Introduction
Pancreatic ductal adenocarcinoma (PDAC) exhibits a
high propensity for metastasis, and is therefore considered to be a
highly lethal human malignancy (1,2) with a
low 5-year survival rate of <5% (3–6).
Previous reports have demonstrated that both transcriptional and
translational downregulation of E-cadherin in parental cells
initiated epithelial-mesenchymal transition (EMT), consequently
promoting metastasis (7–9). The inhibition of adhesion between
epithelial cells (cell-cell adhesion) is known to be an essential
step during the early stage of tumor metastasis, and typically
involves the downregulation of E-cadherin expression (10,11).
The ubiquitin-proteasome pathway is associated with
the degradation of regulatory proteins that influence DNA repair,
signal transduction and apoptosis, and therefore, the proteasome
represents a promising target for cancer therapy (12,13).
MG132 is a well-characterized proteasome inhibitor that induces
caspase-8-dependent osteosarcoma-cell apoptosis (14). Moreover, MG132 was reported to
inhibit the transcriptional activity and expression of the
oncogenic transcription factor forkhead box M1, promoting apoptosis
in several human cancer types (15).
Similarly, the anti-proliferative effect of MG132 and cisplatin
co-administration for the treatment of osteosarcoma xenograft
models was superior to the monotherapeutic effect of cisplatin,
supporting the potential of MG132/cisplatin for the treatment of
patients with osteosarcoma (12).
Additionally, it was discovered that MG132 may upregulate the
expression of nuclear factor erythroid 2-related factor 2 and
increase proteasome activity via the inhibition of diabetic
nephropathy in an animal model (16). Collectively, the aforementioned
results suggest that MG132 affects the growth of numerous solid
tumor types, and should consequently be considered as a therapeutic
target.
E26 transformation-specific family transcription
factor (ESE3) plays a pivotal role in the differentiation and
development programs of numerous epithelial tissue types. ESE3 has
been reported to bind directly to its target genes with the
propensity to regulate EMT, stem-like physiological processes (an
increased ability to form spheres, similar to epithelial stem
cells) (17–19) and tumor progression (17,20).
Therefore, ESE3 is considered as a potential tumor suppressor gene
associated with pancreatic cancer. In addition, ESE3 was shown to
inhibit the migration and invasion capacities of PDAC cells (both
in vitro and in vivo), suggesting that the mechanism
behind its regulation of PDAC metastasis may involve the regulation
of E-cadherin expression levels at the transcriptional level
(21). In summary, the aim of the
present study was to investigate the effect of MG132 on the
expression level of E-cadherin and the accumulation of ESE3, and to
determine its influence on the migration and invasion abilities of
PDAC cells.
Materials and methods
Reagents and short interfering
(si)RNAs
Proteasome inhibitor MG132 (cat. no. SML1135) and
cycloheximide (CHX; cat. no. C104450) were purchased from
Sigma-Aldrich; Merck KGaA. The primary antibodies were purchased
from Abcam and include; ESE3 (cat. no. ab24337), E-cadherin (cat.
no. ab76055) and β-actin (cat. no. E4D9Z; all Cell Signaling
Technology, Inc.) antibodies. To determine the influence of ESE3
expression levels on MG132 activity, ESE3 was knocked down in
PANC-1 and SW1990 cells by the transfection of specific siRNAs
(ESE3#1 forward, 5′-GCCAGUGGCAUGAAAUUCATT-3′ and reverse,
5′-UGAAUUUCAUGCCACUGGCTT-3′; and ESE3#2 forward,
5′-CAGCCGAGCUAUGAGAUAUTT-3′ and reverse,
5′-AUAUCUCAUAGCUCGGCUGTT-3′) and an unrelated silencing sequence
was synthesized as a negative control (siNC forward,
5′-lUUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′). All siRNAs were purchased from Sangon
Biotech Co., Ltd. Stock solutions of MG132 were prepared in DMSO at
a concentration of 10 mg/ml and then diluted in cell culture medium
to a final working concentration of 10 µM.
Cell culture conditions and
pharmacological treatment
The SW1990 and PANC-1 PDAC cell lines were cultured
in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.) containing 10% fetal bovine serum (FBS, Shanghai
ExCell Biology, Inc.) at 37°C, in a humidified culture chamber with
5% CO2. The cells were detached from the culture plates
using trypsin (0.05%) and EDTA (0.5 mM) in phosphate-buffered
saline (PBS). The cell lines were treated with 10 µM MG132 for 0,
2, 4 and 6 h at 37°C. Groups of control cells treated with DMSO
only were evaluated in parallel to the experimental groups.
Cycloheximide (CHX), a protein-synthesis inhibitor, was used to
test the degradation of proteins following MG132 and DMSO treatment
at 50 µg/ml for 4 h. All experiments were repeated three times.
Inhibitory effects of MG132
The half-maximal inhibitory concentration
(IC50) of MG132 on PDAC cells was detected using the
Real-Time Cellular Analysis (RTCA) system (ACEA Biosciences, Inc.).
DMEM (10% FBS) was added to the E-Plate assay (ACEA Biosciences,
Inc.) to determine the background impedance values. Log-phase cells
were collected and counted to achieve a suspension of
4×103 cells/well; the cells were then added to the
E-Plate on a test stand (having been previously incubated at 37°C
and 5% CO2), and left to react at room temperature for
30 min. Real-time dynamic cell-proliferation detection was
performed overnight at 37°C, 5% CO2. The MG132 stock
solution was added to the corresponding concentration gradient
solutions, and real-time dynamic detection was continued. The cell
effect curves and IC50 values were obtained using the
RTCA Software (RTCA Software Lite 2.2.1).
