Introduction
Neuroblastoma (NBL) is a common solid tumor in
children that originates from primordial neural crest cells
(1). Despite multidisciplinary
therapies, patients with high-risk NBL have a dismal prognosis.
Recent genome-wide analyses have revealed anaplastic lymphoma
kinase (ALK) gene alterations, amplification, and activating
heterozygous mutations in the tyrosine kinase domain (TKD) in
approximately 8% of primary NBLs (2–5).
ALK mutations, often detected in heterozygous, are involved
in cell proliferation and invasion and more frequently occur in
high-risk and relapsed cases (1,6), while
homozygous ALK mutations are quite rare (4,7). The
discovery of ALK alterations provides the first tractable
oncogenic target in NBL and facilitates the clinical application of
ALK inhibitors, such as crizotinib (8). Here we present the case of a female
patient with relapsed high-risk NBL harboring a heterozygous
ALK F1245L mutation at the time of the initial diagnosis.
This mutation became homozygous at relapse due to uniparental
disomy (UPD), which was associated with aggressive tumor behavior
after relapse. We further analyzed genetic alterations that may
explain the molecular mechanism for NBL relapse.
Materials and methods
Samples
Biopsy samples at primary and relapse were subjected
to this analysis after receiving written, informed consent
according to protocols approved by the Human Genome, Gene Analysis
Research Ethics Committee of the University of Tokyo and St. Luke's
International Hospital. No matched normal sample was available.
Genomic DNA of each sample was isolated by NucleoSpin DNA RapidLyse
kit (Macherey-Nagel Gmbh & Co., Düren, Germany) according to
manufacturer's protocol.
Direct sequencing
Direct sequencing of ALK was performed according to
manufacturer's protocol with the use of an ABI 3500 Genetic
Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The following set of primers were used:
ALK-24_F: GCTGCCCATGTTTACAGAATGC; ALK-24_R: TCCATCGAGGAACTTGCTAC;
ALK-25_F: CTGGTGTAGCTGCATGTTC; ALK-25_R: CATCCTACATCCAAATGGCTC;
ALK-26_F: AATCCTAGTGATGGCCGTTG; ALK-26_R:
GGAGATGATGTAAGGGACAAGC.
Targeted capture sequencing
Targeted capture was performed using a SureSelect
custom kit (Agilent Technologies, Inc., Santa Clara, CA, USA)
according to the manufacturer's protocol (9–11). The
custom bait library was designed to include: i) all coding exons of
367 genes; ii) untranslated regions and introns of 16 genes
(CD274, CTNNB1, ERG, ETV1, ETV4, EWSR1, FEV, FLI1, FOXO1, FUS,
INO80D, NCOA1, NCOA2, NOTCH1, PAX3, and PAX7) for
detecting breakpoints of structural variations; iii) 110,000 bases
surrounding TERT for detecting TERT rearrangement;
iv) promoter and enhancer regions of FGFR3, MYC and
TERT; v) 11 microRNA genes (MIR100, MIRLET7A1, MIRLET7A2,
MIRLET7A3, MIRLET7B, MIRLET7C, MIRLET7D, MIRLET7E, MIRLET7F1,
MIRLET7F2, MIRLET7G), and vi) 3,527 SNP positions for copy
number analysis. The total of 381 targeted genes and regions
besides the SNP positions were selected to include: i) genes
adopted in more than one of the following existing gene panels:
MSK-IMPACT (12) CMS400 (Thermo
Fisher Scientific, Inc.), FoundationOne (Foundation Medicine,
Cambridge, MA, USA), Human Comprehensive Cancer Panel (Qiagen GmbH,
Hilden, Germany); ii) the most frequently mutated 20 genes in each
type of malignancy according to Catalogue of Somatic Mutations in
Cancer (COSMIC) v78, and iii) genes that were recurrently affected
in pediatric malignancies including neuroblastoma (6,13,14), hepatoblastoma (15,16),
pleuropulmonary blastoma (17,18),
rhabdomyosarcoma (19,20), Ewing's sarcoma (21,22), and
germ cell tumor (23).
Captured targets were subjected to sequencing using
a HiSeq 2000 (Illumina, Inc., San Diego, CA, USA) with a standard
125-bp pairedend read protocol according to the manufacturer's
instructions (24). With mean depths
of 383 and 381 in paired primary-relapse specimens, respectively,
sequence alignment and mutation calling was performed using our
in-house pipeline ‘Genomon v.2.5.0’, as previously described
(25) in which reads that had either
a mapping quality score of <25, a base quality score of <30,
or five or more mismatched bases were excluded from the analysis.
Candidate mutations with i) a variant allele frequency in tumor
samples ≥0.1; and ii) a EBcall (26)
(Empirical Bayesian mutation calling) P ≤1 ×10−10 was
adopted and filtered by excluding i) synonymous mutations and
variants without complete ORF information; ii) known variants
listed in the 1000 Genomes Project (May 2011 release); NCBI SNP
database (dbSNP) build 131; National Heart, Lung, and Blood
Institute (NHLBI) Exome Sequencing Project (ESP) 5400; the Human
Genome Variation Database (HGVD; October 2013 release); or our
in-house SNP database; iii) variants present only in unidirectional
reads; iv) variants occurring in repetitive genomic regions; v)
variants with <5 supporting reads in tumor samples; and vi) all
variants found in nonpaired normal samples (n=30) showing an allele
frequency of >0.0025. Putative structural variations were
manually curated and further filtered by removing those with: i)
<20 supporting reads in tumor samples; ii) a VAF of <0.10 in
tumor samples; and iii) all variants found in normal samples.
