Introduction
Worldwide, head and neck squamous cell carcinomas
(HNSCCs) are highly aggressive tumors in the upper aerodigestive
tract affecting more than half a million patients each year
(1). So, these tumors that affect
the oral cavity, oropharynx, larynx, and hypopharynx are usually
described as squamous cell carcinoma of the upper aerodigestive
tract, being the patient outcomes strongly associated to tumor
stage (2). Patients with advanced
stage tumors (stages III and IV) present frequently recurrences
(3), which lead to a poor prognosis
and quality of life. Cigarette smoking and alcohol consumption have
been established as the main risk factors for these carcinomas
(4). The Human Papillomavirus (HPV)
infection is a well established risk factor for squamous cell
carcinoma of the oropharynx but its role remains unclear in oral
cavity and laryngeal cancer (5).
Surgical intervention, radiotherapy and chemotherapy are the main
treatments for HNSCC. The 5-year survival rate for these patients
is still about 50% even with treatment advances (6).
These tumors are considered the final stage of a
multistep carcinogenic process involving amplifications, deletions,
up and downregulation of oncogenes and tumor suppressor genes,
responsible for the initiation, promotion and progression of
neoplasms (7). In the last years,
important progresses in the understanding of molecular biology of
upper aerodigestive tract cancer have been achieved, which allowed
a better characterization of these tumors; however, the survival of
these patients remains without great improvements. Introducing
molecular biomarkers as predictive factors to determine which
patients will develop recurrences, will fail to response to
treatment or will benefit with target therapies may become a great
help to improve survival and quality of life of the patients. In
this study, we used whole genome copy number alterations (CNAs) and
methylation status in order to identify genomic and epigenetic
signatures able to predict recurrence/metastasis development in
patients with primary upper aerodigestive tract carcinoma treated
with curative intent. Our results represent a step further through
the identification of clinically significant biomarkers with
predictive value for these cancer patients management.
Materials and methods
Study population
The study protocol was approved by the Committee on
Ethics in Research of the Faculty of Medicine of the University of
Coimbra. All patients provided their written consent to participate
in the study after being informed about the research purposes,
following the regulations in the Declaration of Helsinki.
The study cohort includes tumor tissue and tissue
from surgery resection margin (macroscopically tumor-free tissue)
of 21 patients with diagnosis of upper aerodigestive tract
carcinoma. The tissue samples were snap-frozen in liquid nitrogen
within 30 min after resection and stored at −80°C until use. The
patients were recruited between December 2013 and March 2017 from
the Department of Otorhinolaryngology-Head and Neck Surgery of
Coimbra Hospital and University Centre (CHUC), EPE, Portugal.
Diagnosis and staging were performed in accordance with the
American Joint Committee on Cancer TNM staging system (8). Patients were followed-up through
hospital revisits during routine clinical appointments. The
follow-up periods ranged from 6 to 46 months. Details of our study
cohort are listed in Table I. For
the control group, 7 palatine uvulas from patients diagnosed with
sleep apnoea and/or snoring were used (5 males and 2 females, with
ages ranging from 31 to 71 years).
 | Table I.Clinic-pathologic characteristics of
study population (n=21). |
Table I.
Clinic-pathologic characteristics of
study population (n=21).
| Characteristic | n |
|---|
| Sex |
|
|
Male | 20 |
|
Female | 1 |
| Anatomic
subsite |
|
|
Larynx | 8 |
|
Hypopharynx | 2 |
|
Piriform sinus | 2 |
|
Oropharynx | 2 |
|
Epiglottis | 1 |
|
Supraglottis | 1 |
|
Glottis | 1 |
|
Hemilarynx | 1 |
| Vocal
chord | 1 |
|
Pharynx | 1 |
|
Palate/left tonsil | 1 |
| Tobacco |
|
|
Yes | 21 |
| No | 0 |
| Alcohol |
|
|
Yes | 20 |
| No | 1 |
| Age at diagnosis
(Years) |
|
|
<60 | 12 |
|
≥60 | 9 |
| TNM stage |
|
| I | 2 |
| II | 1 |
|
III | 2 |
| IV | 16 |
| Treatment |
|
| Surgery
only | 4 |
| QT
alone | 2 |
| RT
alone | 2 |
| Surgery
+ QT | 2 |
| Surgery
+ RT | 3 |
| Surgery
+ RT + QT | 1 |
| RT +
QT | 3 |
| NA | 4 |
| Vital status |
|
|
Relapses/Metastasis in
follow-up | 8 |
|
Dead-Cancer | 8 |
|
Dead-non-Cancer | 2 |
DNA extraction and HPV typing
DNA from fresh frozen tissues of patients and
controls were extracted using a High Pure PCR Template Preparation
Kit (Roche GmbH), according to the manufacturer's instructions. The
DNAs were quantified by UV spectrophotometric analysis using a
NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Inc.).
