Introduction
Despite recent developments of various screening
methods and procedures for early detection of cancer, colorectal
cancer (CRC) remains one of the most fatal cancer types globally,
with almost 700,000 mortalities reported in 2012 (1) The overall incidence rate of CRC is
decreasing; however, the mortality rate for adults <50 years old
in USA is increasing, which makes it essential to improve
understanding of CRC at the molecular and cellular level (2). A total of three major pathways are
involved in the progression of CRC, including the
cytosine-phosphate-guanine island methylator phenotype (CIMP)
pathway, the microsatellite instability (MSI) pathway and the
chromosomal instability (CIN) pathway (3). The CIMP pathway is associated with the
hypermethylation of gene promoters. In subsets of CIMP-positive
tumors, mutations in BRAF are present (4). BRAF that is activated by mutations is
strongly associated with the CIMP pathway and this association has
been reported in CRC cases (4). The
MSI pathway results from impaired DNA mismatch repair (MMR), which
causes mutations to accumulate (3).
Inactivation of MMR induces the loss of the adenomatous polyposis
coli (APC) gene and mutates cell differentiation genes (5). One of the early events of the CIN
pathway is the loss of APC (5).
APC-mutant cells slowly accumulate to produce a polyp, followed by
the inactivation of additional tumor suppressor genes and the
activation of proto-oncogenes, including c-Src, Myc and KRAS
(5). Mutations in the small GTPase
KRAS induce large adenomas and early carcinomas (6). Subsequently, the loss of SMAD4 and tumor
protein 53 has been identified in carcinomas (6).
Genes encoding signaling molecules in the Wnt/enes
encoding signhave been revealed to be frequently mutated, resulting
in abnormal activation of this signaling cascade. For example,
familial adenomatous polyposis, an autosomal dominant syndrome, is
largely associated with germline mutations in the APC tumor
suppressor gene, which is a negative regulator of the Wnt pathway
(7). Inactivating mutations within
APC have also been identified in >80% of sporadic CRC cases and
50% of patients with CRC with wild-type APC exhibit activating
mutations within the pathwayrgely al which demonstrates a clear
association between colorectal neoplasia and aberrant Wnt signals
(8). These data indicate that the Wnt
pathway is a promising therapeutic target for the treatment of CRC.
The Wnt/promisin pathway regulates numerous target genes, including
Myc, cyclin D1 and vascular endothelial growth factor (9–11).
However, it is noteworthy that the malignant phenotype induced by
APC deficiency was rescued upon Myc deletion in murine intestine,
implying that aberrant Wnt signaling exerts its effect
predominantly through the upregulation of Myc expression (12). Therefore, strategies have been devised
to therapeutically target Myc (13).
For example, inhibitors of proviral integration site for Moloney
murine leukemia virus (PIM) kinases, including PIMi and SMI-4a,
have been developed to decrease Myc protein stability (13). Additionally, 10058-F4 is a chemical
drug that inhibits Myc-Max dimerization and their binding to DNA
(13). A better understanding of the
mechanism underlying Myc deregulation may result in more efficient
targeting of Myc in Myc-dependent CRC.
Cyclic adenosine 3′,5′-monophosphate (cAMP), an
intracellular second messenger, was first identified by Sutherland
and Rall in 1958 (14,15). Since then, its broad impact on
cellular physiology, including its role in inflammation, cell cycle
progression and cytotoxicity has been well characterized (16–18).
Adenylyl cyclase and phosphodiesterases (PDEs) generate and
hydrolyze cAMP (18). Previous
studies using various types of cancer models have indicated that
cancer cells, including B lymphoma cells and CRC cells, exhibit low
levels of intracellular cAMP; conversely, the elevation of cAMP
levels by inhibition of PDE4 activities can result in growth arrest
and/or cell death (19–22). These data indicate that PDE4 serves a
critical role in the regulation of cAMP levels in these cells. PDE4
consists of four different isoforms, PDE4A, 4B, 4C and 4D, and a
previous study reported that PDE4D is a target of microRNA
(miR)-139, whose expression is in turn regulated by the p53 tumor
suppressor (23). The expression of
miR-139 is inversely correlated with that of PDE4D in CRC samples
(23), which indicates that PDE4D is
a potential oncogene. Previous studies demonstrated that aberrant
Myc expression is causally associated with the pathophysiology of
CRC (5); however, how Myc is
regulated by cAMP/PDE4D signaling remains unknown. The present
study aimed to elucidate the mechanism underlying Myc deregulation,
which may result in more effective targeting of Myc in CRC.
Materials and methods
Cell culture and reagents
Human DLD-1 CRC cells (Korean Cell Line Bank; Korean
Cell Line Research Foundation, Seoul, Korea) were maintained at
37°C in 5% CO2 in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (Capricorn Scientific GmbH, Ebsdorfergrund,
Germany).
The following antibodies were used in the present
study: Anti-phosphorylated (p)AKT (1:2,000 dilution; catalog no.
9271; Cell Signaling Technology, Inc., Danvers, MA, USA), anti-AKT
(1:2,000 dilution; catalog no. 9272; Cell Signaling Technology,
Inc.), anti-eukaryotic translation initiation factor 4E-binding
protein 1 (1:2,000 dilution; 4EBP1; catalog no. 9452; Cell
Signaling Technology, Inc.), anti-p4EBP1 (1:2,000 dilution; catalog
no. 9459; Cell Signaling Technology, Inc.), anti-c-Myc (1:2,000
dilution; catalog no. ab32072; Abcam, Cambridge, UK), anti-β-actin
(1:5,000 dilution; catalog no. sc-47778; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), and horseradish peroxidase (HRP)-conjugated
anti-rabbit (1:10,000 dilution; catalog no. A120-101p; Bethyl
Laboratories, Inc., Montgomery, TX, USA) and anti-mouse (1:10,000
dilution; catalog no. A90-116p-33; Bethyl Laboratories, Inc.)
antibodies.
The following chemicals were used: Dimethylsulfoxide
(DMSO) (catalog no. D1370.0100; Duchefa Biochemie, Haarlem,
Netherlands), Forskolin (catalog no. BML-CN100; Enzo Life Sciences,
Inc., Farmingdale, NY, USA), rolipram (catalog no. R1012; AG
Scientific, Inc., San Diego, CA, USA), roflumilast (catalog no.
2675-50; BioVision, Inc., Milpitas, CA, USA), rapamycin (catalog
no. sc3504; Santa Cruz Biotechnology, Inc.), JQ1 (catalog no.
A1910; ApexBio Technology, Houston, TX, USA) and resveratrol
(catalog no. 554325; Merck KGaA, Darmstadt, Germany).
cAMP assays
DLD-1 cells (3×105 cells/well) were
seeded in 6-well plates. Following incubation overnight at 37°C in
a 5% CO2 incubator, the cells were treated with
forskolin (20 or 40 µM) for 2 h at 37°C, following pre-incubation
with rolipram (40 µM) for 6 h or resveratrol (20 µM) for 16 h at
37°C. The concentration of cAMP was determined using a Parameter
cAMP assay kit (catalog no. KGE002B; R&D Systems, Inc.,
Minneapolis, MN, USA), according to the manufacturer's
protocol.
Western blot analysis
DLD-1 (3×105 cells/well) were seeded in
6-well dishes and treated with DMSO, forskolin (10 or 20 µM for 16
h), rolipram (20 or 40 µM for 16 h), roflumilast (40 µM for 16 h),
JQ1 (0.5 or 1 µM for 24 h), rapamycin (25 or 50 nM for 24 h), and
resveratrol (20 µM for ~48–72 h) at 37°C. Following treatment,
cells were harvested by scraping and rinsed with PBS. The cells
were lysed in radioimmunoprecipitation assay buffer (Elpis Biotech,
Inc., Daejeon, Korea) mixed with sodium vanadate (1 mM;
Sigma-Aldrich; Merck KGaA), β-glycerol phosphate (50 mM;
Sigma-Aldrich; Merck KGaA), protease inhibitor (1X; G-Biosciences,
St. Louis, MO, USA), EDTA (5 mM; G-Biosciences) and
β-mercaptoethanol (142 mM; Bioworld Technology, Inc., St. Louis
Park, MN, USA). The lysates were separated on 10 or 15%
polyacrylamide gels, transferred onto polyvinylidene difluoride
membranes and then blocked with 1% bovine serum albumin for 1 h at
room temperature (MP Biomedicals, LLC, Santa Ana, CA, USA)
dissolved in TBS and Tween-20. The membranes were probed with the
aforementioned primary antibodies overnight at 4°C and
HRP-conjugated secondary antibodies for 1 h at room temperature.