Finally, mapping errors were removed by visual inspection on the
Integrative Genomics Viewer (IGV) browser (27).
SNP Array Analysis
SNP array analysis was performed using GeneChip
Human Mapping Cytoscan HD (Affymetrix; Thermo Fisher Scientific,
Inc.). Genome-wide detection of copy number abnormalities or
allelic imbalances was processed using Copy Number Analysis for
Affymetrix GeneChip (CNAG)/Allele-Specific Copy Number using
Anonymous References (AsCNAR) v3.5.1 software (28).
Results
Aggressive clinical course in a
relapsed NBL patient
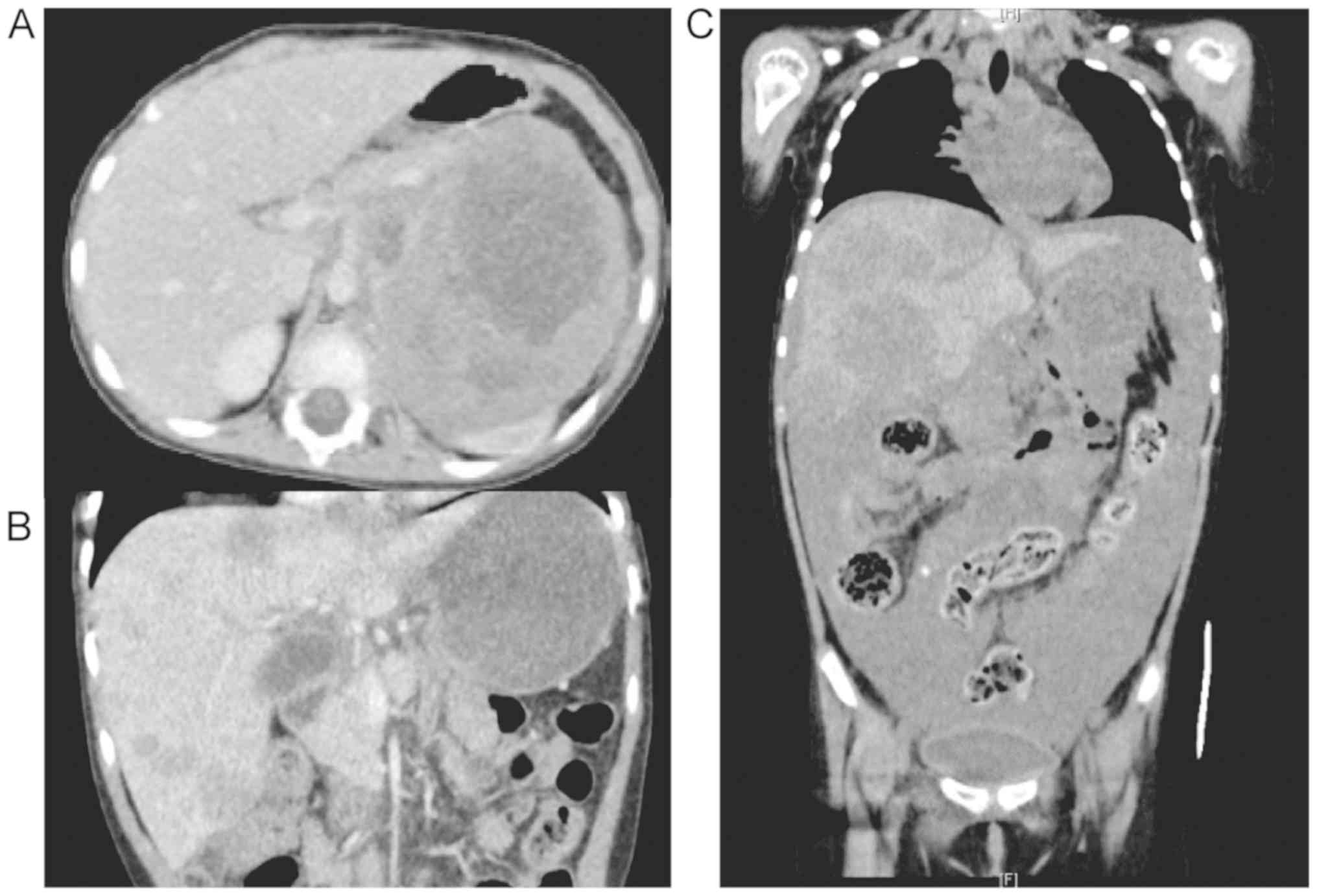
A 3-year-old girl was diagnosed with high-risk NBL
of the left adrenal gland with bone and marrow metastases. Neuron
specific enolase (NSE) and urine vanillylmandelic acid (VMA) levels
were elevated (215 µg/l and 8.4 mg/l, respectively), and
MYCN amplification was present. Immunohistochemical analyses
revealed tumor cells positive for ALK. The patient received five
courses of chemotherapy followed by high dose chemotherapy
(busulfan and melphalan) with autologous hematopoietic stem cell
transplantation, surgical gross total resection of the primary
tumor, and radiation therapy to the tumor bed (19.8 Gy). The
combination therapy successfully induced complete remission.
