Introduction
In the past decades, gynecological cancer has been
the leading cause of cancer mortality in women globally (1). The major types of gynecological cancer
include ovarian serous cystadenocarcinoma (OV), uterine corpus
endometrial carcinoma (UCEC), and cervical squamous cell carcinoma
and endocervical adenocarcinoma (CESC) (2). Despite the various therapeutic methods
that have been developed, including surgical, hormone therapeutic
and chemotherapeutic treatments, the 5-year survival rate of
patients with gynecological cancer has remained poor (3,4). For
example, the 5-year overall survival (OS) of patients with
late-stage OV is <20% (3). The
advanced-stage CESC 5-year OS rate has remained as low as 30%
(4). Of note, the mechanisms
underlying gynecological cancer require further investigation.
Exploration the potential regulators involved in gynecological
cancer progression is urgently needed to identify novel biomarkers
of cancer prognosis and targets for treatment.
Next-generation sequencing (NGS) is an important
tool in the generation of new cancer therapies and diagnostic
methods (5,6). The Cancer Genome Atlas (TCGA) database,
including >30 types of human cancer, is the most widely used NGS
database and has played a crucial role in the discovery of
cancer-associated genes and mutations (7,8). For
example, Sanchez-Vega et al (9) analyzed oncogenic signaling pathways
across 33 human cancer types using TCGA datasets. In gynecological
cancer, a series of key regulators were also identified by using
similar strategies. For instance, Berger et al (10) identified a series of mutated genes
and somatic copy-number alterations in gynecological cancer by
comprehensively analyzing TCGA datasets. Song et al
(11) constructed an aberrant long
noncoding RNA-microRNA-mRNA network in CESC using TCGA datasets.
Comprehensive analysis of TCGA datasets provides novel insights
into the mechanisms involved in tumor progression to allow for the
identification of new biomarkers for human cancer, including
gynecological cancer.
In order to obtain a comprehensive assessment of the
molecular mechanisms underlying gynecological cancer progression, a
bioinformatics analysis of OV, CESC and USEC datasets from TCGA was
conducted to identify hub genes and key pathways in the present
study. In addition, the prognostic value of these key genes in
gynecological cancer was also evaluated.
Materials and methods
TCGA dataset analysis
In the present study, TCGA CESC, OV and UCEC
datasets were downloaded from the cBioPortal system (12). Level 3 RNA sequencing version 2 data
were downloaded from TCGA (https://www.cbioportal.org/). A total of 233 stage I +
II CESC and 68 stage III + IV CESC samples were included in TCGA
CESC dataset. A total of 23 stage I + II OV and 282 stage III + IV
OV samples were included in TCGA OV dataset. A total of 122 stage I
+ II UCEC and 54 stage III + IV UCEC samples were included in TCGA
UCEC dataset. All specimens were independently assessed by two
experienced pathologists according to the 8th edition of the
American Joint Committee on Cancer (AJCC) TNM staging system
(13). Gene expression with
P<0.01 between early-stage (stage I + II) and advanced-stage
(stage III + IV) samples was identified to indicate significantly
differential expression. Hierarchical cluster analysis was
performed, and a hierarchical clustering heat map was generated for
the abnormally expressed genes using CLUSTER version 3.0 (14) and the Tree View system (15).
Protein-protein interaction (PPI)
networks and module analysis
PPI networks were constructed to reveal the
relationships among gynecological cancer progression-associated
genes following two steps. Firstly, the combined score between each
protein-protein pair was calculated using STRING version 11.0
(http://www.string-db.org/); reliable
protein-protein interactions (combined score, >0.4) were
selected for PPI network construction. Secondly, an analysis of the
degrees of each node was performed, and the key nodes (node degree
≥5) in the PPI network were retained using Cytoscape software
(version 3.6.0; http://www.cytoscape.org/).
Gene ontology (GO) and pathway
analysis
The Database for Annotation, Visualization and
Integrated Discovery (DAVID) version 6.8 system (https://david.ncifcrf.gov/tools.jsp) provides a
comprehensive set of functional annotation tools to identify
disease-associated biological processes (16). Therefore, DAVID was used to conduct
GO analysis (17,18). The top 15 associated ‘biological
processes’ (BPs) are shown. The analysis results of molecular
functions and Cellular Component were not shown in this study. BPs
with P<0.05 were considered to be significant.
Survival analysis of differentially
expressed genes (DEGs)
In order to examine whether these DEGS could be the
potential biomarkers for the prognosis of gynecological cancer,
Kaplan-Meier analysis and log-rank tests were conducted using an
online public database, GEPIA (http://gepia.cancer-pku.cn/index.html). Patients with
gynecological cancer were categorized into 2 groups depending on
the expression levels in cancer samples; the median expression of
candidate genes in all tumor samples was selected as the cut-off
point to divide gynecological cancer samples in to high- or
low-expression groups. P<0.05 was considered to indicate a
statistically significant difference.
Statistical analysis
Differences in gene expression between the
individual groups were analyzed using unpaired Student's t-test or
Mann-Whitney U-test. PASW Statistics 23.0 software from SPSS Inc.
was used. P<0.05 was considered to indicate a statistically
significant difference.
Results
Identification of DEGs in the
progression of gynecological cancer
Datasets from TCGA were downloaded to identify DEGs
in OV, CESC and UCEC progression. Gene expression with P<0.01
between low-stage (stage I + II) and advanced-stage (stage III +
IV) samples was identified to indicate differential expression. A
total of 153, 335 and 406 upregulated genes and 646, 153 and 215
downregulated genes were identified in OV, CESC and UCEC
progression, respectively. Hierarchical clustering showed DEGs in
higher-stage compared with lower-stage OV (Fig. 1A), CESC (Fig. 1B) and UCEC (Fig. 1C) samples.