All tumor tissue samples were analyzed for HPV infection as
previous described (9,10).
Array-CGH analysis
High-resolution whole genome analyses were performed
using Agilent SurePrint G3 Human Genome microarray 180 K (Agilent
Technologies), according our previous work (2). DNA of tumor samples was labelled with
Cy5 by random primer labelling. DNA from controls was labelled with
Cy3. Results were analysed using Agilent Genomic Workbench v6.5
software with the following settings: ADM1 as aberration algorithm,
threshold of 6.0, moving average 2 Mb. The results are according to
Human Genome build 19 and include imbalances with at least three
consecutive probes with abnormal log2 ratios.
MS-MLPA analysis
MS-MLPA analyses were performed using MS-MLPA probe
set ME002 (MRC-Holland), which can simultaneously detect CNAs in 38
tumor suppressor genes and aberrant methylation patterns in a
subset of 25 of these genes. All MS-MLPA reactions were performed
according our previous work (11,12).
Three controls selected from the previously analyzed control group,
without CNAs and methylation values below 20%, as well as a
negative control (without DNA), were always included in each
MS-MLPA assay. Binning of the raw data and comparative analyses
were performed using Coffalyser. NET software. For each probe we
determined the specific cutoff values for gain and loss, using 95%
confidence intervals as determined on non-cancer subjects. A copy
number gain was scored when a value exceeded 1.2 and a copy number
loss was scored when a value was lower than 0.8. We considered a
gene promoter as methylated when the methylation dosage ratio was
≥0.20, which means that at least 20% of the DNA was methylated.
These cut-off values were based on our previous works (9–11).
Statistical analysis
Methylation and CNA data were firstly analysed
considering the number of patients that presented genes
alterations. The genes that have less than 10% of patients carrying
an alteration were discarded. For the remaining genes, 7 regarding
methylation and 25 with CNA, a measure of the effect size for
discriminating the groups with and without relapses/metastases was
calculated. The odds-ratio was used for this purpose. Since the
total number of cases is only 21, we selected the four genes that
presented the largest odds-ratio to fit a logistic regression,
aiming to further study the influence of these genes in the
occurrence of relapses/metastaes.
The MS-MLPA data were desribed resorting to a circos
plot, which allows by visual inspection, to identify the 3p region
as the best candidate as a biomarker of relapses/metastases. We
calculate the fraction of deletion and amplification as the
quotient between the number of pair bases altered and the number of
pair bases composing the 3p arm. We then assessed, with a ROC
analysis, this quantitative measure as a potential discrminative
variable of the relapses/metastases group.
Finaly, we integrated the genes selected from the
methylation and the CNA data with the fraction of deletion in a
logistic model. As the number of cases is quite small the classical
forms (e.g. ANOVA test, Naguelkerke pseudo R2, Wald test, etc) to
evaluate the logistic regression tend to give inconsistent results,
instead we adopted a leave-one -out method to compute the accuracy
in the cases not entering in the training set and, in turn, to get
a generalizability measure.
The analyses were carried out using SPSS v24, R
v3.3.2 and Matlab R2018b. The significance level adopt was
0.05.
Results
HPV typification
We did not detect any HPV-positive sample in the 21
patients analyzed.