However, 12 months after the diagnosis, she suffered from local
tumor recurrence at the primary site. Two cycles of topotecan and
cyclophosphamide (TC) and three of ifosfamide, cisplatin, and
etoposide (ICE) gradually reduced the tumor size. After a fourth
course of ICE, the tumor rapidly grew and urgent radiotherapy, with
a combination of vincristine and irinotecan, was required. After
obtaining approval from the institutional review board, crizotinib
was administered for 28 days. Unfortunately, the tumor progressed
and she succumbed to the disease (Fig.
1).
Molecular analysis of paired
diagnosis-relapse samples
After obtaining informed consent from her guardians,
DNA was extracted from biopsy samples of the adrenal tumor both at
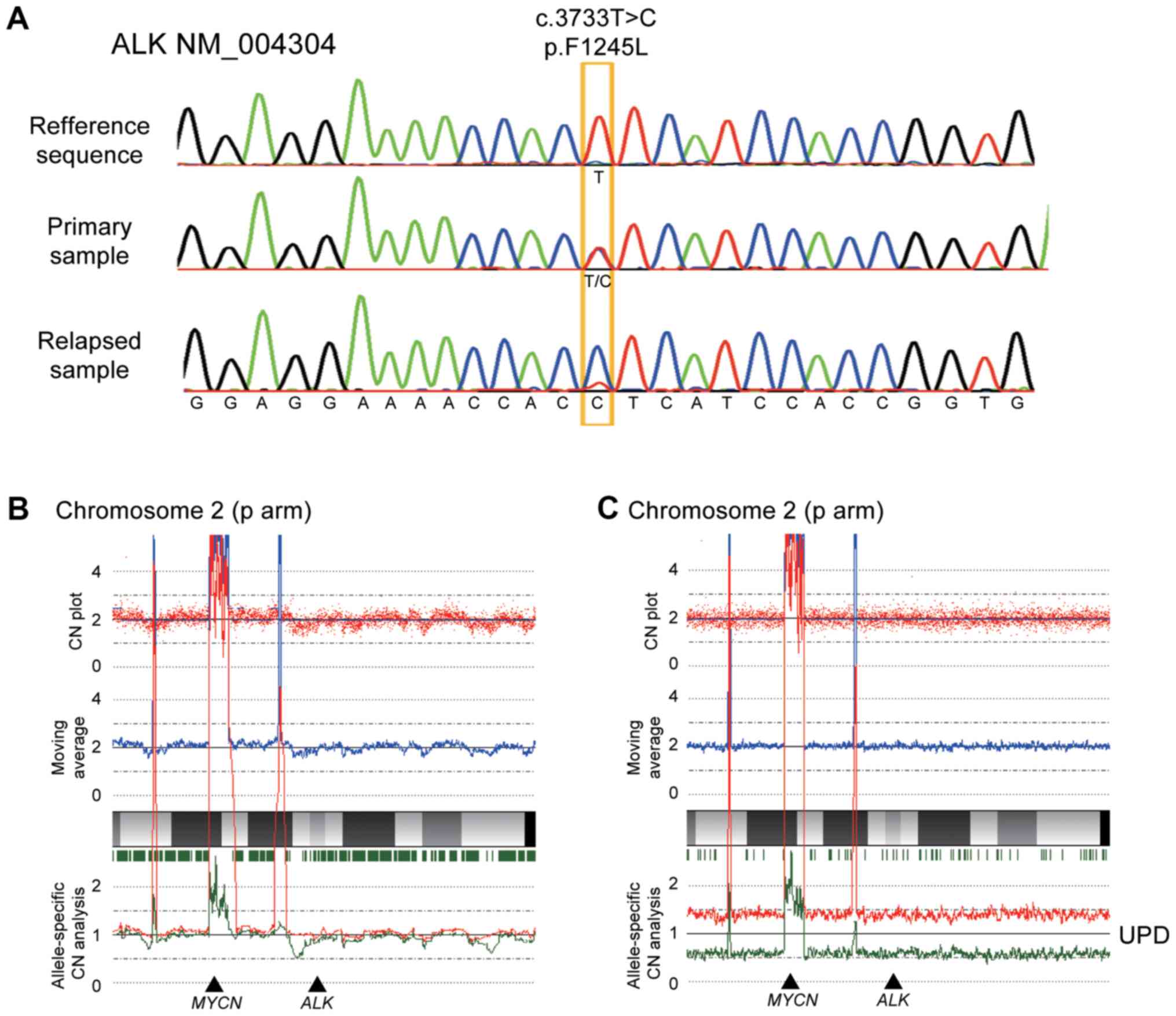
diagnosis and after relapse. Direct sequencing was performed to
detect ALK mutations, and a F1245L mutation (c.T3733C;
TTC>TCC) was identified in both the sample at diagnosis and
relapse, which is a common activating mutation in NBL.