Western blotting
The cells were washed with PBS and then lysed using
RIPA buffer containing 1% protease inhibitor (both Sigma-Aldrich;
Merck KGaA). Equal amounts of protein were then resolved using 10%
SDS-PAGE. Protein concentrations were quantified using the Pierce
protein assay kit (cat. no. UA269551, Thermo Fisher Scientific,
Inc.). The separated proteins (20 µg/lane) were carefully
transferred to polyvinylidene fluoride membranes at 4°C for 2 h.
Subsequently the proteins were blocked using 5% skimmed milk (BD
Biosciences) and probed with the following primary antibodies:
Anti-ESE3 (1:1,000; cat. no. ab24337), anti-E-cadherin (1:1,000;
both Abcam; cat. no. ab76055) and β-actin (1:5,000; cat. no. E4D9Z;
Cell Signaling Technology, Inc.) for 2 h at room temperature.
Following primary incubation, the membranes were incubated with
horseradish peroxidase-conjugated goat anti-rabbit (1:10,000;
ProteinTech Group, Inc; cat. no. SA00001-2)/anti-mouse secondary
antibodies (1:10,000; ProteinTech Group, Inc; cat. no. SA00001-1)
for 1 h at room temperature. Protein quantification was performed
using Image Lab Software (version 5.2.1, Bio-Rad Laboratories,
Inc).
Wound-healing and invasion assays
For the wound-healing assay, the PANC-1 and SW1990
cells (treated with either MG132 or DMSO) were seeded into a
24-well plate prior to wounding. The wounds were created by
scratching across the monolayer using sterile pipette tips. The
cells were induced with DMEM containing 1% FBS. After a 48-h
incubation in a humidified culture chamber (5% CO2) the
cells were imaged using an inverted routine microscope
(magnification, ×40; ECLIPSE Ts2; Nikon Corporation).
For the invasion assay, 1×105 cells were
added to the upper chamber of a transwell insert with DMEM
containing 1% FBS, and filters coated with Matrigel (1:5 dilution,
BD Biosciences) were used. DMEM containing 10% FBS was added to the
lower chamber and used as a chemoattractant. The cells were
subsequently incubated under controlled conditions (37°C and 5%
CO2) for 18 h. A three-step stain set (Corning Inc.) was
used to stain the cells that had migrated to the bottoms of the
filters at room temperature (each step was stained for 5 min).