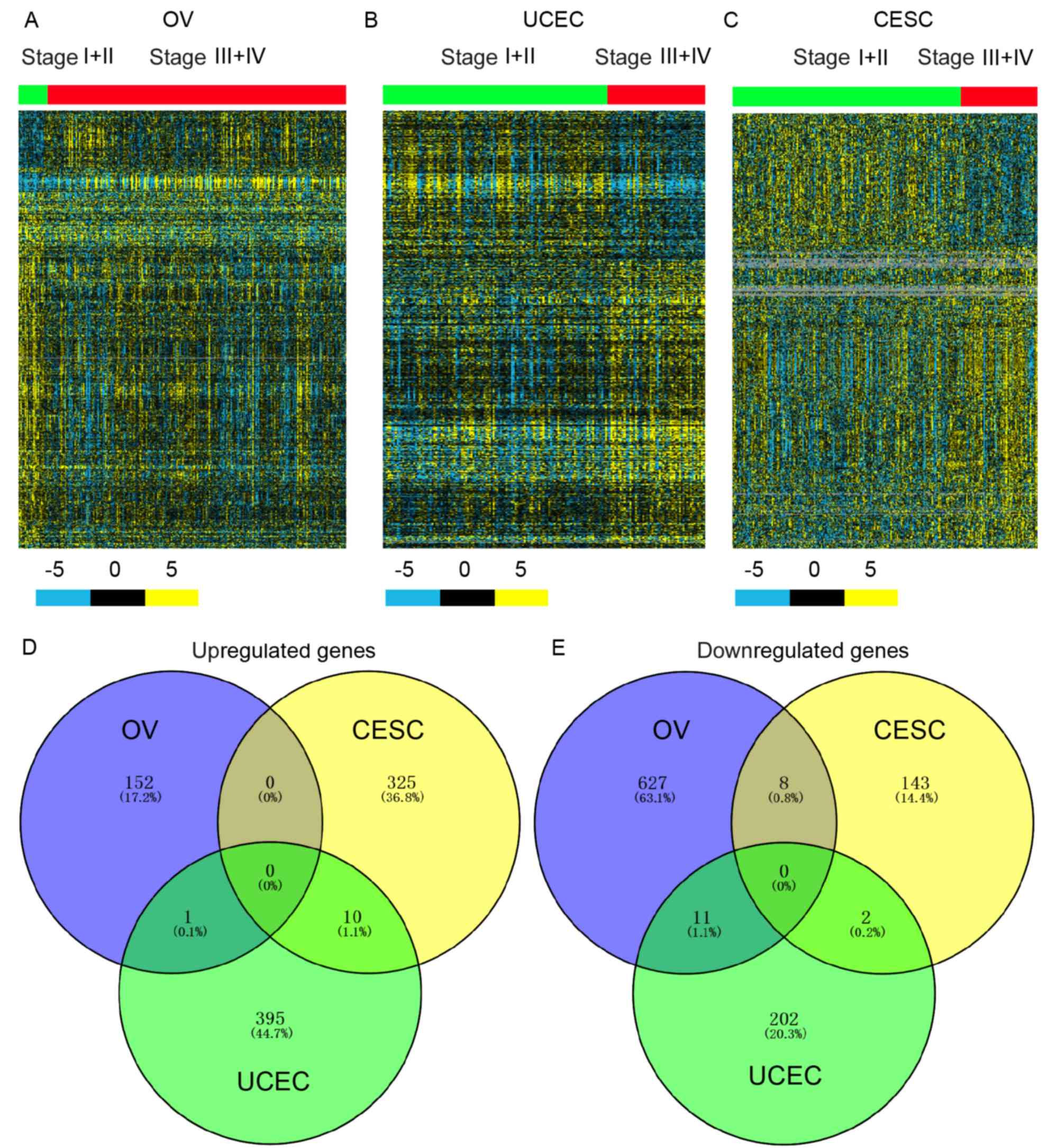 | Figure 1.Identification of DEGs in
gynecological cancer progression. Hierarchical clustering analysis
showing the DEGs (P<0.01) between (A) Stage I + II and Stage III
+ IV OV samples, (B) Stage I + II and Stage III + IV UCEC samples,
and (C) Stage I + II and Stage III + IV CESC samples. The blue,
black and yellow colors refer to 5-, 0- and −5-folds changes in
expression, respectively. Venn diagrams for DEGs whose expression
was significantly (D) upregulated and (E) downregulated in OV, UCEC
and CESC samples. OV, ovarian serous cystadenocarcinoma; CESC,
cervical squamous cell carcinoma and endocervical adenocarcinoma;
UCEC, uterine corpus endometrial carcinoma; DEGs, differentially
expressed genes. |
In order to understand whether common or
cancer-specific genes drive the progression of gynecological
cancer, the dysregulated genes were compared. As shown in Fig. 1, no common dysregulated genes were
observed in OV, CESC and UCEC (Fig. 1C
and D). Meanwhile, only 11 upregulated and 21 downregulated
genes were found in two types of gynecological cancer (Fig. 1C and D). These results suggested that
different genes regulate cancer progression in different types of
gynecological cancer.