Intriguingly, the relapse sample exclusively contained the mutated
allele, whereas the diagnosis sample clearly showed a heterozygous
change (Fig. 2A). In accordance with
this, SNP arrays analysis in the relapse sample revealed acquired
UPD of chromosome 2, including the ALK locus, in which the
wild-type allele was likely to be lost by chromosomal
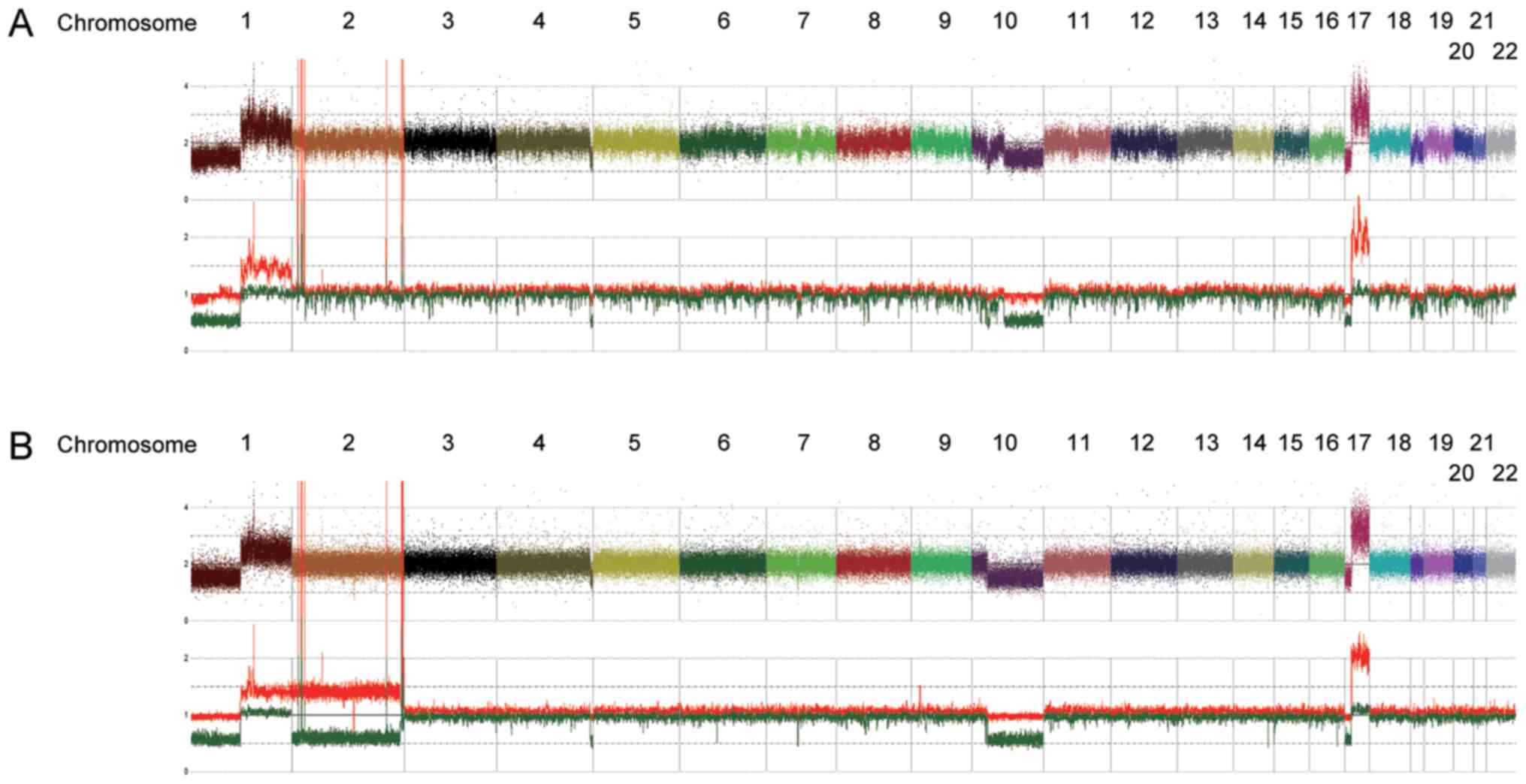
mis-segregation with duplication of the mutated allele (Fig. 2B and C). We also detected several
shared copy number alterations in both the diagnosis and relapse
samples, including a focal amplification of 2p24.2–24.3 comprising
the MYCN locus (Fig. 3). We
further analyzed other genetic alterations, which might have been
associated with disease recurrence and aggressiveness, using
targeted capture sequencing (TCS) for 381 genes related to
pediatric cancers (U-Tokyo Onco-panel v1). Because only tumor
samples were analyzed, candidate mutations were filtered referring
to our previous study (25), where we
have confirmed the somatic mutations detected in TCS by using
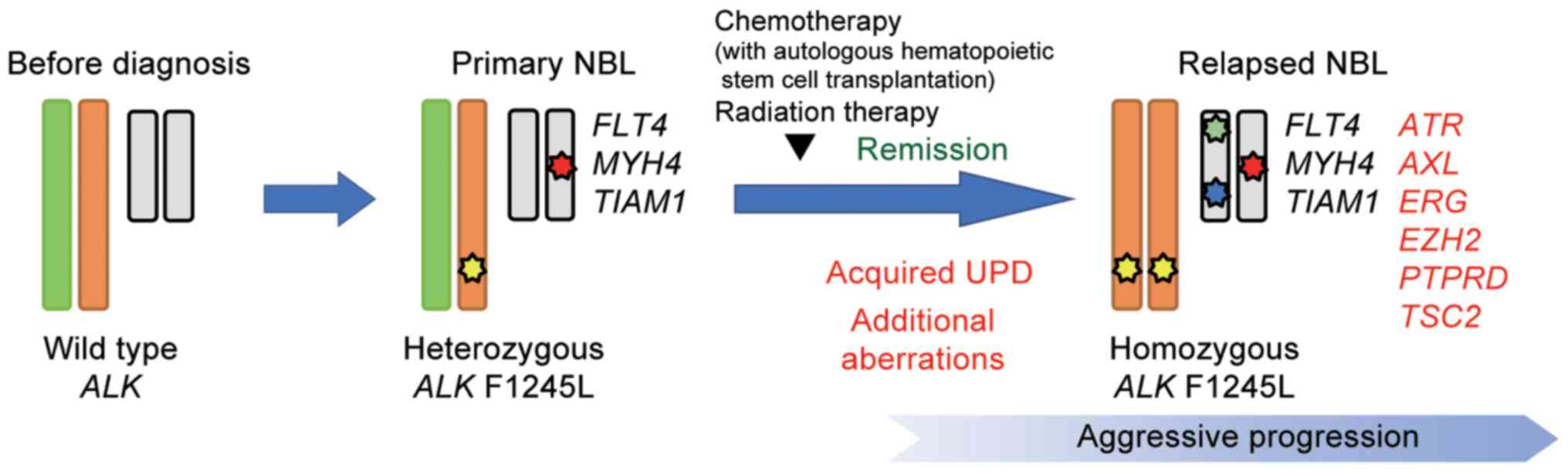
matched normal samples. We identified four and ten variants in the
diagnosis and relapse samples, respectively (Tables I and II). All mutations detected at diagnosis
were also detected in relapse sample. The ALK F1245L
mutation was detected in both the diagnosis (variant allele
frequency (VAF)=0.47) and relapse samples (VAF=0.83), which is
consistent with other results showing duplication of ALK at
relapse. Furthermore, alterations in TSC2 (p.P1092fs),
PTPRD (translocation to chromosome 6), ATR
(p.T1310S), AXL (p.A549S), ERG (p.S315X), and
EZH2 (p.E204X) were acquired at relapse (Fig. 4).
 | Table I.Mutation calls of targeted capture
sequencing. |
Table I.
Mutation calls of targeted capture
sequencing.
| Sample | Chr | Start | End | Ref | Alt | Gene | Mutation type | Amino acid
change | VAF |
|---|
| Primary | 2 | 29436860 | 29436860 | A | G | ALK | SNV |
NM_004304:exon24:c.T3733C:p.F1245L | 0.469 |
| Relapse | 2 | 29436860 | 29436860 | A | G | ALK | SNV |
NM_004304:exon24:c.T3733C:p.F1245L | 0.825 |
| Relapse | 3 | 142253938 | 142253938 | G | C | ATR | SNV |
NM_001184:exon21:c.C3929G:p.T1310S | 0.409 |
| Primary | 5 | 180043943 | 180043943 | C | G | FLT4 | SNV |