Then, 10 different fields of view were randomly selected, and cell
counting was performed using an inverted routine microscope
(magnification, 40×; ECLIPSE Ts2; Nikon Corporation). The mean cell
numbers were calculated and statistical analyses were performed on
these values.
Immunofluorescence
To investigate the expression patterns of ESE3 in
PDAC cells treated with MG132, 2×103 PANC-1 cells were
seeded onto glass microscope slides, washed with PBS, and then
fixed with 4% paraformaldehyde for 10 min at room temperature. The
cells were then permeabilized with 0.3% Triton X-100 in PBS for 5
min at room temperature. To minimize non-specific staining, the
cells were treated with 3% bovine serum albumin (Sigma-Aldrich;
Merck KGaA) in PBS for 1 h. During the immunolabeling step, an
anti-ESE3 antibody (1:100 dilution) was used to stain the cells,
overnight at 4°C. The nuclei of the labeled cells were subsequently
stained with 4′,6-diamidino-2-phenylindole (DAPI) Fluoromount-G
medium (Southern Biotech), and the slides were viewed using a
confocal fluorescence microscope (magnification, ×200; LSN 880;
ZEISS; GmbH).
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 7.0 software (GraphPad Software, Inc.). The unpaired
Student's t-test or Mann-Whitney U test were used to compare the
differences between two groups, and one-way ANOVA followed by the
SNK-q test was used to compare ≥3 groups. The results are presented
as the mean ± standard error of the mean (SEM) and P<0.05 was
considered to indicate a statistically significant result.
Results
Proteasome inhibitor MG132 exhibits
cytotoxic effects and inhibits the migration and invasion of
pancreatic cancer cells
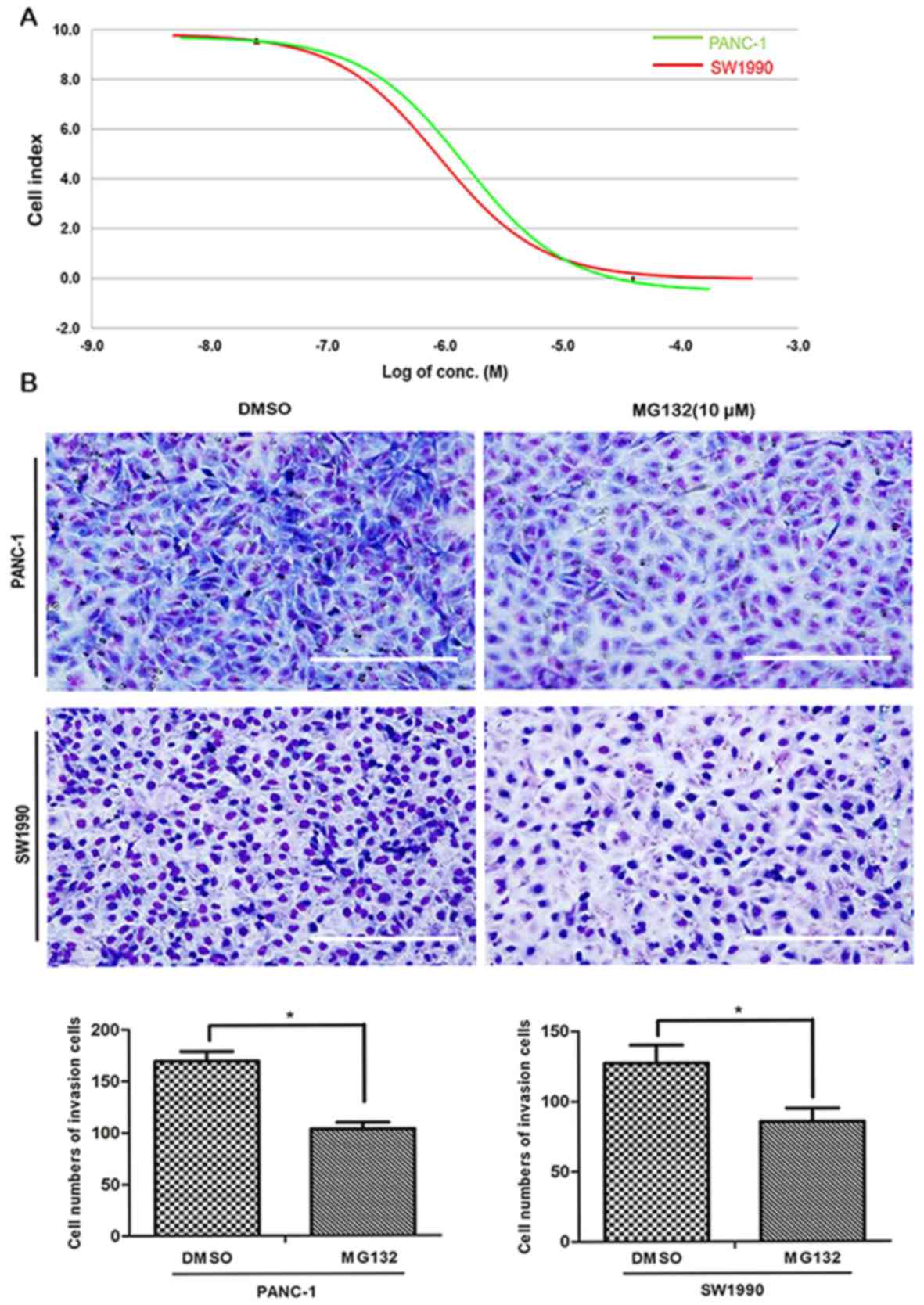
After 48 h of treatment, the IC50 values
of MG132 in PANC-1 and SW1990 PDAC cells were 11.20±0.742 and
11.18±0.787 µM, respectively, according to RTCA (Fig. 1A). Based on this finding, and
consistent with previously reported literature (22), the MG132 concentration was fixed at
10 µM for subsequent experiments.
The effect of MG132 on the invasion cell number of
PANC-1 and SW1990 cells was evaluated using Transwell chambers. The