Bioinformatics analysis of DEGs in
gynecological cancer
Next, bioinformatics analysis was performed on the
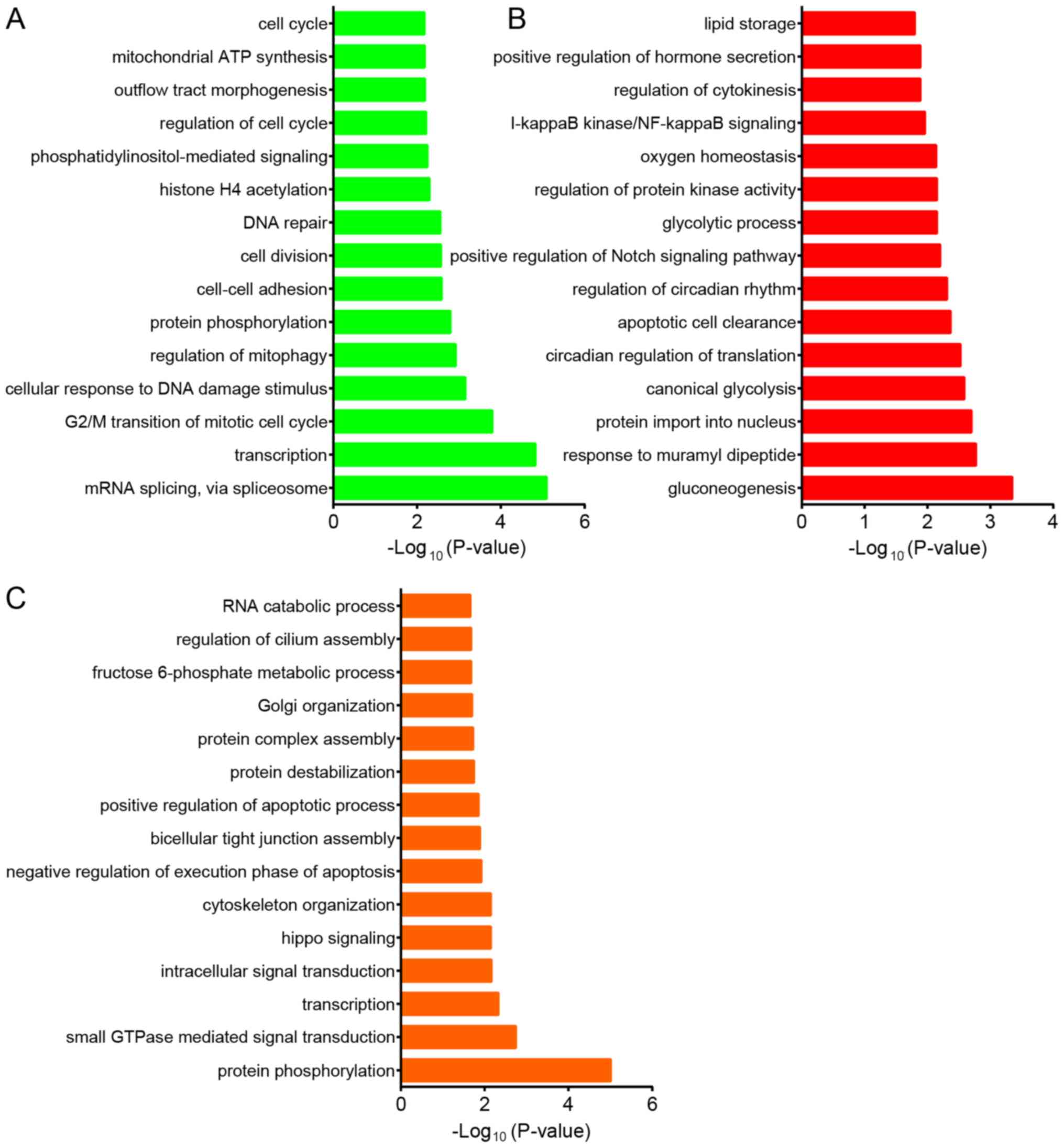
DEGs in gynecological cancer. GO analysis revealed that the DEGs
associated with OV progression were mainly involved in regulating
‘mRNA splicing’, ‘transcription’, ‘G2/M transition of mitotic cell
cycle’, ‘cellular response to DNA damage stimulus’, ‘mitophagy’,
‘protein phosphorylation’, ‘cell-cell adhesion’, ‘cell division’
and ‘DNA repair’ (Fig. 2A). DEGs in
CESC progression were mainly involved in regulating
‘gluconeogenesis’, ‘response to muramyl dipeptide’, ‘protein import
into nucleus’, ‘canonical glycolysis’, ‘circadian regulation of
translation’, ‘apoptotic cell clearance’, ‘positive Notch signaling
pathway’, ‘glycolytic process’, ‘protein kinase activity’ and
‘oxygen homeostasis’ (Fig. 2B). The
study also indicated that DEGs in UCEC were associated with
‘protein phosphorylation’, ‘small GTPase mediated signal
transduction’, ‘transcription’, ‘intracellular signal
transduction’, ‘hippo signaling’, ‘cytoskeleton organization’,
‘negative regulation of execution phase of apoptosis’, ‘bicellular
tight junction assembly’, ‘positive regulation of apoptotic
process’ and ‘protein destabilization’ (Fig. 2C).