Subsequently, the signals were visualized using enhanced
chemiluminescent reagent (EzWestLumi plus; ATTO Corporation, Tokyo,
Japan) and analysis of the relevant protein levels was performed
using Luminograph II image analysis software (WSE-6100; ATTO
Corporation).
Clonogenic assay
To measure clonogenicity, 1×103 DLD-1
cells/well were seeded in 6-well plates, incubated overnight at
37°C and then treated with DMSO (control), 20 µM forskolin and/or
40 µM rolipram/roflumilast for 7 days at 37°C. The visible colonies
that developed were stained with 0.5% crystal violet solution in
25% methanol, and counted with the naked eye at room
temperature.
Soft agar assay
DLD-1 cells (5×103 cells/well) were mixed
with 0.2% top agar (catalog no. A9414-5G; Sigma-Aldrich; Merck
KGaA) and seeded on 0.6% bottom agar in 12-well plates to visualize
their anchorage-independent growth. The treatment conditions (DMSO,
20 µM forskolin and 40 µM rolipram/roflumilast) were maintained for
21 days at 37°C with 5% CO2. The colonies were then
stained with 0.005% crystal violet solution for 1 h at room
temperature. The colonies were counted using a light microscope
(Olympus CKX41; Olympus Corporation, Tokyo, Japan) at ×20
magnification.
Cell counting
DLD-1 cells were seeded on day 0 at a density of
1×105 cells/well into 6-well plates and cultured with
DMSO (control), 20 µM forskolin and 40 µM rolipram/roflumilast for
6 days at 37°C. The cells were detached by trypsinization at 37°C
and washed with PBS for counting with a hemocytometer at ×100
magnification every day for 6 days.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
To quantify mRNA levels of PDE4A, 4B, 4C and 4D,
RT-qPCR was performed and calculated using the
2−ΔΔCq method (20). TATA-box binding protein (TBP) was used
as an internal control. First, 1×106 DLD-1 cells/well
were seeded in 6-well plates overnight at 37°C and subsequently
treated with DMSO, 40 µM forskolin for 2 h, 40 µM rolipram for 6 h,
or a combination of forskolin and rolipram at 37°C. Finally, DLD-1
cells were harvested by scraping. Total RNA from DLD-1 CRC cells
(1×106 cells per sample) was extracted using Tri-RNA
Reagent (Favorgen Biotech Cooperation, Kaohsiung, Taiwan). cDNA was
synthesized using PrimeScript™ RT reagent kit with gDNA Erase
(Takara Bio, Inc., Otsu, Japan), according to the manufacturer's
protocol. qPCR was then performed using a CFX connect™ Realtime
system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and TOPreal™
qPCR 2X PreMIX (SYBR® Green with low ROX; Enzynomics,
Inc., Daejeon, Korea), according to the manufacturer's protocols.
The primer sequences for PDE4A, 4B and 4D were as described
previously (19,23,24). The
primer sequences for PDE4C and TBP were as follows: PDE4C forward,
5′-AGTCCCATGTGTGACAAGCA-3′, and reverse,
5′-TCTGGTTGTCGAGGGGTAAG-3′; and TBP forward,
5′-TATAATCCCAAGCGGTTTGCTGCG-3′, and reverse,
5′-AATTGTTGGTGGGTGAGCACAAGG-3′. The following PCR amplification
cycles were used: 95°C for 15 min, and 40 cycles of 95°C for 15
sec, 59°C for 15 sec and 27°C for 30 sec.
Statistical analysis
A non-parametric Mann-Whitney U test was performed
using Excel 1810 software 2018 (Microsoft Corporation, Redmond, WA,
USA) and one-way analysis of variance (ANOVA) followed by Tukey's
post hoc test was carried out using One-way ANOVA with post-hoc
Tukey HSD test calculator (http://www.astatsa.com). P<0.05 was considered to
indicate a statistically significant difference. Data are presented
as the mean ± standard deviation. All experiments were
independently repeated a minimum of three times.
Results
PDE4D is a major regulator of cAMP
expression levels in DLD-1 cells
Considering that previous studies demonstrated a
potential role of cAMP signaling in the survival of CRC cells
(22,25), the present study investigated how
intracellular cAMP expression levels are affected by forskolin, a
chemical activator of adenylyl cyclase. Forskolin alone
demonstrated an insignificant effect (Fig. 1A), which indicates that cAMP-degrading
PDEs are present. It was identified that treatment with rolipram, a
selective PDE4 inhibitor, in combination with forskolin, was
associated with a significant increase in cAMP levels, which
indicates that PDE4 is a major hydrolyzer of cAMP. Notably,
treatment with rolipram alone did not increase cAMP expression
levels, which may be due to low basal activities of adenylyl
cyclase. Subsequently, RT-qPCR was performed, which identified that
the PDE4D mRNA level was significantly increased, compared with
PDE4A, 4B or 4C, which indicates that PDE4D is primarily
responsible for cAMP degradation in DLD-1 CRC cells (Fig. 1B). However, other cAMP PDEs, including
PDE3 and PDE7, may serve a role.