number of PANC-1 and SW1990 cells passing through the polycarbonate
membrane at 18 h was reduced by 10 µM MG132, compared with that of
the DMSO-treated control group (Fig.
1B). The mean number of invading PANC-1 cells was 105 for the
treatment group, compared with 169 for the control group, and the
mean number of invading SW1990 cells was 85 for the treatment
group, compared with 128 for the control group (P<0.05). The
wound-healing assay revealed that the migration of PANC-1 and
SW1990 cells was significantly reduced following MG132 treatment.
The wound areas for PANC-1 cells were ~12.0±4.1% at 48 h for the
MG132-treated group, compared with ~47.2±3.8% for the control group
(P<0.01; Fig. 1C). SW1990-cell
wound healing was also significantly suppressed, with wound areas
of ~27.6±2.0% at 48 h for the MG132-treated group, compared with
~43.9±2.8% for the control group (P<0.005; Fig. 1C). The results of the present study
suggest that MG132 is able to inhibit the invasion and migration
properties of PANC-1 and SW1990 cells.
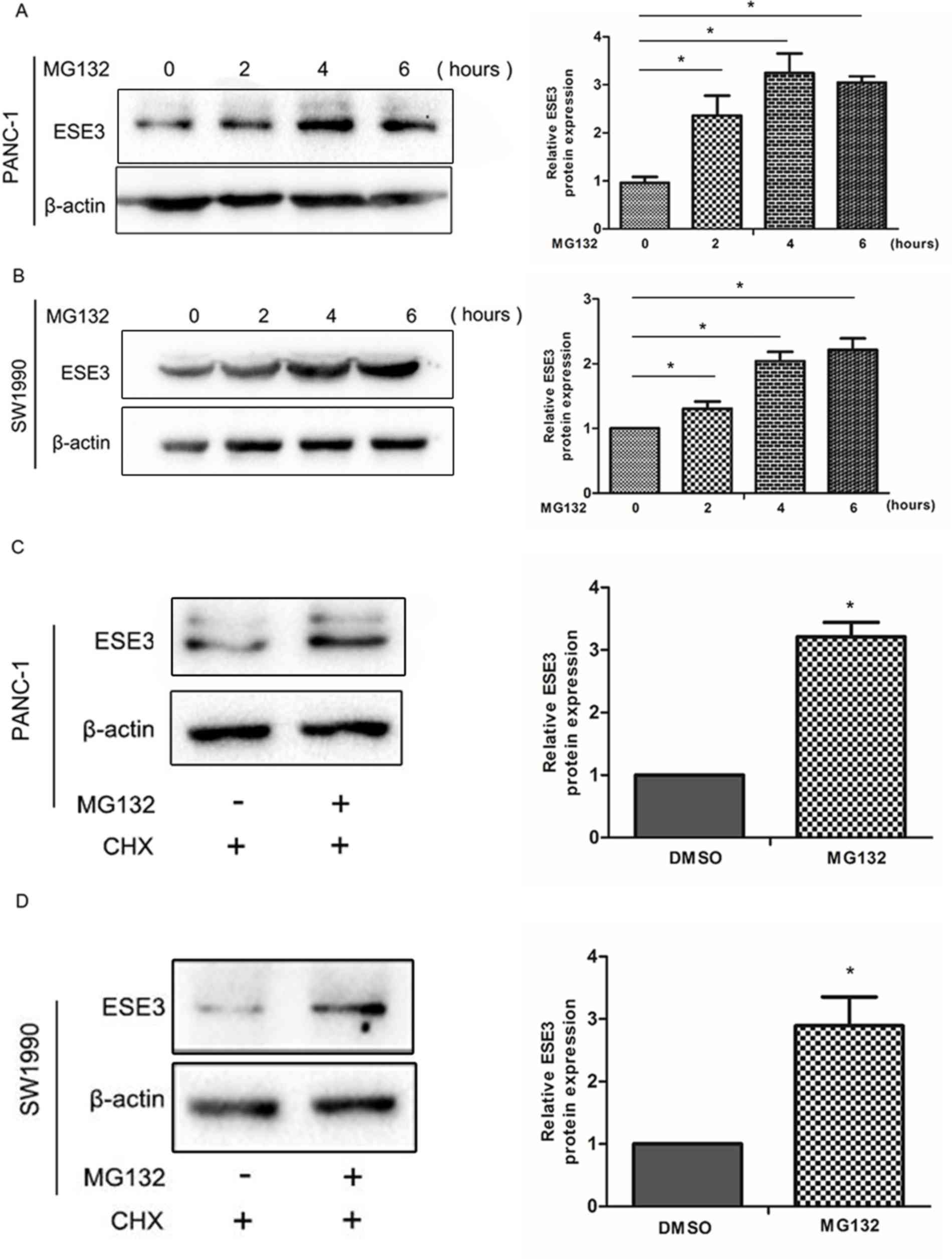
Degradation of ESE3 via the
ubiquitin-proteasome pathway is inhibited by MG132
To investigate the potential mechanism of
MG132-associated suppression of PDAC cell invasion and migration,
western blotting and immunofluorescence experiments were performed.
Following the treatment of pancreatic cell lines (PANC-1 and
SW1990) with 10 µM MG132 for various time periods (0–6 h), the ESE3
expression levels increased in a time-dependent manner (Fig. 2A and B; P<0.05). To determine
whether the observed increase involved an MG132-dependent pathway,
10 µM MG132 was added to PANC-1 and SW1990 cells for 2 h, followed
by treatment with CHX (50 µg/ml) for 4 h to inhibit ESE3 protein
synthesis. Western blotting determined that ESE3 exhibited high
expression levels following MG132 treatment (Fig. 2C and D; P<0.05). Thus, it was
demonstrated that MG132 increased ESE3 levels via the inhibition of
its proteasomal degradation.
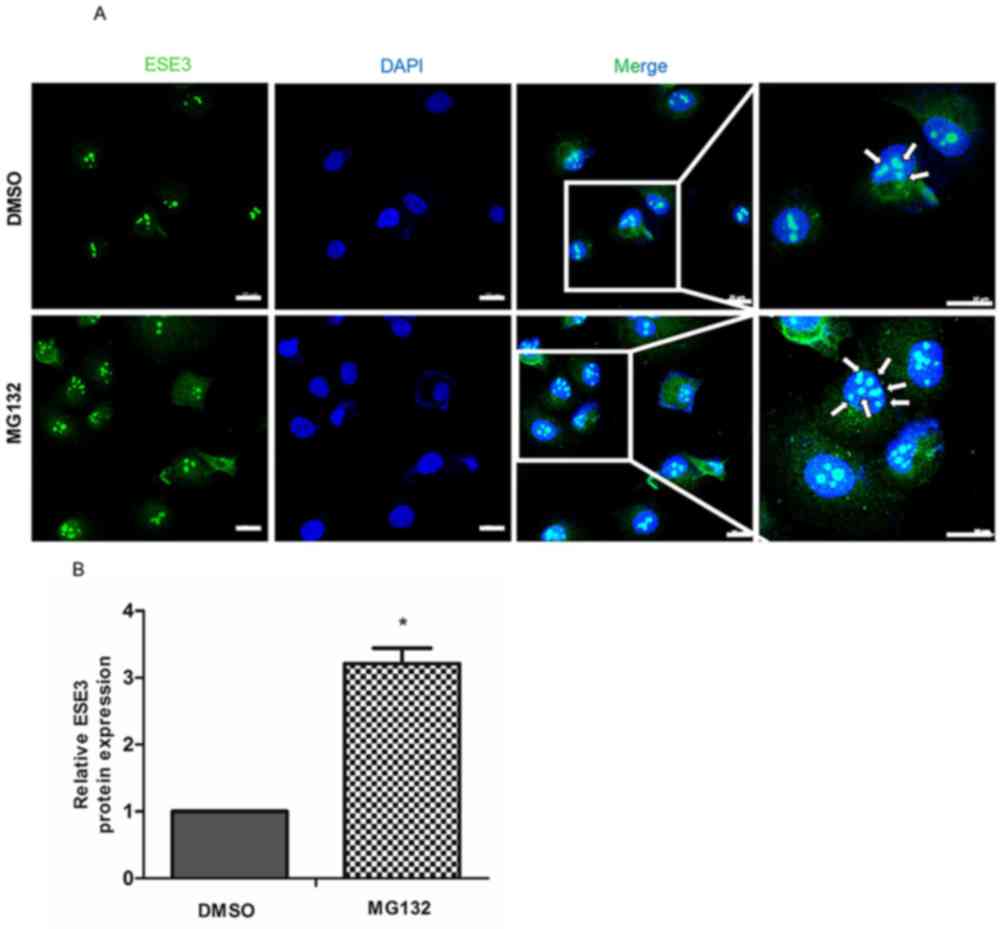
MG132 increases ESE3 nuclear
translocation
Previous studies have determined that ESE3 is
primarily localized in the nucleus of normal epithelial esophageal
cells (20,23,24).