Construction of progression-associated
gene-mediated PPI networks in gynecological cancer
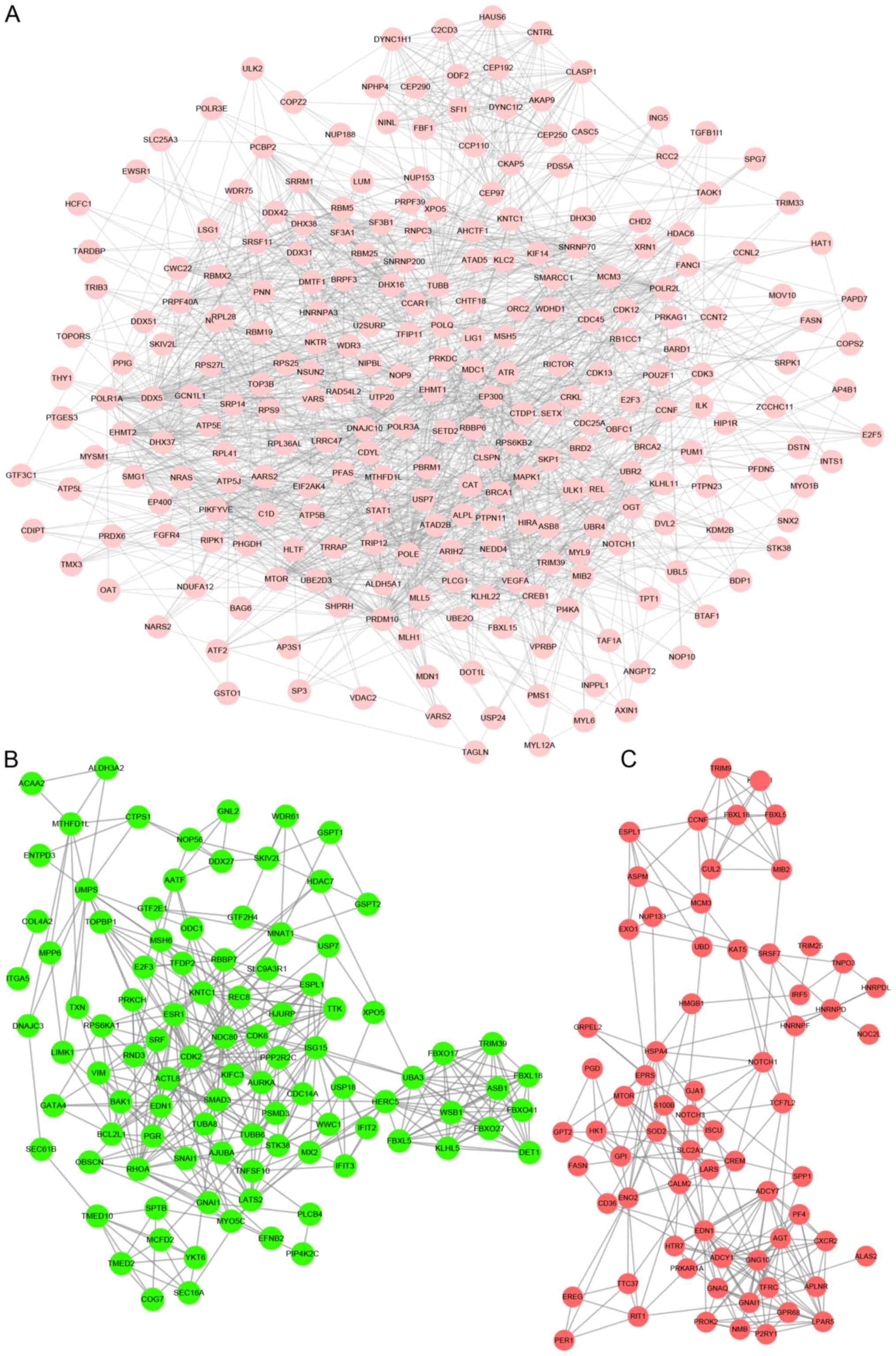
As presented in Fig.
3, the OV progression-associated PPI networks included 258
proteins and 1,872 edges (Fig. 3A).
The top 10 hub genes with highest degrees involved in OV
progression were identified, including EHMT1, EHMT2, BRCA1,
PRDM10, CKAP5, SNRNP70, ATR, MTOR, SETD2 and MIB2. The
UCEC progression-associated PPI networks included 99 proteins and
375 edges (Fig. 3B). The top 10 hub
genes with the highest degrees involved in UCEC progression were
identified, including RHOA, ISG15, LATS2, ACTL8, CDK2, SPTB,
TTK, EDN1, FBXO41 and RBBP7. The CESC
progression-associated PPI networks included 69 proteins and 228
edges (Fig. 3C). The top 10 hub
genes with the highest degrees involved in CESC progression were
identified, including EDN1, GNG10, AGT, EPRS, HSPA4, RIT1, CUL2,
GNAI1, GPI and GPR68.
Prognostic significance of
progression-associated genes in gynecological cancer
Furthermore, Kaplan-Meier curve analysis was
conducted to determine the association between
progression-associated gene expression and OS in gynecological
cancer, using TCGA datasets. The median expression of
progression-associated genes was selected as the cut-off to divide
the gynecological cancer cases into high- and low-expression
groups.