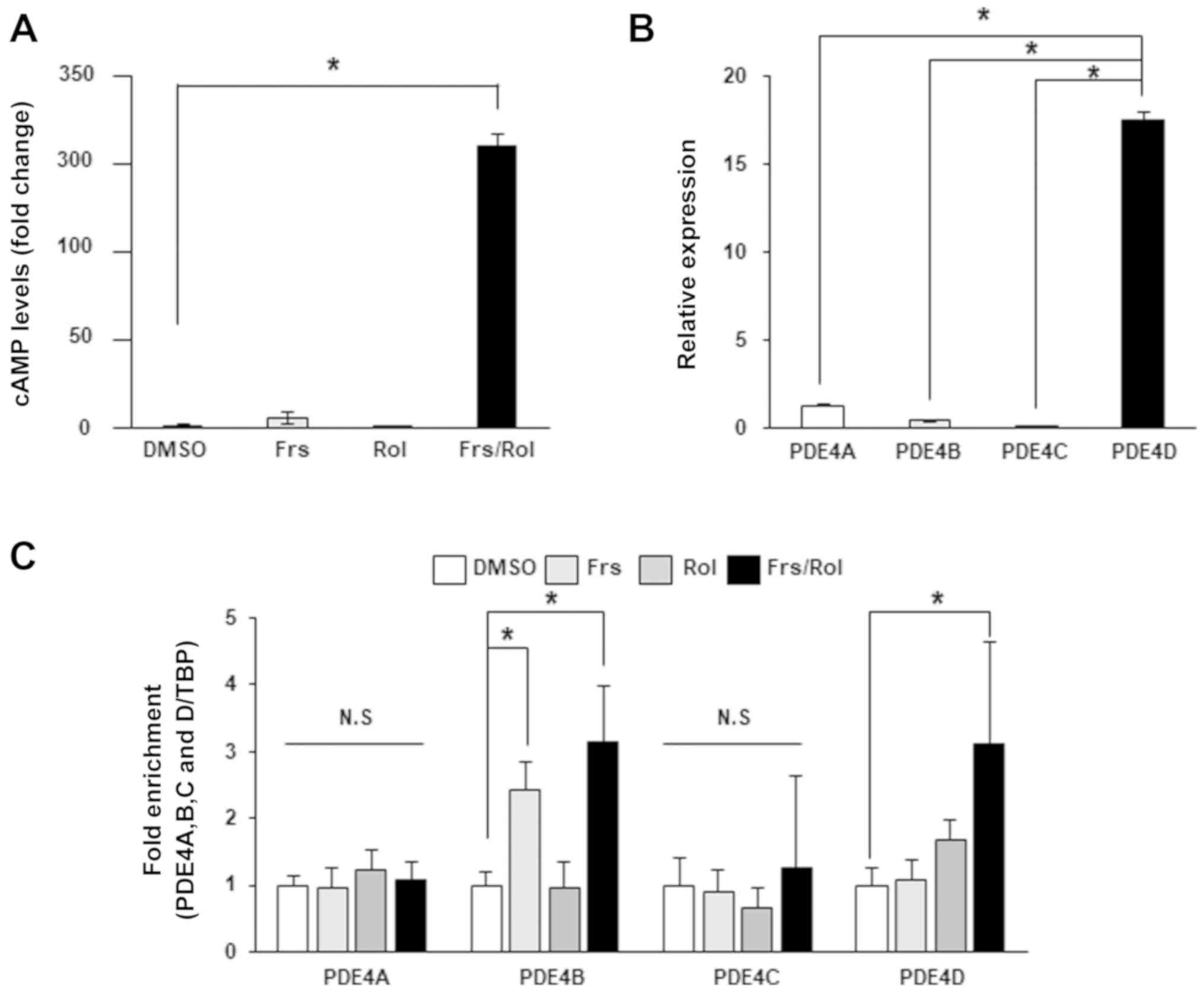 | Figure 1.Intracellular cAMP levels are
modulated by PDE4D in DLD-1 CRC cells. (A) Intracellular cAMP
expression levels were measured in PDE4D-high DLD-1 cells following
the addition of DMSO, 40 µM Frs for 2 h, 40 µM Rol for 6 h, or a
combination of Frs and Rol. Data are presented relative to
treatment with DMSO. (B) Expression levels of PDE4A, 4B, 4C and 4D
were analyzed by reverse transcription-quantitative polymerase
chain reaction. (C) PDE4D mRNA levels were analyzed in DLD-1 CRC
following treatment with DMSO, 40 µM Frs for 2 h, 40 µM Rol for 6
h, or a combination of Frs and Rol. Data are presented as the mean
± standard deviation. *P<0.05, according to one-way analysis of
variance. cAMP, cyclic adenosine 3′,5′-monophosphate; PDE,
phosphodiesterase; CRC, colorectal cancer; Frs, forskolin; Rol,
rolipram; TBP, TATA-box binding protein; DMSO, dimethyl
sulfoxide. |
A previous study characterized the cAMP response
elements (CREs) in the PDE4D promoter region using human airway
smooth muscle cells (26). To
investigate whether the expression of PDE4D is responsive to cAMP
in DLD-1 cells, the present study exposed cells to forskolin,
rolipram or a combination of forskolin and rolipram to modulate
intracellular cAMP levels. The administration of single agents
exhibited no significant effect on PDE4D expression; however,
co-treatment with forskolin and rolipram significantly increased
the expression level of PDE4D by >3-fold (Fig. 1C). This data is associated with cAMP
concentrations upon treatment with these drugs, with
forskolin/rolipram combination, but not forskolin or rolipram
treatment alone, resulting in increased cAMP levels, indicating
that PDE4D expression is regulated by cAMP in DLD-1 cells. The
expression levels of other PDE4 isoforms, including PDE4A, 4B and
4C, were significantly reduced compared with PDE4D; however, their
expression levels in response to forskolin, rolipram or a
combination were also examined. As demonstrated in Fig. 1C, PDE4B mRNA levels significantly
increased upon exposure to forskolin or forskolin/rolipram. This
result agrees with a previous study, which demonstrated that CREs
exist in the promoter region of PDE4B, but not PDE4A (27). To the best of our knowledge, the
presence or absence of CREs in the PDE4C promoter region remains to
be characterized.
Suppression of the AKT/mTOR/Myc
signaling pathway by cAMP enhances oncogenic properties of DLD-1
cells
The aforementioned data demonstrated that only a
combination of forskolin with rolipram significantly increased cAMP
concentrations, whereas either agent alone exhibited no significant
effect (Fig. 1). Subsequently, the
present study examined whether the increase in cAMP levels by
combinatory treatment affects the phenotype in DLD-1 cells.
Clonogenic and soft agar assays demonstrated that treatment with
forskolin/rolipram significantly inhibited colony formation, cell
proliferation and anchorage-independent growth, which indicates
that cAMP exhibits an inhibitory effect on the malignant properties
of these cells (Fig. 2A-C).
Roflumilast, an FDA-approved PDE4 inhibitor (28), when combined with forskolin, exhibited
the same effect as rolipram (Fig.
2D-F). In summary, the data demonstrated that high cAMP
concentrations can suppress the oncogenic properties of DLD-1
cells, which indicates that these cells may typically exhibit low
cAMP levels.
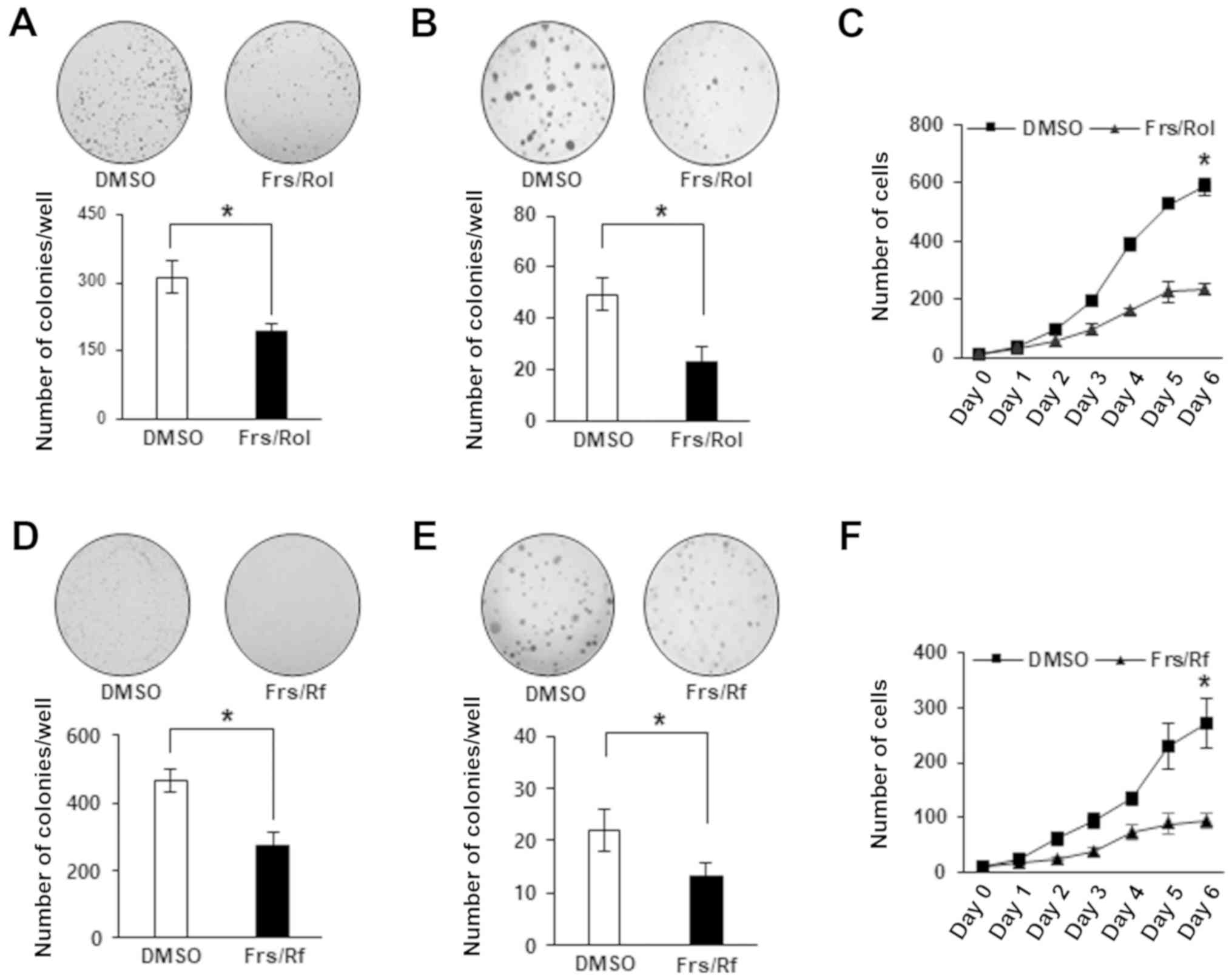 | Figure 2.Cyclic adenosine
3′,5′-monophosphate/phosphodiesterase 4D signals affect the
oncogenic phenotypes of DLD-1 cells. (A) For colony formation
assays, DLD-1 cells were seeded in 6-well plates followed by
treatment with DMSO, or a combination of 20 µM Frs and 40 µM Rol
for 7 days. The colonies were stained with 0.5% crystal violet and
counted at ×100 magnification. (B) For soft agar assays, DLD-1
cells were seeded in soft agar and treated with DMSO, or a
combination of 20 µM Frs and 40 µM Rol. Following 21 days, 0.005%