Hence, the nuclear expression of ESE3 was investigated using
immunofluorescence in PDAC cells treated with MG132. Fluorescent
staining was performed to visualize the expression of ESE3
(Fig. 3A). Arrowheads indicate the
nuclear expression of ESE3 (Fig.
3A), and the number of arrowheads was determined and compared
with the DMSO-treated control group. The nuclear translocation of
ESE3 was increased in PANC-1 cells treated with MG132 (Fig. 3A; P<0.05), yet no significant
increase was observed in SW1990 cells.
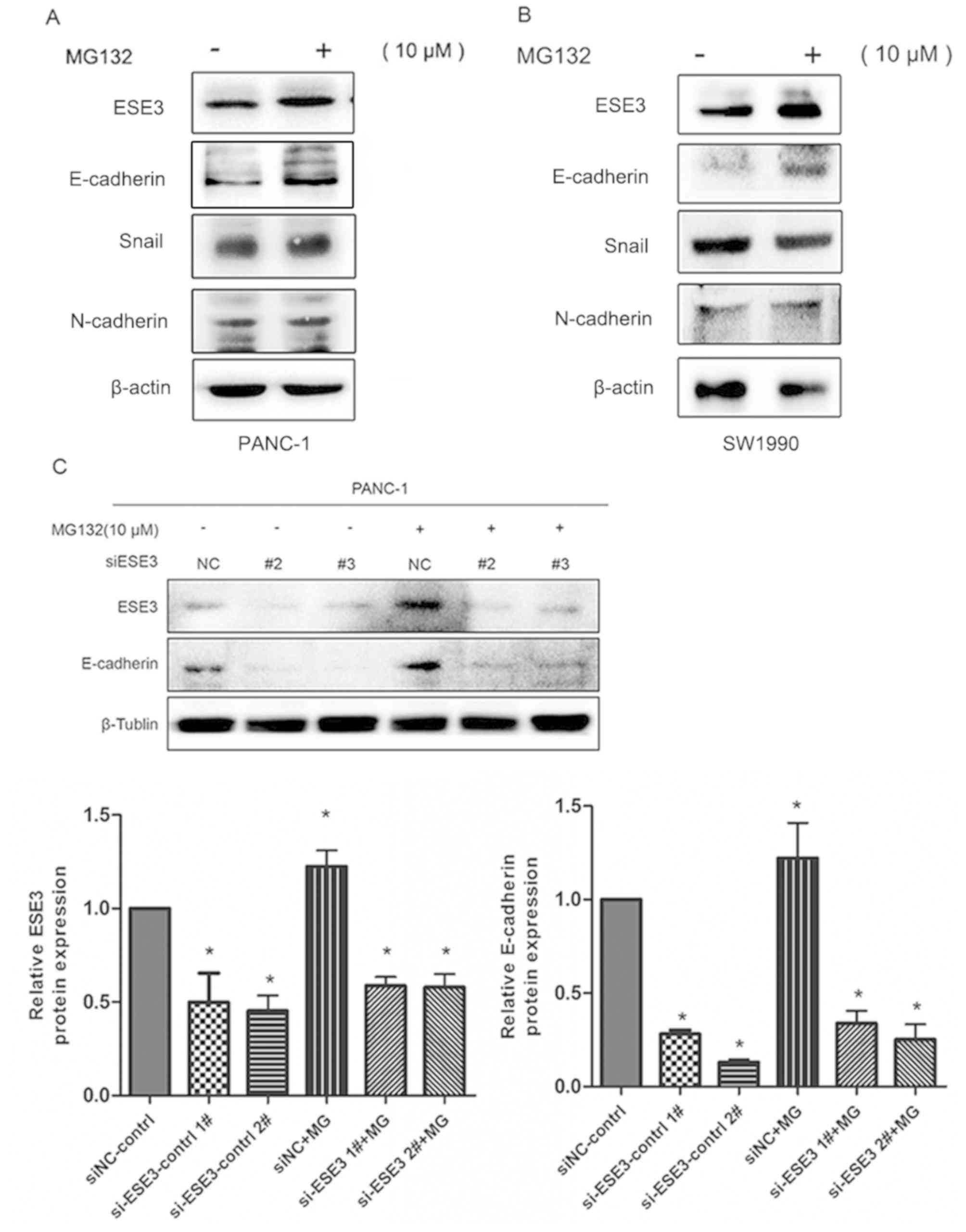
MG132 upregulates E-cadherin
expression levels via the upregulation of ESE3
The results of the present study indicated that
treatment with MG132 was associated with increased expression of
ESE3 in PDAC cells. To investigate the factors affecting the
migration and invasion of PDAC cells down-stream of ESE3,
E-cadherin [a classical EMT marker and a direct target of ESE3;
(21)] expression levels were
determined to be increased in both SW1990 and PANC-1 cell lines,
following MG132 treatment (Fig. 4A and
B). However, similar changes were not detected in other
EMT-related markers such as N-cadherin or Snail. Additionally, in
PANC-1 cells transfected with siESE3, the upregulation of
E-cadherin expression caused by MG132 was significantly inhibited
(Fig. 4C; P<0.05). This suggested
that MG132 directly stimulates E-cadherin expression via the
increased expression of ESE3.