CNAs detection by array-CGH
The genomic characterization of tumor samples
through whole genome array-CGH revealed several copy number gains
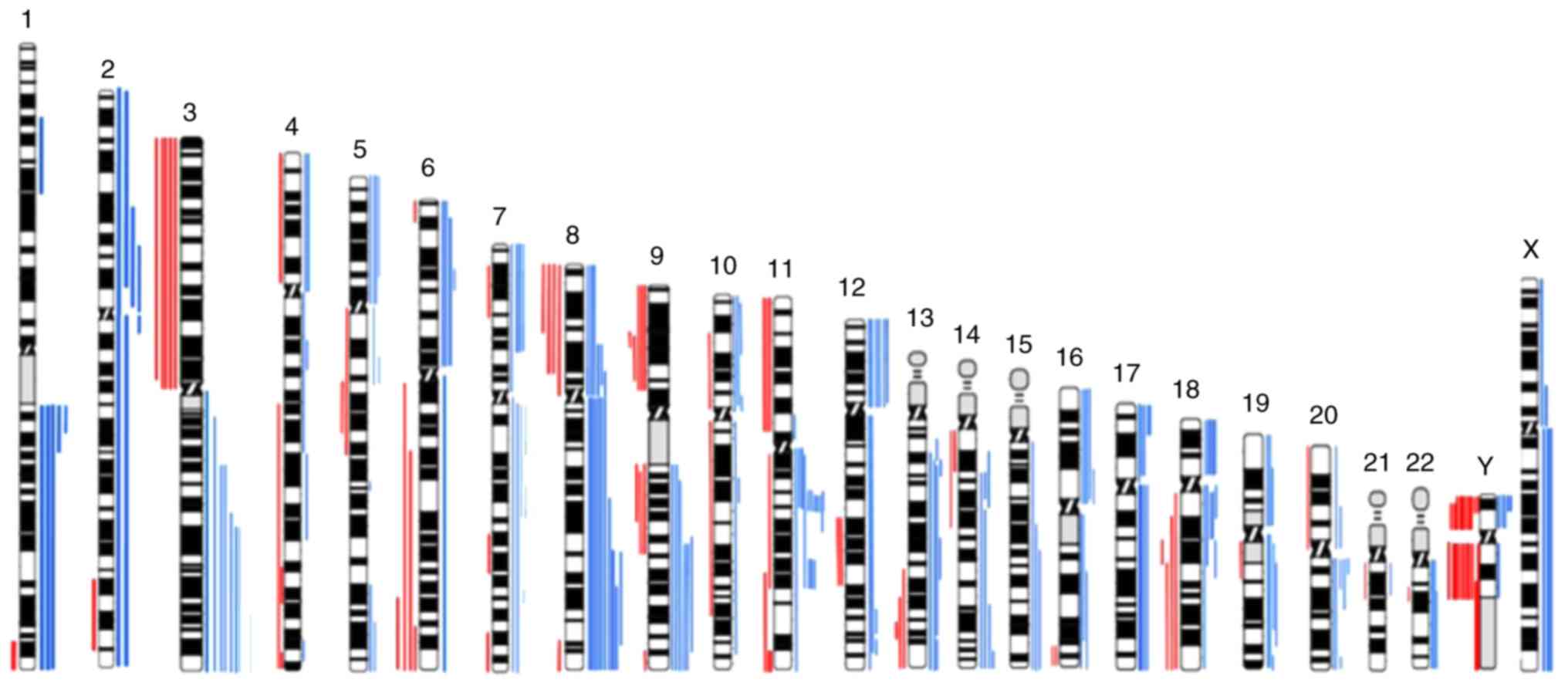
and losses in all chromosomes (Fig.
1), being the most frequent copy number gains observed at 3q,
7p, 8q, 9q, 11q13, 12p and 18p and the most frequent copy number
losses observed at 3p, 8p 9p and Y (Fig.
1). This aCGH analysis demonstrated that gains of chromosomal
segments were more common than losses.