NM_182925:exon22:c.G3053C:p.S1018T | 0.425 |
| Relapse | 5 | 180043943 | 180043943 | C | G | FLT4 | SNV |
NM_182925:exon22:c.G3053C:p.S1018T | 0.496 |
| Relapse | 7 | 148525847 | 148525847 | C | A | EZH2 | Stopgain |
NM_001203247:exon6:c.G610T:p.E204X | 0.387 |
| Relapse | 16 | 2129420 | 2129420 | − | G | TSC2 | Frameshift |
NM_000548:exon28:c.3276dupG:p.
P1092fs | 0.245 |
| Primary | 17 | 10361019 | 10361019 | C | A | MYH4 | Stopgain |
NM_017533:exon16:c.G1615T:p.E539X | 1 |
| Relapse | 17 | 10361019 | 10361019 | C | A | MYH4 | Stopgain |
NM_017533:exon16:c.G1615T:p.E539X | 0.7 |
| Relapse | 19 | 41754659 | 41754660 | CG | TC | AXL | SNV |
NM_021913:exon14:c.CG1644_1645TC:
p.A549S | 0.429 |
| Primary | 21 | 32493125 | 32493125 | C | G | TIAM1 | SNV |
NM_003253:exon29:c.G4337C:p.R1446T | 0.506 |
| Relapse | 21 | 39772318 | 39772318 | G | T | ERG | Stopgain |
NM_001291391:exon8:c.C944A:p.S315X | 0.43 |
| Relapse | 21 | 32493125 | 32493125 | C | G | TIAM1 |
SNV |
NM_003253:exon29:c.G4337C:p.R1446T | 0.552 |
| Table II.Structural variant call of targeted
capture sequencing. |
Table II.
Structural variant call of targeted
capture sequencing.
| Sample | Chr Gene 1 | Position Gene
1 | Direction Gene
1 | Chr Gene 2 | Position Gene
2 | Direction Gene
2 | Variant type | Gene 1 | Gene 2 | VAF |
|---|
| Relapse | 6 | 138269463 | − | 9 | 8338848 | − | Translocation | − | PTPRD | 0.5745 |
Discussion
The ALK mutation hotspots are located within
the intracellular TKD at codons F1174, R1275, and F1245, accounting
for 85% of the ALK mutations found in NBL (2–5). Germline
ALK mutations, such as R1275, have also been identified as
the major cause of hereditary NBL; however, their penetrance has
been considered relatively low (29–31). In
contrast, other hotspot mutations (F1174 and F1245) are rarely
observed in familial cases. These F1174 and F1245 germline
mutations have been linked to severe neurocognitive defects and
abnormalities (30), which is
consistent with reports that mutations in F1174 and F1245 have
strong effects on the nonphosphorylated ALK TKD activity (5). Because both F1174 and F1245 are located
in the region inhibiting ligand-independent activation, they impair
self-inhibition and enhance the tyrosine kinase activity (32).