ESE3-knockdown and low E-cadherin
expression reverse the inhibitory effect of MG132 on the migration
and invasion abilities of PANC-1 and SW1990 cells
To assess the influence of ESE3 expression on the
migration and invasion capacities of PANC-1 cells treated with
MG132, ESE3 expression was knocked down using siESE3 and the effect
was analyzed using Transwell and wound-healing assays. The results
showed that knockdown of ESE3 expression significantly reduced the
inhibitory effect of MG132 on PANC-1 and SW1990 cell invasiveness
(Fig. 5A and B) and migration
(Fig. 5C and D). These findings
suggest that ESE3 is an important target under MG132 treatment
conditions.
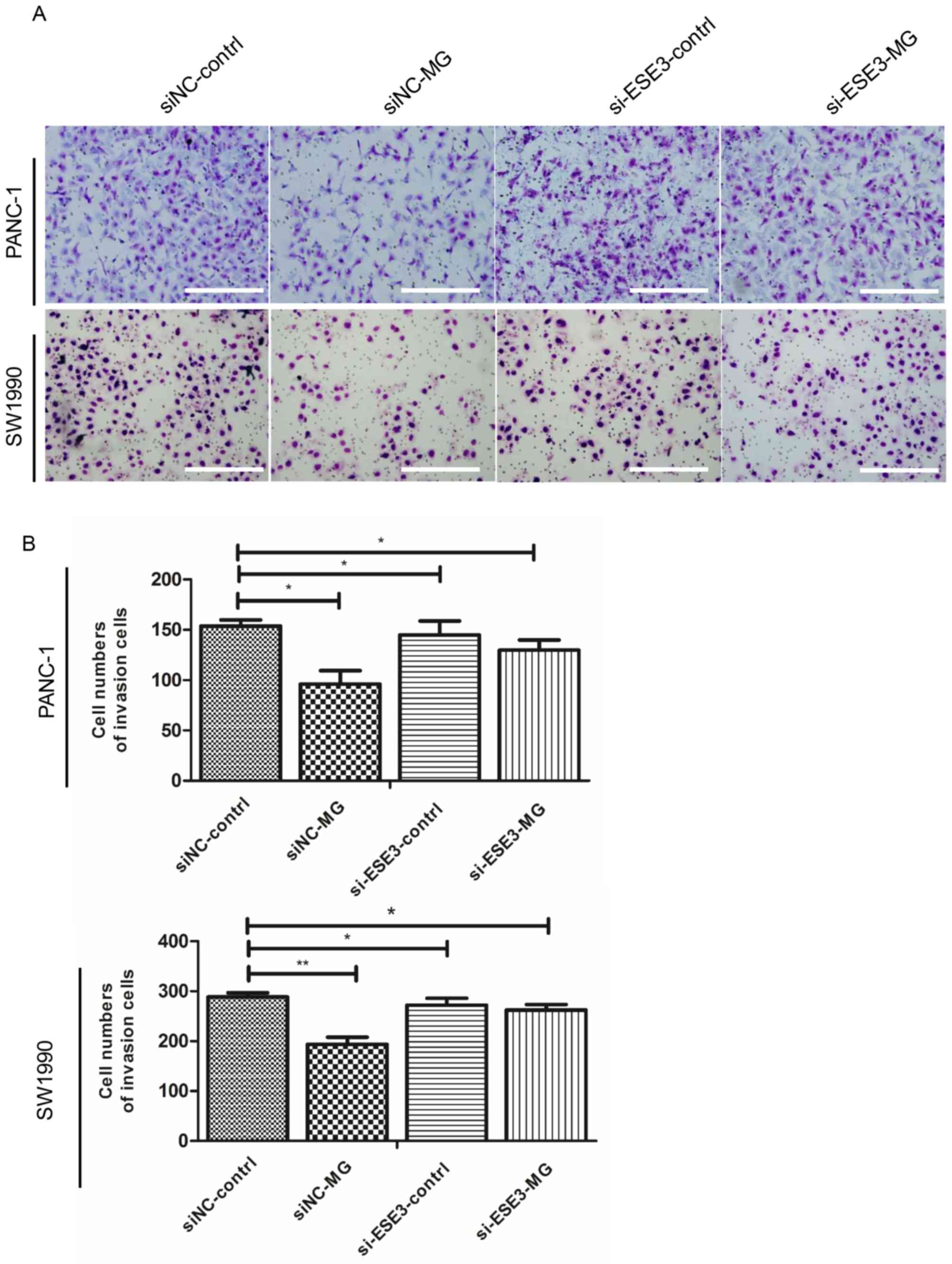 | Figure 5.Knockdown of ESE3 and the expression
level of E-cadherin inhibits the migration and invasion of PANC-1
cells. PANC-1 cells were transfected with preselected siRNAs
targeting ESE3. (A) Invasion was inhibited in both the
si-ESE3-control and si-ESE3-MG groups with or without MG132
treatment (10 µM, 48 h), respectively. Results and representative
images are exhibited for PANC-1 and SW1990 cells treated in the
presence and absence of MG132. (B) Number of cells that flowed
through the Transwell chamber. Bars represent the migration index
of each treatment. *P<0.05 and **P<0.01 vs. the control
group. All experiments were repeated three times; scale bar, 200
µm. ESE3, ETS homologous factor; siNC, short interfering negative
control; MG, MG132. Knockdown of ESE3 and the expression level of
E-cadherin inhibits the migration and invasion of PANC-1 cells.