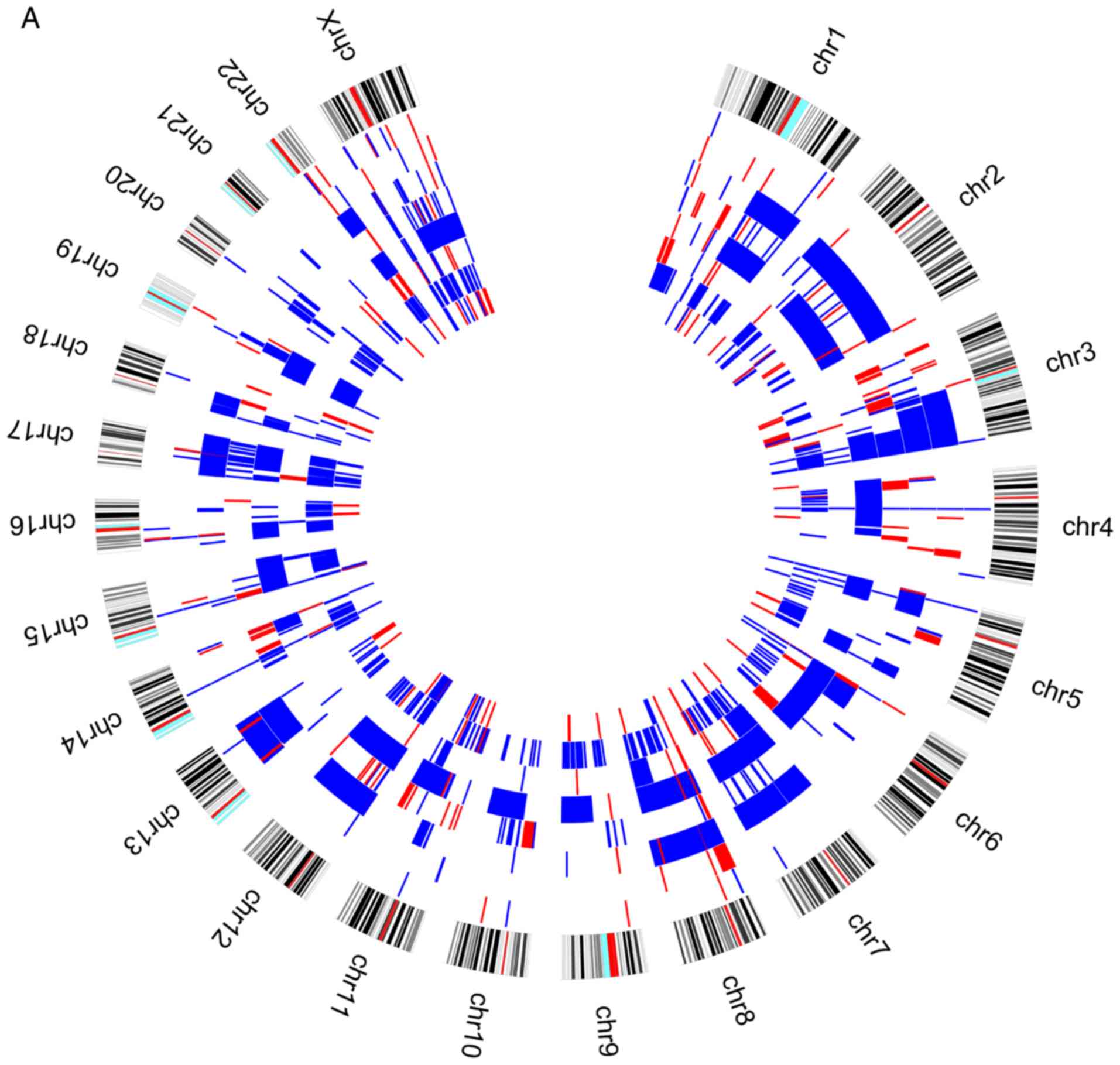
When we analysed the genomic profile of the two
groups of patients according to the presence or absence of
relapses/metastases during the patients follow-up, we observed that
losses at 3p chromosomes are more frequent in patients without
relapses/metastases (Fig. 2).