We detected a heterozygous ALK F1245 mutation
at diagnosis that became homozygous at relapse in the present case.
Homozygous ALK mutations have been rarely reported as far
(4,7).
This duplication at relapse may lead to growth advantage of tumor
cells and be associated with disease progression. In fact, acquired
UPD, resulting in the duplication of an oncogenic mutation, has
been reported for various neoplasms. We have previously shown that
the aggressive transformation of juvenile myelomonocytic leukemia
is associated with the duplication of the oncogenic KRAS due
to acquired UPD (33). Further, a
previous report has revealed that the homozygous ALK F1174S
mutation due to acquired UPD during the disease course may be
responsible for the very rapid progress of NBL and resistance to
chemotherapy (7). An enhanced
oncogenic potential has also been reported in homozygous
EGFR mutations on TKD in patients with non-small cell lung
cancer, particularly in metastatic cases (34). Thus, homozygous TKD mutations are
associated with an aggressive tumor progression in patients. In
addition to the strong effects of the F1245 mutation on the ALK TKD
activity, the bi-allelic presence of the ALK oncogenic
mutation due to acquired UPD may have conferred further
aggressiveness to the tumor during the relapse in this case.
In our comprehensive analysis, in addition to the
ALK F1245 mutation and MYCN amplification, we also
detected somatic oncogenic alterations comprising PTPRD
(chromosomal rearrangement) and TSC2 (p.P1092fs) at relapse.
PTPRD is a member of a large family of protein tyrosine
phosphatases and functions as a tumor suppressor by destabilizing
aurora kinase A (35). The low
expression of PTPRD is associated with high-risk NBL with
MYCN amplification (36).
TSC2 is a tumor suppressor gene that is responsible for
tuberous sclerosis, which is an autosomal dominant disorder
characterized by the formation of hamartomas in various organs. The
inactivation of TSC2 results in TSC1-TSC2 dissociation and markedly
impairs the ability of TSC2 to inhibit mTOR signals, leading to
cell proliferation and disease progression (37). A TSC2 mutation has not been
reported in patients with NBL. However, because alterations of
PTPRD and TSC2 occurred in a heterozygous setting,
the functional relevance of these genes in the tumor progression of
our case remains unclear. We also identified a truncated mutation
of EZH2 (p.E204X), which is an essential component of the
polycomb repressive complex 2. The pathogenic role of the truncated
mutation of EZH2 is unclear because most EZH2
mutations reported in NBL cases were gain of function mutations
mediating the epigenetic silencing of a tumor suppressor (38).
In the present case, all detected single nucleotide
variants were observed in the major clone (VAF >0.35). None of
acquired alterations at relapse were detected in primary sample
even as the minor clone (VAF <0.35), although there was a
technical limitation on our TCS, which cannot detect mutations with
low allele frequency (VAF <0.05). Because no minor variants were
detected, intratumor heterogeneity was not defined in the present
case. Intratumor heterogeneity leads to selective outgrowth of
clones that have a phenotypic advantage (39). The occurrence or presence of
bi-allelic change of the ALK mutation due to acquired UPD
and other acquired mutations such as TSC2 and PTPRD
might be involved in tumor relapse and chemo-resistance in the
present cases.
We report a patient with relapsed NBL harboring
ALK F1245 mutation, which was heterozygous at diagnosis and
became homozygous at relapse with resistance to crizotinib. This
duplication of ALK mutation was extremely rare, and may be
associated with aggressive behavior after relapse in this case. As
previously reported, the ALK F1245 mutations would be
suggested to be relatively crizotinib-resistant (5). Although a clinical benefit of ALK
inhibitors in patients with NBL has not been confirmed, more potent
next-generation ALK inhibitors have been developed and clinical
trials to estimate their efficacy are currently underway
(NCT03107988) (40,41). In future, a treatment based on the
ALK mutation status will be promising.
Acknowledgements
The authors would like to thank Ms. M Matsumura, Ms.
K Yin, and Ms. F Saito (The University of Tokyo) for their
technical assistance; Dr T Okunushi and Dr M Hino (Chiba
University) for collecting samples; and Dr K Yoshida, Dr Y Fujii,
Dr K Kataoka (Kyoto University), and Dr Y Shiraishi (The University
of Tokyo) for help with SNP array and next-generation
sequencing.
Funding
The present study was supported by KAKENHI (grant
no. 17H04224; received by JT) from the Japan Society of Promotion
of Science and Project for Cancer Research and Therapeutic
Evolution (P-CREATE) (grant no. 16cm0106509h001; received by JT) by
the Japan Agency for Medical Research and Development (AMED).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SK, DH, MSeki, MSekig, JT and AM wrote the
manuscript; SK, DH, MSeki, YY, AD, SH, YH, and AM collected and
analyzed data; SK, MSekig, and SM performed experiments; MK, SO, JT
and AM conceived the study; JT designed the study. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the University of Tokyo and St. Luke's International
Hospital.
Patient consent for publication
Prior written informed consent was obtained from the
patient's guardians.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cheung NK and Dyer MA: Neuroblastoma:
Developmental biology, cancer genomics and immunotherapy. Nat Rev
Cancer. 13:397–411. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
George RE, Sanda T, Hanna M, Fröhling S,
Luther W II, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, et al:
Activating mutations in ALK provide a therapeutic target in
neuroblastoma. Nature. 455:975–978. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Janoueix-Lerosey I, Lequin D, Brugières L,
Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A,
Schleiermacher G, Pierron G, et al: Somatic and germline activating
mutations of the ALK kinase receptor in neuroblastoma. Nature.