crystal violet was added and the colonies were counted at ×20
magnification. (C) For cell counting, DLD-1 cells were counted at
×100 magnification every day for 6 days while treating with either
DMSO, or a combination of 20 µM Frs and 40 µM Rol. (D) For colony
formation assays, DLD-1 cells were seeded in 6-well plates followed
by treatment with DMSO, or a combination of 20 µM Frs and 40 µM Rf
for 7 days. The colonies were counted at ×100 magnification
following staining with 0.5% crystal violet. (E) For soft agar
assays, DLD-1 cells were seeded in soft agar and treated with DMSO,
or a combination of 20 µM Frs and 40 µM Rf. After 21 days, 0.005%
crystal violet was added and the colonies were counted at ×20
magnification. (F) For cell counting, DLD-1 cells were counted at
×100 magnification every day for 6 days, while treated with either
DMSO, or a combination of 20 µM Frs and 40 µM Rf. Data are
presented as the mean ± standard deviation. *P<0.05 vs. DMSO,
according to Mann-Whitney U test. Frs, forskolin; Rol, rolipram;
Rf, Roflumilast; DMSO, dimethyl sulfoxide. |
Subsequently, the present study aimed to identify
the signaling pathways that mediate the tumor-suppressive
activities of cAMP. It has previously been demonstrated that the
AKT/mTOR pathway and Myc, a well-characterized downstream target,
regulate the survival of CRC cells (29–31).
Additionally, the activities of AKT/mTOR signaling and the
expression of Myc have been revealed to be regulated by cAMP in
different types of cells, including human diffuse large B cell
lymphoma cell lines (19,32,33). The
present study hypothesized that a downregulation of the
AKT/mTOR/Myc axis by increased cAMP levels may inhibit the survival
of DLD-1 CRC cells. To test this, DLD-1 CRC cells were treated with
forskolin, rolipram or forskolin/rolipram, and the expression
levels of pAKT, p4EBP1 and Myc were measured by western blot
analysis. Treatment with either agent alone exhibited no
significant effect on pAKT, p4EBP1 and Myc expression levels, which
were markedly downregulated by simultaneous administration of
forskolin and rolipram (Fig. 3A-C).
These results support the aforementioned observed intracellular
cAMP levels upon the addition of these agents both when used
separately and in combination. Specifically, co-treatment with
forskolin and rolipram resulted in a significant increase in cAMP
levels while either agent alone exhibited no significant effect
(Fig. 1A), which indicates that cAMP
inhibits the AKT/mTOR/Myc signaling pathway, which is associated
with the regulation of the malignant properties of DLD-1 CRC cells.
To confirm cAMP's negative impact on this signaling pathway,
western blot analysis was also performed using another PDE4
inhibitor roflumilast, which demonstrated a similar effect on pAKT,
p4EBP1 and Myc expression levels, compared with rolipram (Fig. 3D). Treatment with
forskolin/roflumilast decreased the expression levels of pAKT,
p4EBP1 and Myc. In summary, these data indicate that the oncogenic
phenotype of DLD-1 cells is predominantly suppressed by cAMP
through inhibition of the AKT/mTOR/Myc pathway; however, the
possibility that cAMP modulates the survival of DLD-1 cells via
other signaling pathways cannot be excluded.
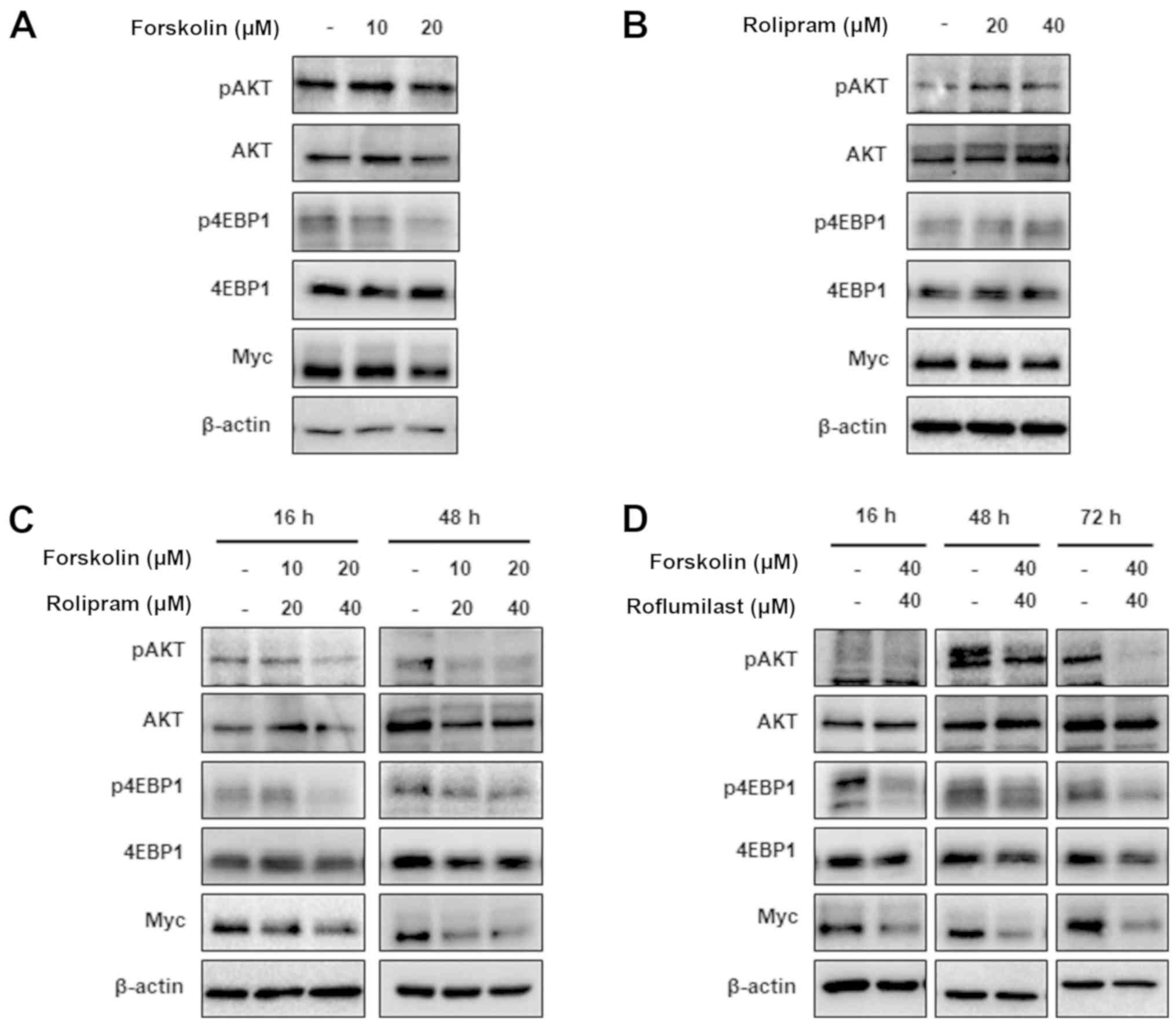 | Figure 3.Cyclic adenosine
3′,5′-monophosphate/PDE4D signaling regulates the activities of the
AKT/mammalian target of rapamycin/Myc pathway. Western blot assays
were performed to examine the phosphorylation levels of AKT and
4EBP1, and the expression of Myc following treatment with (A)
forskolin (0–20 µM for 16 h), (B) rolipram (0–40 µM for 16 h), (C)
a combination of forskolin and rolipram, or (D) a combination of
forskolin and roflumilast in PDE4D-high DLD-1 colorectal cancer
cells. Western blots for β-actin confirm equal loading. The
expression levels of Myc, pAKT and p4EBP1 were markedly decreased
upon combined treatment with forskolin and rolipram/roflumilast,
while the addition of single agents exhibited a minimal effect.