PANC-1 cells were transfected with preselected siRNAs targeting
ESE3. (C) Migration was inhibited in both the si-ESE3-control and
si-ESE3-MG groups with or without MG132 treatment (10 µM, 48 h),
respectively. Results and representative images are exhibited for
PANC-1 and SW1990 cells treated in the presence and absence of
MG132. (D) Values were calculated relative to the closure distance
of the cell monolayers. *P<0.05 and **P<0.01 vs. the control
group. All experiments were repeated three times; scale bar, 200
µm. ESE3, ETS homologous factor; siNC, short interfering negative
control; MG, MG132. |
Discussion
It has been reported that during the early stages of
tumor metastasis, it is important to inhibit cell-cell adhesion
between epithelial cells (25–27).
Indeed, inhibition of the cellular adhesion mediated by E-cadherin
during this period represents a major step in the treatment of
primary tumors, and serves as a classical EMT marker (28–30). As
a nuclear factor, ESE3 may play a role in this process by directly
binding to the promoter of the E-cadherin gene in PDAC cells,
thereby stimulating its expression (21).
In the current study, it was determined that the
proteasome inhibitor MG132 inhibited PDAC cell invasion and
migration in vitro, which has not been previously reported.
To determine the molecular mechanism underlying this process, the
expression level of the EMT marker E-cadherin (but not snail or
N-cadherin) was evaluated in PDAC cells. E-cadherin expression
increased following treatment with MG132. Further investigation
indicated that MG132 increased the accumulation of ESE3 by
inhibiting its proteasomal degradation. An immunofluorescence assay
was performed to investigate MG132-associated promotion of
translocation to the nucleus in PANC-1 cells; however, no
significant increase was observed in SW1990 cells. Data from the
present study indicate that MG132 activated E-cadherin by
influencing both the expression level and translocation of ESE3.
The addition of CHX demonstrated that the increase in ESE3 levels
was not influenced by an increase in protein synthesis. Thus, MG132
suppressed the invasion and migration of two PDAC cell lines by
increasing ESE3 expression levels, via the suppression of the
relevant proteasome pathway, and its down-stream target, the
E-cadherin gene.
Subsequently, the central role of ESE3 in the
inhibition of migration and invasion in PDAC cells (which was
increased by MG132) was further investigated. ESE3 was knocked down
by transfection of PDAC cells with siESE3. The rescue experiment
demonstrated that siESE3 reduced MG132-induced inhibition of
migration and invasion in PDAC cell lines. This conclusion was
further supported by the discovery that MG132 promoted
E-cadherin-induced PDAC migration and invasion through the
accumulation of ESE3.
Future studies should focus on discovering the
specific signaling pathway by which MG132 influences the expression
of ESE3, as this may further explain the difference in
translocation of ESE3 to the nucleus between PANC-1 cells and
SW1990 cells, following MG132 treatment.
Overall, the findings of the present study improve
our understanding of the biological function of MG132 in PDAC
metastasis. The current results suggest that MG132 may be combined
with other chemotherapeutic drugs for the treatment PDAC, in order
to block the signaling pathway that downregulates ESE3 expression.
By inhibiting PDAC metastasis, this strategy may result in improved
patient outcome.
Acknowledgements
Not applicable.
Funding
The present was supported by Major Anticancer
Technologies R&D Program of Tianjin (grant. no. 12ZCDZSY16700)
and the National Science Foundation of China (grant nos. 81502067,
31470951, 81302082, 81272685, 31301151, 81172355, 31471340,
31470957, 81472264 and 81401957).
Availability of data and material
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
FJ and TZ conceived and designed the research. The
experiments and data analysis were performed by FJ and DX. MY and
TZ contributed materials, reagents and analysis tools. The
manuscript was written by FJ.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Roy R, Zurakowski D, Wischhusen J,
Frauenhoffer C, Hooshmand S, Kulke M and Moses MA: Urinary TIMP-1
and MMP-2 levels detect the presence of pancreatic malignancies. Br
J Cancer. 111:1772–1779. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roe JS, Hwang CI, Somerville TDD, Milazzo
JP, Lee EJ, Da Silva B, Maiorino L, Tiriac H, Young CM, Miyabayashi
K, et al: Enhancer reprogramming promotes pancreatic cancer
metastasis. Cell. 170:875–888.e20. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Knudsen ES, O'Reilly EM, Brody JR and
Witkiewicz AK: Genetic diversity of pancreatic ductal
adenocarcinoma and opportunities for precision medicine.
Gastroenterology. 150:48–63. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang C, Li N, Li Z, Chang A, Chen Y, Zhao
T, Li Y, Wang X, Zhang W, Wang Z, et al: Tumour-derived Interleukin
35 promotes pancreatic ductal adenocarcinoma cell extravasation and
metastasis by inducing ICAM1 expression. Nat Commun. 8:140352017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
DeSantis CE, Miller KD, Goding Sauer A,
Jemal A and Siegel RL: Cancer statistics for African Americans,
2019. CA Cancer J Clin. 69:211–233. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kotiyal S and Bhattacharya S: Events of
molecular changes in epithelial-mesenchymal transition. Crit Rev
Eukaryot Gene Expr. 26:163–171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Amawi H, Ashby CR, Samuel T, Peraman R and
Tiwari AK: Polyphenolic nutrients in cancer chemoprevention and
metastasis: Role of the epithelial-to-mesenchymal (EMT) pathway.