455:967–970. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Y, Takita J, Choi YL, Kato M, Ohira
M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, et al:
Oncogenic mutations of ALK kinase in neuroblastoma. Nature.
455:971–974. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bresler SC, Weiser DA, Huwe PJ, Park JH,
Krytska K, Ryles H, Laudenslager M, Rappaport EF, Wood AC, McGrady
PW, et al: ALK mutations confer differential oncogenic activation
and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer
Cell. 26:682–694. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pugh TJ, Morozova O, Attiyeh EF,
Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M,
Kiezun A, et al: The genetic landscape of high-risk neuroblastoma.
Nat Genet. 45:279–284. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martinsson T, Eriksson T, Abrahamsson J,
Caren H, Hansson M, Kogner P, Kamaraj S, Schönherr C, Weinmar J,
Ruuth K, et al: Appearance of the novel activating F1174S ALK
mutation in neuroblastoma correlates with aggressive tumor
progression and unresponsiveness to therapy. Cancer Res. 71:98–105.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mossé YP, Lim MS, Voss SD, Wilner K,
Ruffner K, Laliberte J, Rolland D, Balis FM, Maris JM, Weigel BJ,
et al: Safety and activity of crizotinib for paediatric patients
with refractory solid tumours or anaplastic large-cell lymphoma: A
children's oncology group phase 1 consortium study. Lancet Oncol.
14:472–480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kataoka K, Nagata Y, Kitanaka A, Shiraishi
Y, Shimamura T, Yasunaga J, Totoki Y, Chiba K, Sato-Otsubo A, Nagae
G, et al: Integrated molecular analysis of adult T cell
leukemia/lymphoma. Nat Genet. 47:1304–1315. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Haferlach T, Nagata Y, Grossmann V, Okuno
Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T,
et al: Landscape of genetic lesions in 944 patients with
myelodysplastic syndromes. Leukemia. 28:241–247. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suzuki H, Aoki K, Chiba K, Sato Y,
Shiozawa Y, Shiraishi Y, Shimamura T, Niida A, Motomura K, Ohka F,
et al: Mutational landscape and clonal architecture in grade II and
III gliomas. Nat Genet. 47:458–468. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cheng DT, Mitchell TN, Zehir A, Shah RH,
Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, et al:
Memorial sloan kettering-integrated mutation profiling of
actionable cancer targets (MSK-IMPACT): A hybridization
capture-based next-generation sequencing clinical assay for solid
tumor molecular oncology. J Mol Diagn. 17:251–264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Molenaar JJ, Koster J, Zwijnenburg DA, van
Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J,
Westerman BA, van Arkel J, et al: Sequencing of neuroblastoma
identifies chromothripsis and defects in neuritogenesis genes.
Nature. 483:589–593. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Valentijn LJ, Koster J, Zwijnenburg DA,
Hasselt NE, van Sluis P, Volckmann R, van Noesel MM, George RE,
Tytgat GA, Molenaar JJ and Versteeg R: TERT rearrangements are
frequent in neuroblastoma and identify aggressive tumors. Nat
Genet. 47:1411–1414. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eichenmüller M, Trippel F, Kreuder M, Beck
A, Schwarzmayr T, Häberle B, Cairo S, Leuschner I, von Schweinitz
D, Strom TM and Kappler R: The genomic landscape of hepatoblastoma
and their progenies with HCC-like features. J Hepatol.
61:1312–1320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sumazin P, Chen Y, Treviño LR, Sarabia SF,
Hampton OA, Patel K, Mistretta TA, Zorman B, Thompson P, Heczey A,
et al: Genomic analysis of hepatoblastoma identifies distinct
molecular and prognostic subgroups. Hepatology. 65:104–121. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seki M, Yoshida K, Shiraishi Y, Shimamura
T, Sato Y, Nishimura R, Okuno Y, Chiba K, Tanaka H, Kato K, et al:
Biallelic DICER1 mutations in sporadic pleuropulmonary blastoma.
Cancer Res. 74:2742–2749. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pugh TJ, Yu W, Yang J, Field AL, Ambrogio
L, Carter SL, Cibulskis K, Giannikopoulos P, Kiezun A, Kim J, et
al: Exome sequencing of pleuropulmonary blastoma reveals frequent
biallelic loss of TP53 and two hits in DICER1 resulting in
retention of 5p-derived miRNA hairpin loop sequences. Oncogene.
33:5295–5302. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seki M, Nishimura R, Yoshida K, Shimamura
T, Shiraishi Y, Sato Y, Kato M, Chiba K, Tanaka H, Hoshino N, et
al: Integrated genetic and epigenetic analysis defines novel
molecular subgroups in rhabdomyosarcoma. Nat Commun. 6:75572015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shern JF, Chen L, Chmielecki J, Wei JS,
Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, et
al: Comprehensive genomic analysis of rhabdomyosarcoma reveals a
landscape of alterations affecting a common genetic axis in
fusion-positive and fusion-negative tumors. Cancer Discov.
4:216–231. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Crompton BD, Stewart C, Taylor-Weiner A,
Alexe G, Kurek KC, Calicchio ML, Kiezun A, Carter SL, Shukla SA,
Mehta SS, et al: The genomic landscape of pediatric Ewing sarcoma.