PDE4D, phosphodiesterase 4D; p, phosphorylated; 4EBP1, 4E-binding
protein 1. |
mTOR-Myc axis serves a critical role
in regulating the malignant phenotype of DLD-1 cells
To further characterize the mediators that regulate
the oncogenic properties of DLD-1 cells, the present study used
rapamycin and JQ1, chemical inhibitors of mTOR and bromo- and
extra-terminal domain (BET) proteins (34), respectively. Treatment with rapamycin
decreased the expression levels of Myc and p4EBP1, which indicates
that Myc is a downstream target of mTOR signaling (Fig. 4A). Additionally, the inhibition of the
mTOR/Myc axis by rapamycin was associated with a significant
decrease in the number of colonies and the number of cells in
proliferation assays (Fig. 4B-D). As
expected, JQ1 also decreased the expression level of Myc and
demonstrated a similar effect on colony number and cell number,
compared with rapamycin (Fig. 4),
which indicates that mTOR and Myc are critical mediators of the
antitumor effect of cAMP in DLD-1 cells.
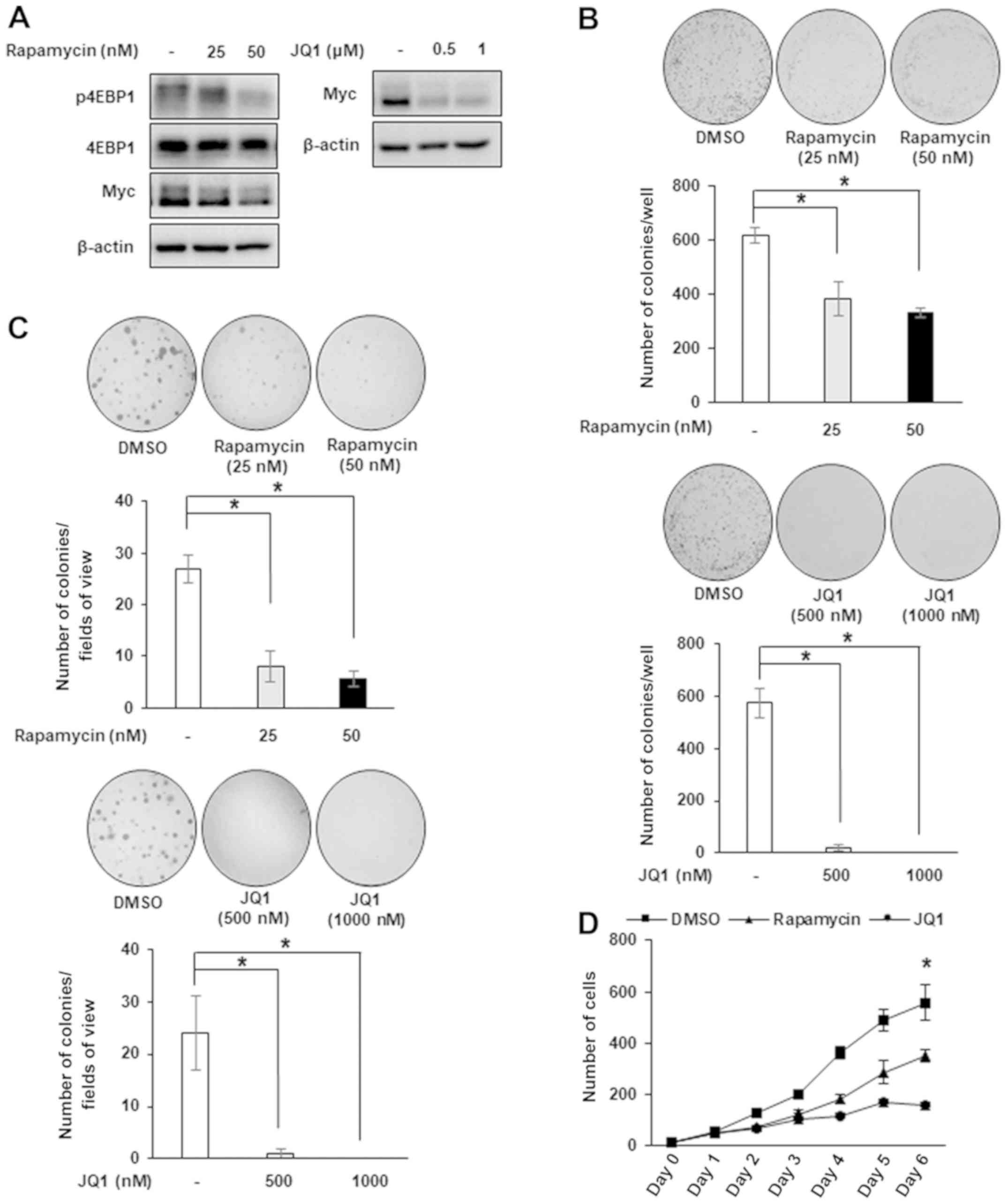 | Figure 4.JQ1 and rapamycin duplicate the
inhibitory effect of cyclic adenosine 3′,5′-monophosphate in DLD-1
cells. (A) DLD-1 cells were treated with rapamycin or JQ1 for 24 h
to analyze the expression levels of p4EBP1 and Myc by western blot
analysis. (B) Colony formation assay and (C) soft agar assay were
performed following treatment with DMSO, rapamycin (0–50 nM) or JQ1
(0–1,000 nM) and counted with the naked eye and at ×20
magnification. *P<0.05, according to one-way analysis of
variance. (D) Cell counting was performed following treatment with
DMSO, rapamycin (0–50 nM) or JQ1 (0–1,000 nM). Statistical
significance for each drug-treated group was compared with DMSO
treatment. *P<0.05, according to one-way analysis of variance.
Data are presented as the mean ± standard deviation. p,
phosphorylated; 4EBP1, 4E-binding protein 1; DMSO, dimethyl
sulfoxide. |
Resveratrol reproduces the antitumor
effect of PDE4 inhibitors
Based on previous studies that demonstrated that
resveratrol, a type of natural phenol, inhibits PDE4 activities
(35,36), the present study investigated whether
resveratrol could exhibit similar effects to the selective chemical
inhibitors rolipram and roflumilast. Following treatment with
resveratrol and/or forskolin, the intracellular levels of cAMP were
measured. Combined treatment with resveratrol and forskolin
significantly increased the expression level of cAMP in DLD-1
cells, compared with the control; however, the addition of either
agent alone did not exhibit any significant effect (Fig. 5A). This confirms that resveratrol
exhibits PDE4-inhibitor activities. Consistent with the antitumor
effect of cAMP, an increase in cAMP levels following the
administration of resveratrol and forskolin was associated with
decreased levels of p4EBP1 and Myc (Fig.
5B). This in turn was associated with suppression of cell
proliferation and colony formation in DLD-1 cells (Fig. 5C and D). These data indicate that
resveratrol may be an effective therapeutic agent for CRC with high
PDE4 activity.