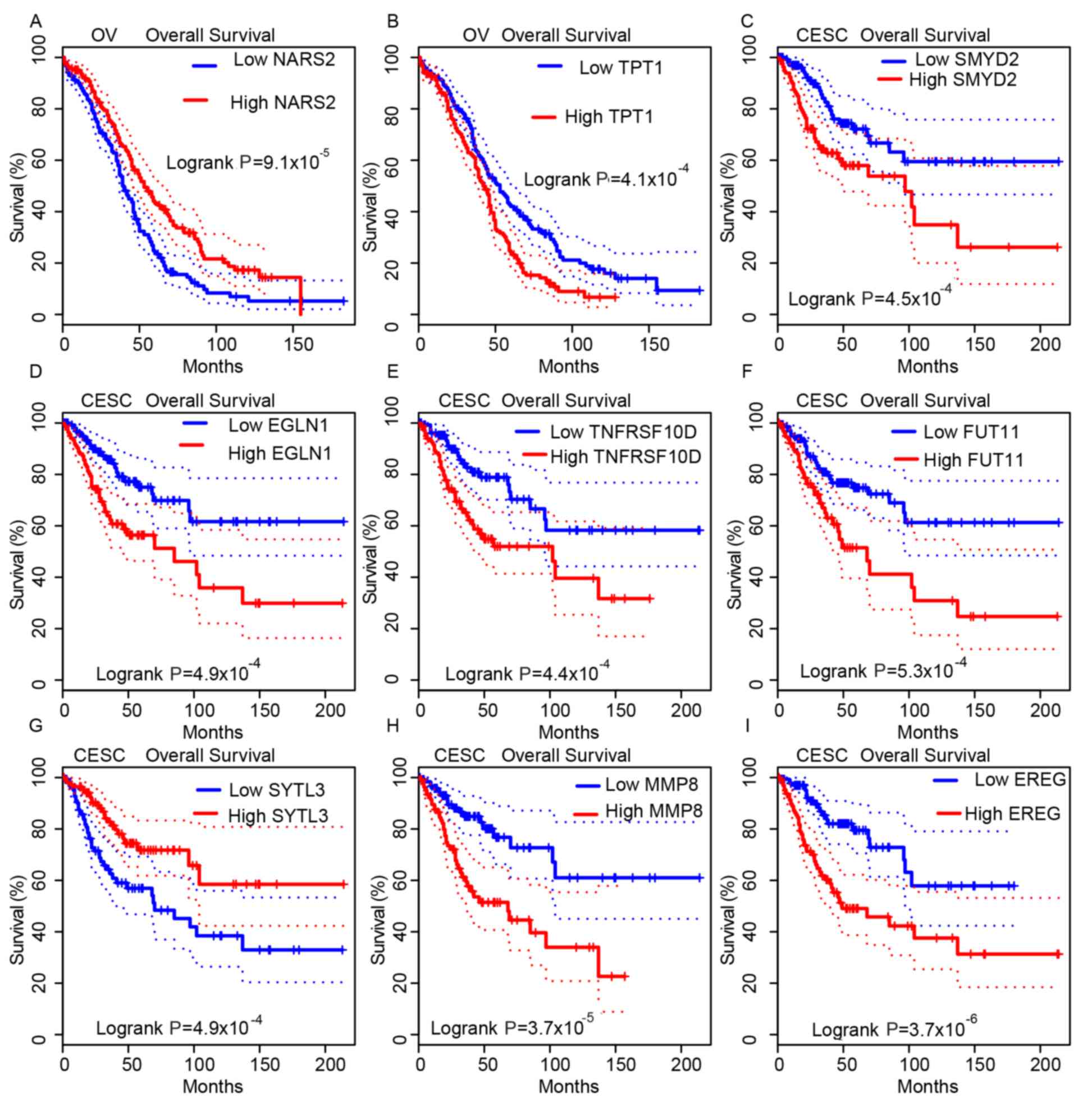
Higher expression of NARS2 and lower
expression of TPT1 were indicated to be associated with a
longer OS time in patients with OV (Fig.
4A and B). Meanwhile, the OS times in SMYD2-high,
EGLN1-high, TNFRSF10D-high, FUT11-high,
SYTL3-low, MMP8-high and EREG-high expression
groups in patients with CESC were significantly shorter compared
with their opposing expression groups (Fig. 4C-I). In patients with UCEC, this
study indicated that higher expression of SLC5A1, TXN, KDM4B,
TXNDC11, HSDL2 and COX16, and lower expression of
MGAT4A, DAGLA, ELOVL7, THRB and PCOLCE2 were
associated with longer OS times (Fig.
5A-K). These analyses indicated that progression-associated
genes could serve as biomarkers for gynecological cancer.