Nutrients. 9(pii): E9112017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mao XY, Li QQ, Gao YF, Zhou HH, Liu ZQ and
Jin WL: Gap junction as an intercellular glue: Emerging roles in
cancer EMT and metastasis. Cancer Lett. 381:133–137. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Petit CS, Barreau F, Besnier L, Gandille
P, Riveau B, Chateau D, Roy M, Berrebi D, Svrcek M, Cardot P, et
al: Requirement of cellular prion protein for intestinal barrier
function and mislocalization in patients with inflammatory bowel
disease. Gastroenterology. 143:122–132.e15. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moirangthem A, Bondhopadhyay B, Mukherjee
M, Bandyopadhyay A, Mukherjee N, Konar K, Bhattacharya S and Basu
A: Simultaneous knockdown of uPA and MMP9 can reduce breast cancer
progression by increasing cell-cell adhesion and modulating EMT
genes. Sci Rep. 6:219032016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun F, Zhang Y, Xu L, Li S, Chen X, Zhang
L, Wu Y and Li J: Proteasome inhibitor MG132 enhances
cisplatin-induced apoptosis in osteosarcoma cells and inhibits
tumor growth. Oncol Res. 26:655–664. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Crawford LJ and Irvine AE: Targeting the
ubiquitin proteasome system in haematological malignancies. Blood
Rev. 27:297–304. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan XB, Yang DS, Gao X, Feng J, Shi ZL and
Ye Z: Caspase-8 dependent osteosarcoma cell apoptosis induced by
proteasome inhibitor MG132. Cell Biol Int. 31:1136–1143. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gartel AL: A new target for proteasome
inhibitors: FoxM1. Expert Opin Investig Drugs. 19:235–242. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cui W, Bai Y, Luo P, Miao L and Cai L:
Preventive and therapeutic effects of MG132 by activating Nrf2-ARE
signaling pathway on oxidative stress-induced cardiovascular and
renal injury. Oxid Med Cell Longev. 2013:3060732013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Albino D, Longoni N, Curti L, Mello-Grand
M, Pinton S, Civenni G, Thalmann G, D'Ambrosio G, Sarti M, Sessa F,
et al: ESE3/EHF controls epithelial cell differentiation and its
loss leads to prostate tumors with mesenchymal and stem-like
features. Cancer Res. 72:2889–2900. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Longoni N, Kunderfranco P, Pellini S,
Albino D, Mello- Grand M, Pinton S, D'Ambrosio G, Sarti M, Sessa F,
Chiorino G, et al: Aberrant expression of the neuronal-specific
protein DCDC2 promotes malignant phenotypes and is associated with
prostate cancer progression. Oncogene. 32:2315–2324, 2324.e1-e4.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Albino D, Civenni G, Rossi S, Mitra A,
Catapano CV and Carbone GM: The ETS factor ESE3/EHF represses IL-6
preventing STAT3 activation and expansion of the prostate cancer
stem-like compartment. Oncotarget. 7:76756–76768. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang L, Xing J, Cheng R, Shao Y, Li P, Zhu
S and Zhang S: Abnormal localization and tumor suppressor function
of epithelial tissue-specific transcription factor ESE3 in
esophageal squamous cell carcinoma. PLoS One. 10:e01263192015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao T, Jiang W, Wang X, Wang H, Zheng C,
Li Y, Sun Y, Huang C, Han ZB, Yang S, et al: ESE3 inhibits
pancreatic cancer metastasis by upregulating E-cadherin. Cancer
Res. 77:874–885. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
He M, Qiao Z, Wang Y, Kuai Q, Li C, Wang
Y, Jiang X, Wang X, Li W, He M, et al: chidamide inhibits aerobic
metabolism to induce pancreatic cancer cell growth arrest by
promoting Mcl-1 degradation. PLoS One. 11:e01668962016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Seth A and Watson DK: ETS transcription
factors and their emerging roles in human cancer. Eur J Cancer.
41:2462–2478. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jain N, Morgan CE, Rife BD, Salemi M and
Tolbert BS: Solution structure of the HIV-1 intron splicing
silencer and its interactions with the UP1 domain of heterogeneous
nuclear ribonucleoprotein (hnRNP) A1. J Biol Chem. 291:2331–2344.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kahlert UD, Joseph JV and Kruyt FAE: EMT-
and MET-related processes in nonepithelial tumors: Importance for
disease progression, prognosis, and therapeutic opportunities. Mol
Oncol. 11:860–877. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim KS, Kim J, Oh N, Kim MY and Park KS:
ELK3-GATA3 axis modulates MDA-MB-231 metastasis by regulating
cell-cell adhesion-related genes. Biochem Biophys Res Commun.
498:509–515. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang M, Hu C, Tong D, Xiang S, Williams
K, Bai W, Li GM, Bepler G and Zhang X: Ubiquitin-specific peptidase
10 (USP10) deubiquitinates and stabilizes MutS homolog 2 (MSH2) to
regulate cellular sensitivity to DNA damage. J Biol Chem.
291:10783–10791. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sanchez-Tillo E, Lazaro A, Torrent R,
Cuatrecasas M, Vaquero EC, Castells A, Engel P and Postigo A: ZEB1
represses E-cadherin and induces an EMT by recruiting the SWI/SNF
chromatin-remodeling protein BRG1. Oncogene. 29:3490–3500. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matos ML, Lapyckyj L, Rosso M, Besso MJ,
Mencucci MV, Briggiler CI, Giustina S, Furlong LI and Vazquez-Levin
MH: Identification of a novel human E-cadherin splice variant and
assessment of its effects upon EMT-related events. J Cell Physiol.
232:1368–1386. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Feldkoren B, Hutchinson R, Rapoport Y,
Mahajan A and Margulis V: Integrin signaling potentiates
transforming growth factor-beta 1 (TGF-β1) dependent
down-regulation of E-cadherin expression-important implications for
epithelial to mesenchymal transition (EMT) in renal cell carcinoma.
Exp Cell Res. 355:57–66. 2017. View Article : Google Scholar : PubMed/NCBI
|