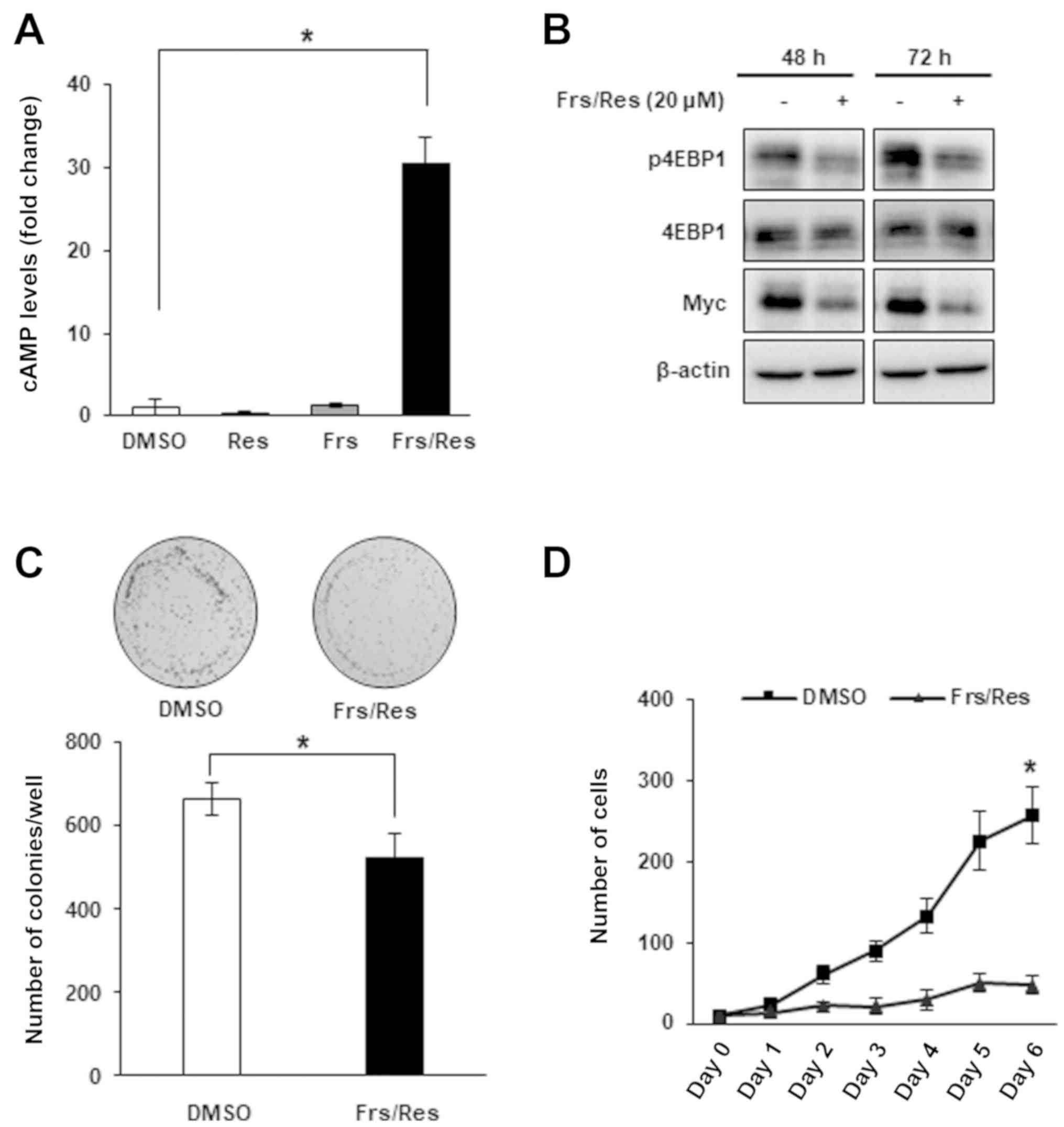 | Figure 5.Res reproduces the effect of rolipram
and roflumilast in DLD-1 cells. (A) DLD-1 colorectal cancer cells
were treated with DMSO, 20 µM Res for 14 h, 20 µM Frs for 2 h or a
combination of Frs and Res, and the intracellular cAMP levels were
measured. Data are presented as fold changes relative to the cAMP
levels in cells treated with DMSO. Co-treatment of Res and Frs
significantly increased cAMP levels while treatment of either agent
alone exhibited no significant effect *P<0.05, according to
one-way analysis of variance. (B) Expression levels of Myc and
p4EBP1 were analyzed following treatment with either DMSO, or a
combination of 20 µM Frs and 20 µM Res for 48 or 72 h. (C) Colony
formation was analyzed following treatment with either DMSO, or a
combination of 20 µM Frs and 20 µM Res and counted with the naked
eye. *P<0.05, according to Mann-Whitney U test. (D) Cell
proliferation was analyzed following treatment with either DMSO, or
a combination of 20 µM Frs and 20 µM Res. *P<0.05 vs. Frs/Res,
according to Mann-Whitney U test. Data are presented as the mean ±
standard deviation. Res, resveratrol; Frs, forskolin; cAMP, cyclic
adenosine 3′,5′-monophosphate; p, phosphorylated; 4EBP1, 4E-binding
protein 1; DMSO, dimethyl sulfoxide. |
Discussion
To the best of our knowledge, for the first time the
present study demonstrated that aberrant PDE4D expression may
contribute to the malignant phenotype of DLD-1 CRC cells. Notably,
the mTOR-Myc axis was identified to serve a critical role in
mediating the effect of PDE4D on the oncogenic properties of these
cells. Increased expression levels of cAMP following treatment with
PDE4 inhibitors suppressed mTOR signaling and Myc expression.
Additionally, the mTOR inhibitor, rapamycin, and BET bromodomain
inhibitor, JQ1, exhibited similar tumor suppressive effects,
compared with PDE4 inhibitors. cAMP, an important second messenger,
regulates diverse processes, including cell survival and death, in
a cell type- and context-dependent manner. It has been demonstrated
that numerous types of cancer cells prefer low levels of cAMP for
their growth and survival, which can be reversed using inhibitors
of cAMP-specific PDEs (25,37,38). The
present study is in agreement with these previous results, which
demonstrates that cancer cells rely on low cAMP levels.
Previous studies have reported that regulation of
mTOR phosphorylation/activity depends on the cell type. For
example, an increase of cAMP levels led to inhibition of mTOR
activity in 3T3-L1 adipocytes and B lymphoma cells (19,39), as
has been observed in CRC cells. However, mTOR activity was enhanced
by cAMP in PCCL3 rat thyroid cells, which was demonstrated by an
increase in ribosomal protein S6 kinase β1 and 4EBP1
phosphorylation (40). It would be
beneficial to examine what determines the cell type-dependent
regulation of mTOR activity by cAMP. The present results, which
revealed a downregulation of mTOR and Myc by roflumilast, may
exhibit clinical implications. Roflumilast was FDA-approved for the
treatment of chronic obstructive pulmonary disease in 2011 and a
clinical trial has successfully been performed to test its efficacy
against aggressive B-cell lymphoma (41). The present study indicates that this
drug may be repurposed to suppress Myc and CRC development in a
clinical setting.
PDE4D has been identified to affect tumor
progression in a number of ways in different types of cancer. For
example, PDE4D interacts with focal adhesion kinase via the
receptor for activated C kinase 1 scaffold protein to regulate cell
invasion in BRAF-mutated melanoma (42). In medulloblastoma, inhibition of PDE4D
downregulates Hedgehog (Hh) signaling to suppress tumor growth,
indicating that PDE4D can be a therapeutic target in tumors with
high Hh activities (43).