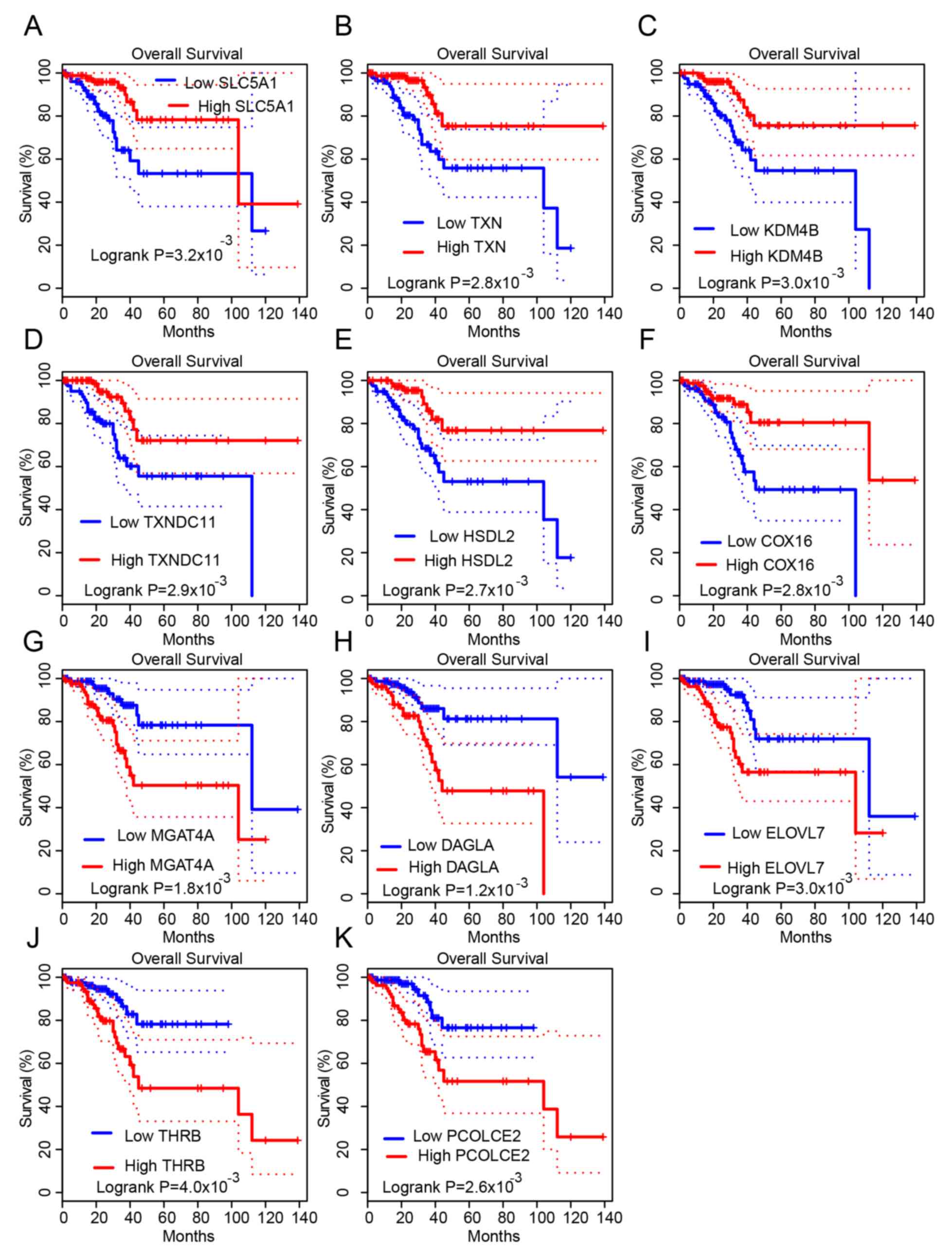 | Figure 5.Progression-related genes are
associated with overall survival time in UCEC. It was revealed that
higher expression levels of (A) SLC5A1, (B) TXN, (C) KDM4B, (D)
TXNDC11, (E) HSDL2 and (F) COX16, and lower expression levels of
(G) MGAT4A, (H) DAGLA, (I) ELOVL7, (J) THRB and (K) PCOLCE2, were
associated with longer OS times in patients with UCEC. UCEC,
uterine corpus endometrial carcinoma. |
Discussion
Gynecological cancer, including OV, CESC and UCEC,
are a leading cause of cancer mortality in women (19). In the present study, TCGA datasets
were analyzed to identify gynecological cancer
progression-associated genes. A total of 153, 335 and 406
upregulated, and 646, 153 and 215 downregulated genes were
associated with OV, CESC and UCEC progression, respectively. In
addition, OV, CESC and UCEC progression-associated PPI networks
were constructed to reveal the associations among these genes.
Furthermore, Kaplan-Meier curve analysis showed that
progression-related genes, such as SMYD2, EGLN1, TNFRSF10D,
SLC5A1 and TXN, could serve as prognostic biomarkers for
gynecological cancer.
Previous studies have reported certain drivers that
are involved in gynecological cancer (20–22). By
using TCGA datasets, Berger et al (10) identified various mutated genes in
gynecological cancer. CT45 was identified as a
chemosensitivity mediator and immunotherapy target in ovarian
cancer (20). BRCA1 and
BRCA2 are considered to be key regulators of ovarian
development and function (21,22).
However, the underlying mechanisms regulating cancer progression
require further investigation. The present study identified 799
dysregulated genes in OV, 488 dysregulated genes in CESC, and 621
dysregulated genes in UCEC. Only a small number of genes were
observed to be dysregulated in more than one gynecological cancer,
suggesting that different mechanisms underlie cancer progression in
different types of gynecological cancer.
Bioinformatics analyses were also performed, and
showed that OV progression-associated genes were involved in
regulating mRNA splicing and cell proliferation-associated BPs.
mRNA splicing had been demonstrated to regulate the progression of
OV. For example, Snail driving alternative splicing of CD44 by
ESRP1 enhances metastasis of OV (23). CESC progression-associated genes were
involved in regulating a series of metabolism-related BPs, such as
glycolysis and oxygen homeostasis. Glycolysis played a crucial role
for the supplication of energy and precursors for human cancer
(24). It was also indicated that
DEGs in UCEC were associated with protein phosphorylation and small
GTPase-mediated signal transduction. In addition, PPI networks were
constructed in this study. A few genes were identified to be key
regulators in gynecological cancer progression, such as
EHMT1 and EHMT2 in OV, EDN1 and GNG10
in CESC, and RHOA and ISG15 in UCEC.
