via the prostaglandin E2-EP2
pathway. Mol Med Rep. 11:4454–4462. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xue WP, Bai SM, Luo M, Bi ZF, Liu YM and
Wu SK: Phase I clinical trial of nasopharyngeal radiotherapy and
concurrent celecoxib for patients with locoregionally advanced
nasopharyngeal carcinoma. Oral Oncol. 47:753–757. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cerchietti LC, Bonomi MR, Navigante AH,
Castro MA, Cabalar ME and Roth BM: Phase I/II study of selective
cyclooxygenase-2 inhibitor celecoxib as a radiation sensitizer in
patients with unresectable brain metastases. J Neurooncol.
71:73–81. 2005. View Article : Google Scholar : PubMed/NCBI
|