Myc has been demonstrated to control several
important aspects of cancer cell biology, including promoting cell
proliferation, inducing resistance to chemotherapeutic drug-induced
apoptosis and it is associated with invasion/metastasis and
metabolic reprogramming (44). As
demonstrated by cell counting and soft agar assays, increased cAMP
due to PDE4D inhibition suppressed cancer cell proliferation and
invasion in vitro. Testing the efficacy of PDE4D inhibitors
in vivo using animal models may provide improved insight
into the role of PDE4D in the pathogenesis of colon cancer. GEBR-7b
and GEBR-32a are two newly developed PDE4D inhibitors (45,46). These
compounds have demonstrated memory-enhancing activities in animal
models and may be used in the therapies of neurodegenerative
disorders, including Alzheimer's disease (46). Additionally, GEBR-7b has been used to
prevent tamoxifen resistance in ER-positive breast cancer in
vivo (47); however, the
tumor-suppressive effect of these inhibitors has not been
investigated in colon cancer, which requires further studies.
It has been demonstrated that PDE4D is aberrantly
expressed in patients with prostate cancer and tamoxifen-resistant
breast cancer cells (47,48). Although a more systematic approach is
required to reach any substantial conclusion, the RT-qPCR data
indicated that DLD-1 cells highly express PDE4D. This indicates
that CRC cells and patients with CRC may also exhibit abnormal
PDE4D levels, which may potentially affect the pathogenesis of the
disease. The mechanisms underlying PDE4D overexpression in CRC
remain to be elucidated. However, recent data indicated that
downregulation of miR-139-5p may serve a role in elevated levels of
PDE4D. Firstly miRNA-139-5p induced by the p53 tumor suppressor has
been demonstrated to target PDE4D in cancer cells (23). Additionally, the expression of
miR-139-5p was markedly reduced in CRC tissues, compared with
adjacent non-cancerous tissues (49).
Lastly, the present study revealed that the expression levels of
miR-139-5p and PDE4D were inversely correlated in CRC tissue
samples. Further studies may improve the understanding regarding
the mechanisms underlying PDE4D overexpression in CRC and other
types of cancer.
Protein kinase A (PKA) and exchange protein
activated by cAMP (EPAC) are the main effectors of cAMP (50); however, it is unclear whether the
anti-proliferative effect of cAMP in DLD-1 cells is dependent on
PKA and/or EPAC. Notably, the cytotoxic effects of cAMP in normal
and malignant B cells are independent of PKA and EPAC (21). Additionally, activation of cAMP
signaling by loss of PDE4D mediates resistance to the
chemotherapeutic drug Triapine via EPAC in the SW480 human colon
adenocarcinoma cell line (51). These
data indicate that cAMP signaling is performed in a cell type- and
context-dependent manner. It would be beneficial to examine
downstream target molecules of cAMP that mediate its
tumor-suppressive effect in DLD-1 cells.
Resveratrol is a natural polyphenolic compound
present in red wine and other food products. It is an antioxidant
with potential antitumor and anti-aging properties (35). In a murine aging model, treatment with
resveratrol reversed aging-associated metabolic abnormalities,
including diet-induced obesity and impaired glucose tolerance,
which has been identified to be reproduced by a PDE4 inhibitor
rolipram (35). The present study
demonstrated that resveratrol can suppress the malignant phenotype
of DLD-1 cells with high expression of PDE4D. Previous data,
together with the present results, clearly indicate that PDE4D is a
promising target for the treatment of patients with cancer with
aberrant expression of PDE4D.
Acknowledgements
Not applicable.
Funding
This work was supported by Basic Science Research
Program through the National Research Foundation of Korea funded by
the Ministry of Science, ICT and Future Planning
(NRF-2013R1A1A2008838 and NRF-2016R1A2B4011758) to SWK.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
DUK designed and performed the experiments, analyzed
the data and wrote the manuscript. JN performed experiments and
analyzed the data. MDC performed statistical analysis of all data
and assisted with revision of the manuscript. SWK designed and
supervised the study, analyzed the data and wrote the manuscript.
All the authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Arnold M, Sierra MS, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global patterns and trends in
colorectal cancer incidence and mortality. Gut. 66:683–691. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A:
Colorectal cancer mortality rates in adults aged 20 to 54 years in
the united states, 1970–2014. JAMA. 318:572–574. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Worthley DL and Leggett BA: Colorectal
cancer: Molecular features and clinical opportunities. Clin Biochem
Rev. 31:31–38. 2010.PubMed/NCBI
|
|
4
|
Hinoue T, Weisenberger DJ, Pan F, Campan
M, Kim M, Young J, Whitehall VL, Leggett BA and Laird PW: Analysis
of the association between CIMP and BRAF in colorectal cancer by
DNA methylation profiling. PLoS One. 4:e83572009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mundade R, Imperiale TF, Prabhu L, Loehrer
PJ and Lu T: Genetic pathways, prevention, and treatment of
sporadic colorectal cancer. Oncoscience. 1:400–406. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Walther A, Johnstone E, Swanton C, Midgley
R, Tomlinson I and Kerr D: Genetic prognostic and predictive
markers in colorectal cancer. Nat Rev Cancer. 9:489–499. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fodde R: The APC gene in colorectal
cancer. Eur J Cancer. 38:867–871. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schneikert J and Behrens J: The canonical
Wnt signalling pathway and its APC partner in colon cancer
development. Gut. 56:417–425. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Easwaran V, Lee SH, Inge L, Guo L,
Goldbeck C, Garrett E, Wiesmann M, Garcia PD, Fuller JH, Chan V, et
al: Beta-Catenin regulates vascular endothelial growth factor
expression in colon cancer. Cancer Res. 63:3145–3153.
2003.PubMed/NCBI
|
|
10
|
Herbst A, Jurinovic V, Krebs S, Thieme SE,
Blum H, Göke B and Kolligs FT: Comprehensive analysis of β-catenin
target genes in colorectal carcinoma cell lines with deregulated
Wnt/β-catenin signaling. BMC Genomics. 15:742014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morin PJ: Beta-catenin signaling and
cancer. Bioessays. 21:1021–1030. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sansom OJ, Meniel VS, Muncan V, Phesse TJ,
Wilkins JA, Reed KR, Vass JK, Athineos D and Clevers H: Myc
deletion rescues Apc deficiency in the small intestine. Nature.
446:676–679. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sewastianik T, Prochorec-Sobieszek M,
Chapuy B and Juszczyński P: MYC deregulation in lymphoid tumors:
Molecular mechanisms, clinical consequences and therapeutic
implications. Biochim Biophys Acta. 1846:457–467. 2014.PubMed/NCBI
|
|
14
|
Rall TW and Sutherland EW: Formation of a
cyclic adenine ribonucleotide by tissue particles. J Biol Chem.
232:1065–1076. 1958.PubMed/NCBI
|
|
15
|
Sutherland EW and Rall TW: Fractionation
and characterization of a cyclic adenine ribonucleotide formed by
tissue particles. J Biol Chem. 232:1077–1091. 1958.PubMed/NCBI
|
|
16
|
Daniel PB, Walker WH and Habener JF:
Cyclic AMP signaling and gene regulation. Annu Rev Nutr.
18:353–383. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fimia GM and Sassone-Corsi P: Cyclic AMP
signalling. J Cell Sci. 114:1971–1972. 2001.PubMed/NCBI
|
|
18
|
Sassone-Corsi P: The cyclic AMP pathway.