Over the past decades, efforts have been made to
identify accurate biomarkers for gynecological cancer. For
instance, upregulation of TRIM44 predicts poor prognosis in
epithelial ovarian cancer (25), and
LYL1 amplification predicts a shorter survival time of patients
with UCEC (26). However, the
prognosis of patients with gynecological cancer remains poor. In
this study, Kaplan-Meier curve analysis was conducted to determine
the prognostic value of progression-associated gene expression in
gynecological cancer. It was revealed that the dysregulation of
NARS2 and TPT1 in OV, the dysregulation of SMYD2,
EGLN1, TNFRSF10D, FUT11, SYTL3, MMP8 and EREG in CESC,
and the dysregulation of DC11, HSDL2, COX16, MGAT4A, DAGLA,
ELOVL7, THRB and PCOLCE2 in UCSC were associated with OS
time. In previous studies, SMYD2 (which encodes SET and MYND
domain containing 2 protein) was found to be an oncogene in various
types of cancer, including triple negative breast cancer (27), hepatocellular carcinoma (28) and pancreatic cancer (29). FUT11 (fucosyltransferase 11)
was identified as a novel prognostic marker for clear cell renal
cell carcinoma (30). Genetic
polymorphisms in MMP8 (encoding matrix metalloproteinase-8)
have been reported to be associated with breast cancer (31), bladder cancer (32) and malignant melanoma risk (33,34).
HSDL2 (hydroxysteroid dehydrogenase-like 2) serves as an
oncogene in ovarian cancer by promoting cell proliferation and cell
motility (35). However, the
majority of these genes were for the first time reported to be
involved in human cancer progression, and these analyses suggested
that these progression-associated genes could serve as biomarkers
for gynecological cancer.
In the present study, 799 dysregulated genes were
identified in OV, 488 dysregulated genes in CESC and 621
dysregulated genes in UCEC. Bioinformatics analysis revealed that
mRNA splicing and cell proliferation-associated BPs played
important roles in OV progression. In addition, metabolism-related
BPs played important roles in CESC progression, and protein
phosphorylation and small GTPase-mediated signal transduction
played important roles in UCEC progression. OV, CESC and UCEC
progression-associated PPI networks were also constructed to reveal
the association among these genes. Furthermore, Kaplan-Meier curve
analysis showed that progression-related genes were associated with
OS time. Finally, NARS2 and TPT1 in OV, SMYD2,
EGLN1, TNFRSF10D, FUT11, SYTL3, MMP8 and EREG in CESC,
and DC11, HSDL2, COX16, MGAT4A, DAGLA, ELOVL7, THRB and
PCOLCE2 in UCSC were identified as hub genes in cancer
progression. Therefore, it is suggested that the present study may
assist in the identification of novel mechanisms underlying cancer
progression and new biomarkers for gynecological cancer prognosis
and therapy.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and material
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XZ and YW were involved in the conception and design
of the research and drafting the manuscript. XZ participated in the
acquisition of data, analysis and interpretation of data and
statistical analysis. YW participated in the design of the study
and performed the statistical analysis. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent to publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hutchinson L: Gynecological cancer: True
progress in ovarian cancer or just the tip of the iceberg? Nat Rev
Clin Oncol. 9:652012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jha A, Khan Y, Mehdi M, Karim MR, Mehmood
Q, Zappa A, Rebholz-Schuhmann D and Sahay R: Towards precision
medicine: Discovering novel gynecological cancer biomarkers and
pathways using linked data. J Biomed Semantics. 8:402017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li H, Zhang W, Sun X, Chen J, Li Y, Niu C,
Xu B and Zhang Y: Overexpression of kinesin family member 20A is
associated with unfavorable clinical outcome and tumor progression
in epithelial ovarian cancer. Cancer Manag Res. 10:3433–3450. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang S and Chen X: Identification of
potential biomarkers in cervical cancer with combined public mRNA
and miRNA expression microarray data analysis. Oncol Lett.
16:5200–5208. 2018.PubMed/NCBI
|
|
5
|
Krishnan P, Ghosh S, Wang B, Heyns M,
Graham K, Mackey JR, Kovalchuk O and Damaraju S: Profiling of small
nucleolar RNAs by next generation sequencing: Potential new players
for breast cancer prognosis. PLoS One. 11:e01626222016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yadav SS, Li J, Lavery HJ, Yadav KK and
Tewari AK: Next-generation sequencing technology in prostate cancer
diagnosis, prognosis, and personalized treatment. Urol Oncol.
33:267 e1–e13. 2015. View Article : Google Scholar
|
|
7
|
Weisenberger DJ: Characterizing DNA
methylation alterations from the cancer genome atlas. J Clin
Invest. 124:17–23. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cancer Genome Atlas Research Network, ;
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA,
Ellrott K, Shmulevich I, Sander C and Stuart JM: The cancer genome
atlas pan-cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sanchez-Vega F, Mina M, Armenia J, Chatila
WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia
S, et al: Oncogenic signaling pathways in the cancer genome atlas.
Cell. 173:321–337 e10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Berger AC, Korkut A, Kanchi RS, Hegde AM,
Lenoir W, Liu W, Liu Y, Fan H, Shen H, Ravikumar V, et al: A
Comprehensive pan-cancer molecular study of gynecologic and breast
cancers. Cancer Cell. 33:690–705 e9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song J, Ye A, Jiang E, Yin X, Chen Z, Bai
G, Zhou Y and Liu J: Reconstruction and analysis of the aberrant
lncRNA-miRNA-mRNA network based on competitive endogenous RNA in
CESC. J Cell Biochem. 119:6665–6673. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hagemann IS, Cole LL, Cosin JA, Gress DM,
Mutch DG and Olawaiye AB: Controversies in gynecologic cancer
staging: An AJCC cancer staging manual, eighth edition perspective.
AJSP Rev Rep. 23:118–128. 2018.
|
|
14
|
de Hoon MJ, Imoto S, Nolan J and Miyano S:
Open source clustering software. Bioinformatics. 20:1453–1454.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Page RD: TreeViewGlasgow University;
Glasgow, UK: 2001
|
|
16
|
Huang DW, Sherman BT, Tan Q, Kir J, Liu D,
Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC and Lempicki RA:
DAVID Bioinformatics Resources: Expanded annotation database and
novel algorithms to better extract biology from large gene lists.