Cancer Discov. 4:1326–1341. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tirode F, Surdez D, Ma X, Parker M, Le
Deley MC, Bahrami A, Zhang Z, Lapouble E, Grossetête-Lalami S,
Rusch M, et al: Genomic landscape of Ewing sarcoma defines an
aggressive subtype with co-association of STAG2 and TP53 mutations.
Cancer Discov. 4:1342–1353. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Taylor-Weiner A, Zack T, O'Donnell E,
Guerriero JL, Bernard B, Reddy A, Han GC, Al Dubayan S,
Amin-Mansour A, Schumacher SE, et al: Genomic evolution and
chemoresistance in germ-cell tumours. Nature. 540:114–118. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sato Y, Yoshizato T, Shiraishi Y, Maekawa
S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki
H, et al: Integrated molecular analysis of clear-cell renal cell
carcinoma. Nat Genet. 45:860–867. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Seki M, Kimura S, Isobe T, Yoshida K, Ueno
H, Nakajima-Takagi Y, Wang C, Lin L, Kon A, Suzuki H, et al:
Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute
lymphoblastic leukemia. Nat Genet. 49:1274–1281. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shiraishi Y, Sato Y, Chiba K, Okuno Y,
Nagata Y, Yoshida K, Shiba N, Hayashi Y, Kume H, Homma Y, et al: An
empirical Bayesian framework for somatic mutation detection from
cancer genome sequencing data. Nucleic Acids Res. 41:e892013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Robinson JT, Thorvaldsdóttir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yamamoto G, Nannya Y, Kato M, Sanada M,
Levine RL, Kawamata N, Hangaishi A, Kurokawa M, Chiba S, Gilliland
DG, et al: Highly sensitive method for genomewide detection of
allelic composition in nonpaired, primary tumor specimens by use of
affymetrix single-nucleotide-polymorphism genotyping microarrays.
Am J Hum Genet. 81:114–126. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mossé YP, Laudenslager M, Longo L, Cole
KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P,
et al: Identification of ALK as a major familial neuroblastoma
predisposition gene. Nature. 455:930–935. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
de Pontual L, Kettaneh D, Gordon CT,
Oufadem M, Boddaert N, Lees M, Balu L, Lachassinne E, Petros A,
Mollet J, et al: Germline gain-of-function mutations of ALK disrupt
central nervous system development. Hum Mutat. 32:272–276. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bourdeaut F, Ferrand S, Brugières L,
Hilbert M, Ribeiro A, Lacroix L, Bénard J, Combaret V, Michon J,
Valteau-Couanet D, et al: ALK germline mutations in patients with
neuroblastoma: A rare and weakly penetrant syndrome. Eur J Hum
Genet. 20:291–297. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee CC, Jia Y, Li N, Sun X, Ng K, Ambing
E, Gao MY, Hua S, Chen C, Kim S, et al: Crystal structure of the
ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J.
430:425–437. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kato M, Yasui N, Seki M, Kishimoto H,
Sato-Otsubo A, Hasegawa D, Kiyokawa N, Hanada R, Ogawa S, Manabe A,
et al: Aggressive transformation of juvenile myelomonocytic
leukemia associated with duplication of oncogenic KRAS due to
acquired uniparental disomy. J Pediatr. 162:1285–1288, 1288.e1.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ma ES, Wong CL, Siu D and Chan WK:
Amplification, mutation and loss of heterozygosity of the EGFR gene
in metastatic lung cancer. Int J Cancer. 120:1828–1831. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Meehan M, Parthasarathi L, Moran N,
Jefferies CA, Foley N, Lazzari E, Murphy D, Ryan J, Ortiz B, Fabius
AW, et al: Protein tyrosine phosphatase receptor delta acts as a
neuroblastoma tumor suppressor by destabilizing the aurora kinase a
oncogene. Mol Cancer. 11:62012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nair P, De Preter K, Vandesompele J,
Speleman F and Stallings RL: Aberrant splicing of the PTPRD gene
mimics microdeletions identified at this locus in neuroblastomas.
Genes Chromosomes Cancer. 47:197–202. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ma L, Chen Z, Erdjument-Bromage H, Tempst
P and Pandolfi PP: Phosphorylation and functional inactivation of
TSC2 by Erk implications for tuberous sclerosis and cancer
pathogenesis. Cell. 121:179–193. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang C, Liu Z, Woo CW, Li Z, Wang L, Wei
JS, Marquez VE, Bates SE, Jin Q, Khan J, et al: EZH2 mediates
epigenetic silencing of neuroblastoma suppressor genes CASZ1, CLU,
RUNX3, and NGFR. Cancer Res. 72:315–324. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Burrell RA, McGranahan N, Bartek J and
Swanton C: The causes and consequences of genetic heterogeneity in
cancer evolution. Nature. 501:338–345. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sakamoto H, Tsukaguchi T, Hiroshima S,
Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N and
Aoki Y: CH5424802, a selective ALK inhibitor capable of blocking
the resistant gatekeeper mutant. Cancer Cell. 19:679–690. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Infarinato NR, Park JH, Krytska K, Ryles
HT, Sano R, Szigety KM, Li Y, Zou HY, Lee NV, Smeal T, et al: The
ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to
crizotinib in alk-driven neuroblastoma. Cancer Discov. 6:96–107.
2016. View Article : Google Scholar : PubMed/NCBI
|