Cold Spring Harb Perspect Biol. 4(a011148)2012.PubMed/NCBI
|
|
19
|
Kim SW, Rai D and Aguiar RC: Gene set
enrichment analysis unveils the mechanism for the phosphodiesterase
4B control of glucocorticoid response in B-cell lymphoma. Clin
Cancer Res. 17:6723–6732. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Smith PG, Wang F, Wilkinson KN, Savage KJ,
Klein U, Neuberg DS, Bollag G, Shipp MA and Aguiar RC: The
phosphodiesterase PDE4B limits cAMP-associated PI3K/AKT-dependent
apoptosis in diffuse large B-cell lymphoma. Blood. 105:308–316.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsunoda T, Ota T, Fujimoto T, Doi K,
Tanaka Y, Yoshida Y, Ogawa M, Matsuzaki H, Hamabashiri M, Tyson DR,
et al: Inhibition of phosphodiesterase-4 (PDE4) activity triggers
luminal apoptosis and AKT dephosphorylation in a 3-D colonic-crypt
model. Mol Cancer. 11:462012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cao B, Wang K, Liao JM, Zhou X, Liao P,
Zeng SX, He M, Chen L, He Y, Li W and Lu H: Inactivation of
oncogenic cAMP-specific phosphodiesterase 4D by miR-139-5p in
response to p53 activation. Elife. 5(e15978)2016.
|
|
24
|
Lakics V, Karran EH and Boess FG:
Quantitative comparison of phosphodiesterase mRNA distribution in
human brain and peripheral tissues. Neuropharmacology. 59:367–374.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McEwan DG, Brunton VG, Baillie GS, Leslie
NR, Houslay MD and Frame MC: Chemoresistant KM12C colon cancer
cells are addicted to low cyclic AMP levels in a phosphodiesterase
4-regulated compartment via effects on phosphoinositide 3-kinase.
Cancer Res. 67:5248–5257. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Le Jeune IR, Shepherd M, Van Heeke G,
Houslay MD and Hall IP: Cyclic AMP-dependent transcriptional
up-regulation of phosphodiesterase 4D5 in human airway smooth
muscle cells. Identification and characterization of a novel PDE4D5
promoter. J Biol Chem. 277:35980–35989. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takahashi M, Terwilliger R, Lane C, Mezes
PS, Conti M and Duman RS: Chronic antidepressant administration
increases the expression of cAMP-specific phosphodiesterase 4A and
4B isoforms. J Neurosci. 19:610–618. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lipworth BJ: Phosphodiesterase-4
inhibitors for asthma and chronic obstructive pulmonary disease.
Lancet. 365:167–175. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Francipane MG and Lagasse E: mTOR pathway
in colorectal cancer: An update. Oncotarget. 5:49–66. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pandurangan AK: Potential targets for
prevention of colorectal cancer: A focus on PI3K/Akt/mTOR and Wnt
pathways. Asian Pac J Cancer Prev. 14:2201–2205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sipos F, Firneisz G and Műzes G:
Therapeutic aspects of c-MYC signaling in inflammatory and
cancerous colonic diseases. World J Gastroenterol. 22:7938–7950.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pirson I, Coulonval K, Lamy F and Dumont
JE: c-Myc expression is controlled by the mitogenic cAMP-cascade in
thyrocytes. J Cell Physiol. 168:59–70. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Williamson EA, Burgess GS, Eder P,
Litz-Jackson S and Boswell HS: Cyclic AMP negatively controls c-myc
transcription and G1 cell cycle progression in p210 BCR-ABL
transformed cells: Inhibitory activity exerted through cyclin D1
and cdk4. Leukemia. 11:73–85. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Di Costanzo A, Del Gaudio N, Migliaccio A
and Altucci L: Epigenetic drugs against cancer: An evolving
landscape. Arch Toxicol. 88:651–668. 2014. View Article : Google Scholar
|
|
35
|
Park SJ, Ahmad F, Philp A, Baar K,
Williams T, Luo H, Ke H, Rehmann H, Taussig R, Brown AL, et al:
Resveratrol ameliorates aging-related metabolic phenotypes by
inhibiting cAMP phosphodiesterases. Cell. 148:421–433. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tsunoda T, Ishikura S, Doi K, Matsuzaki H,
Iwaihara Y and Shirasawa S: Resveratrol induces luminal apoptosis
of human colorectal cancer HCT116 cells in three-dimensional
culture. Anticancer Res. 34:4551–4555. 2014.PubMed/NCBI
|
|
37
|
Goldhoff P, Warrington NM, Limbrick DD Jr,
Hope A, Woerner BM, Jackson E, Perry A, Piwnica-Worms D and Rubin
JB: Targeted inhibition of cyclic AMP phosphodiesterase-4 promotes
brain tumor regression. Clin Cancer Res. 14:7717–7725. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ogawa R, Streiff MB, Bugayenko A and Kato
GJ: Inhibition of PDE4 phosphodiesterase activity induces growth
suppression, apoptosis, glucocorticoid sensitivity, p53, and
p21(WAF1/CIP1) proteins in human acute lymphoblastic leukemia
cells. Blood. 99:3390–3397. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Scott PH and Lawrence JC Jr: Attenuation
of mammalian target of rapamycin activity by increased cAMP in
3T3-L1 adipocytes. J Biol Chem. 273:34496–34501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Blancquaert S, Wang L, Paternot S,
Coulonval K, Dumont JE, Harris TE and Roger PP: cAMP-dependent
activation of mammalian target of rapamycin (mTOR) in thyroid
cells. Implication in mitogenesis and activation of CDK4. Mol
Endocrinol. 24:1453–1468. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kelly K, Mejia A, Suhasini AN, Lin AP,
Kuhn J, Karnad AB, Weitman S and Aguiar RC: Safety and
pharmacodynamics of the PDE4 inhibitor roflumilast in advanced
B-cell malignancies. Clin Cancer Res. 23:1186–1192. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Delyon J, Servy A, Laugier F, André J,
Ortonne N, Battistella M, Mourah S, Bensussan A, Lebbé C and Dumaz
N: PDE4D promotes FAK-mediated cell invasion in BRAF-mutated
melanoma. Oncogene. 36:3252–3262. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ge X, Milenkovic L, Suyama K, Hartl T,
Purzner T, Winans A, Meyer T and Scott MP: Phosphodiesterase 4D
acts downstream of Neuropilin to control Hedgehog signal
transduction and the growth of medulloblastoma. Elife.
4:e070682015. View Article : Google Scholar
|
|
44
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bruno O, Fedele E, Prickaerts J, Parker
LA, Canepa E, Brullo C, Cavallero A, Gardella E, Balbi A,
Domenicotti C, et al: GEBR-7b, a novel PDE4D selective inhibitor
that improves memory in rodents at non-emetic doses. Br J
Pharmacol. 164:2054–2063. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ricciarelli R, Brullo C, Prickaerts J,
Arancio O, Villa C, Rebosio C, Calcagno E, Balbi M, van Hagen BT,
Argyrousi EK, et al: Memory-enhancing effects of GEBR-32a, a new
PDE4D inhibitor holding promise for the treatment of Alzheimer's
disease. Sci Rep. 7:463202017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mishra RR, Belder N, Ansari SA, Kayhan M,
Bal H, Raza U, Ersan PG, Tokat ÜM, Eyüpoğlu E, Saatci Ö, et al:
Reactivation of cAMP pathway by PDE4D inhibition represents a novel
druggable axis for overcoming tamoxifen resistance in ER-positive
breast cancer. Clin Cancer Res. 24:1987–2001. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Böttcher R, Dulla K, van Strijp D, Dits N,
Verhoef EI, Baillie GS, van Leenders GJ, Houslay MD, Jenster G and
Hoffmann R: Human PDE4D isoform composition is deregulated in
primary prostate cancer and indicative for disease progression and
development of distant metastases. Oncotarget. 7:70669–70684. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Li Q, Liang X, Wang Y, Meng X, Xu Y, Cai
S, Wang Z, Liu J and Cai G: miR-139-5p inhibits the
epithelial-mesenchymal transition and enhances the chemotherapeutic
sensitivity of colorectal cancer cells by downregulating BCL2. Sci
Rep. 6:271572016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Garcia-Morales V, Luaces-Regueira M and
Campos-Toimil M: The cAMP effectors PKA and Epac activate
endothelial NO synthase through PI3K/Akt pathway in human
endothelial cells. Biochem Pharmacol. 145:94–101. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Miklos W, Heffeter P, Pirker C, Hager S,
Kowol CR, van Schoonhoven S, Stojanovic M, Keppler BK and Berger W:
Loss of phosphodiesterase 4D mediates acquired triapine resistance
via Epac-Rap1-Integrin signaling. Oncotarget. 7:84556–84574. 2016.
View Article : Google Scholar : PubMed/NCBI
|