Nucleic Acids Res. 35((Web Server Issue)): W169–W175. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
The Gene Ontology Consortium, . The gene
ontology resource: 20 years and still GOing strong. Nucleic Acids
Res. 47(D1): D330–D338. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Abu-Shawer O, Abu-Shawer M, Hirmas N,
Alhouri A, Massad A, Alsibai B, Sultan H, Hammo H, Souleiman M,
Shebli Y and Al-Hussaini M: Hematologic markers of distant
metastases and poor prognosis in gynecological cancers. BMC Cancer.
19:1412019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Coscia F, Lengyel E, Duraiswamy J,
Ashcroft B, Bassani-Sternberg M, Wierer M, Johnson A, Wroblewski K,
Montag A, Yamada SD, et al: Multi-level proteomics identifies CT45
as a chemosensitivity mediator and immunotherapy target in ovarian
cancer. Cell. 175:159–170 e16. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
George J, Alsop K, Etemadmoghadam D,
Hondow H, Mikeska T, Dobrovic A, deFazio A; Australian Ovarian
Cancer Study Group, ; Smyth GK, Levine DA, et al: Nonequivalent
gene expression and copy number alterations in high-grade serous
ovarian cancers with BRCA1 and BRCA2 mutations. Clin Cancer Res.
19:3474–3484. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McLaughlin JR, Rosen B, Moody J, Pal T,
Fan I, Shaw PA, Risch HA, Sellers TA, Sun P and Narod SA: Long-term
ovarian cancer survival associated with mutation in BRCA1 or BRCA2.
J Natl Cancer Inst. 105:141–148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen L, Yao Y, Sun L, Zhou J, Miao M, Luo
S, Deng G, Li J, Wang J and Tang J: Snail driving alternative
splicing of CD44 by ESRP1 enhances invasion and migration in
epithelial ovarian cancer. Cell Physiol Biochem. 43:2489–2504.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pusapati RV, Daemen A, Wilson C, Sandoval
W, Gao M, Haley B, Baudy AR, Hatzivassiliou G, Evangelista M and
Settleman J: mTORC1-dependent metabolic reprogramming underlies
escape from glycolysis addiction in cancer cells. Cancer Cell.
29:548–562. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu S, Yin H, Ji H, Zhu J and Ma R:
Overexpression of TRIM44 is an independent marker for predicting
poor prognosis in epithelial ovarian cancer. Exp Ther Med.
16:3034–3040. 2018.PubMed/NCBI
|
|
26
|
Kim SI, Lee JW, Lee N, Lee M, Kim HS,
Chung HH, Kim JW, Park NH, Song YS and Seo JS: LYL1 gene
amplification predicts poor survival of patients with uterine
corpus endometrial carcinoma: Analysis of the Cancer genome atlas
data. BMC Cancer. 18:4942018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li LX, Zhou JX, Calvet JP, Godwin AK,
Jensen RA and Li X: Lysine methyltransferase SMYD2 promotes triple
negative breast cancer progression. Cell Death Dis. 9:3262018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zuo SR, Zuo XC, He Y, Fang WJ, Wang CJ,
Zou H, Chen P, Huang LF, Huang LH, Xiang H and Liu SK: Positive
expression of SMYD2 is associated with poor prognosis in patients
with primary hepatocellular carcinoma. J Cancer. 9:321–330. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reynoird N, Mazur PK, Stellfeld T, Flores
NM, Lofgren SM, Carlson SM, Brambilla E, Hainaut P, Kaznowska EB,
Arrowsmith CH, et al: Coordination of stress signals by the lysine
methyltransferase SMYD2 promotes pancreatic cancer. Genes Dev.
30:772–785. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zodro E, Jaroszewski M, Ida A, Wrzesiński
T, Kwias Z, Bluyssen H and Wesoly J: FUT11 as a potential biomarker
of clear cell renal cell carcinoma progression based on
meta-analysis of gene expression data. Tumour Biol. 35:2607–2617.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang K, Zhou Y, Li G, Wen X, Kou Y, Yu J,
He H, Zhao Q, Xue F, Wang J and Zhao X: MMP8 and MMP9 gene
polymorphisms were associated with breast cancer risk in a Chinese
Han population. Sci Rep. 8:134222018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wieczorek E, Reszka E, Wasowicz W,
Grzegorczyk A, Konecki T, Sosnowski M and Jablonowski Z: MMP7 and
MMP8 genetic polymorphisms in bladder cancer patients. Cent
European J Urol. 66:405–410. 2014.PubMed/NCBI
|
|
33
|
Dębniak T, Jakubowska A, Serrano-Fernández
P, Kurzawski G, Cybulski C, Chauhan SR, Laxton RC, Maleszka R,
Lubinski J and Ye S: Association of MMP8 gene variation with an
increased risk of malignant melanoma. Melanoma Res. 21:464–468.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Palavalli LH, Prickett TD, Wunderlich JR,
Wei X, Burrell AS, Porter-Gill P, Davis S, Wang C, Cronin JC,
Agrawal NS, et al: Analysis of the matrix metalloproteinase family
reveals that MMP8 is often mutated in melanoma. Nat Genet.
41:518–520. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun Q, Zhang Y, Su J, Li T and Jiang Y:
Role of hydroxysteroid dehydrogenase-like 2 (HSDL2) in human
ovarian cancer. Med Sci Monit. 24:3997–4008. 2018. View Article : Google Scholar : PubMed/NCBI
|