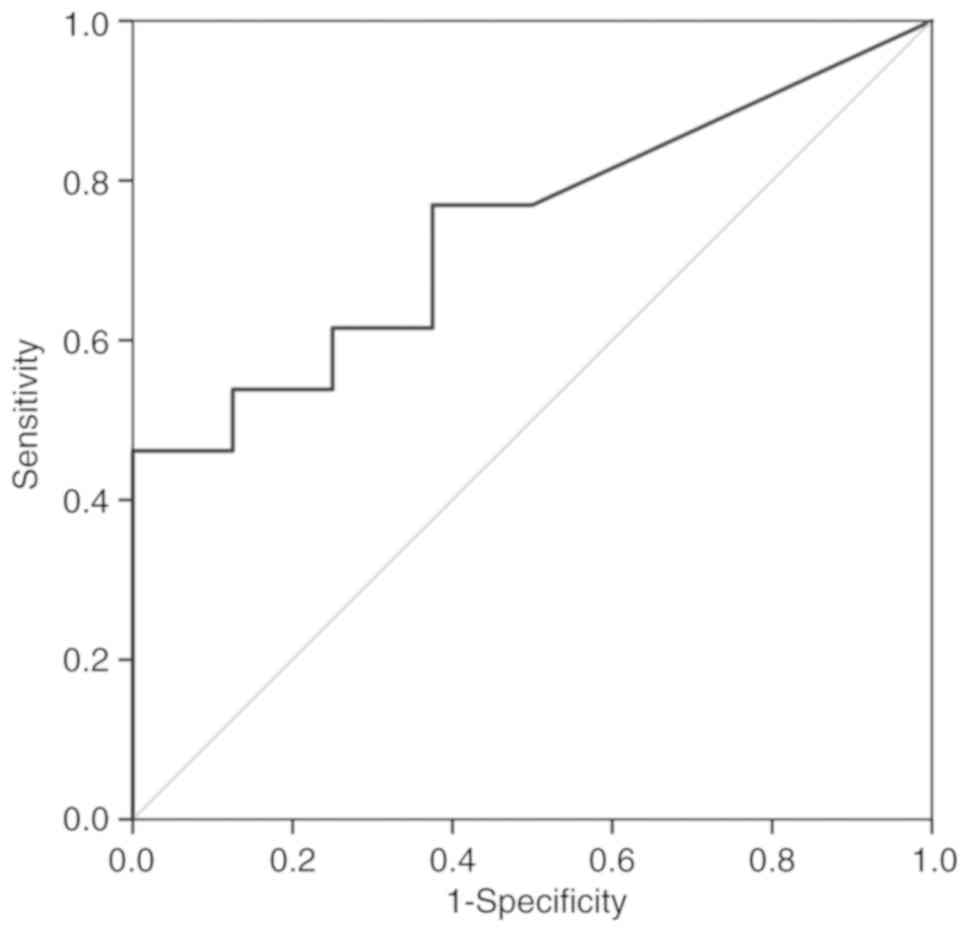
A ROC analysis over the fraction of deletion in 3p
was performed in order to verify if the genomic differences
observed in this chromosome arm could be useful to predict the risk
of these patients develop relapses or metastases. This analysis
showed that the gains observed in 3p can not discriminate the two
groups of patients (AUC=0.510; P=0.942). However, considering the
losses in this region, even without statistical significance
(P=0.070), the AUC is 0.740 [IC95%(0.529; 0.952)], which suggest
that this variable has discriminative power (Fig. 3). This result highlighted 3p losses
as a putative biomarker of reduced risk of relapses/metastases
development during follow-up of upper aerodigestive tract cancer
patients, after the treatment of primary tumor.
The fraction of deletion in 3p was transformed into
a bynary variable using as cut point the value that maximizes the
Youden index of the ROC anaysis. This variable describing the
amount of deletion in 3p is significantly associated with
relapses/metastases (P=0.042) and corroborates the previous
observation that 3p deletion reduces the chance of
relapses/metastases.
CNAs and methylation detection by
MS-MLPA
Several genetic and epigenetic alterations were
detected in the upper aerodigestive tract tumor and non-tumor
tissue samples by MS-MLPA (Fig. 4A and
D). The genetic alterations more frequently altered in
non-tumor samples were loss of MSH6 (2p16.3), VHL
(3p25.3), and CHFR (12q24.33) genes and gain at GATA5
(20q13.33) gene (Fig. 4A-C). The
genetic alterations more frequently altered in tumor samples were
loss of VHL, CDKN2A (9p21.3), CREM (10p11.21),
ATM (11q22.3) and CADM1 (11q23) genes and gain at
GATA5, CDK6 (7q21.2), PTCH1 (9q22.32), CD44
(11p13), RB1 (13q14.2), THBS1 (15q14) and TSC2
(16p13.3) genes (Fig. 4D-F). In
agreement with array-CGH analysis, the CNV analysis through MS-MLPA
assay confirmed the presence of a greater number of gains in
relation to losses in these samples.
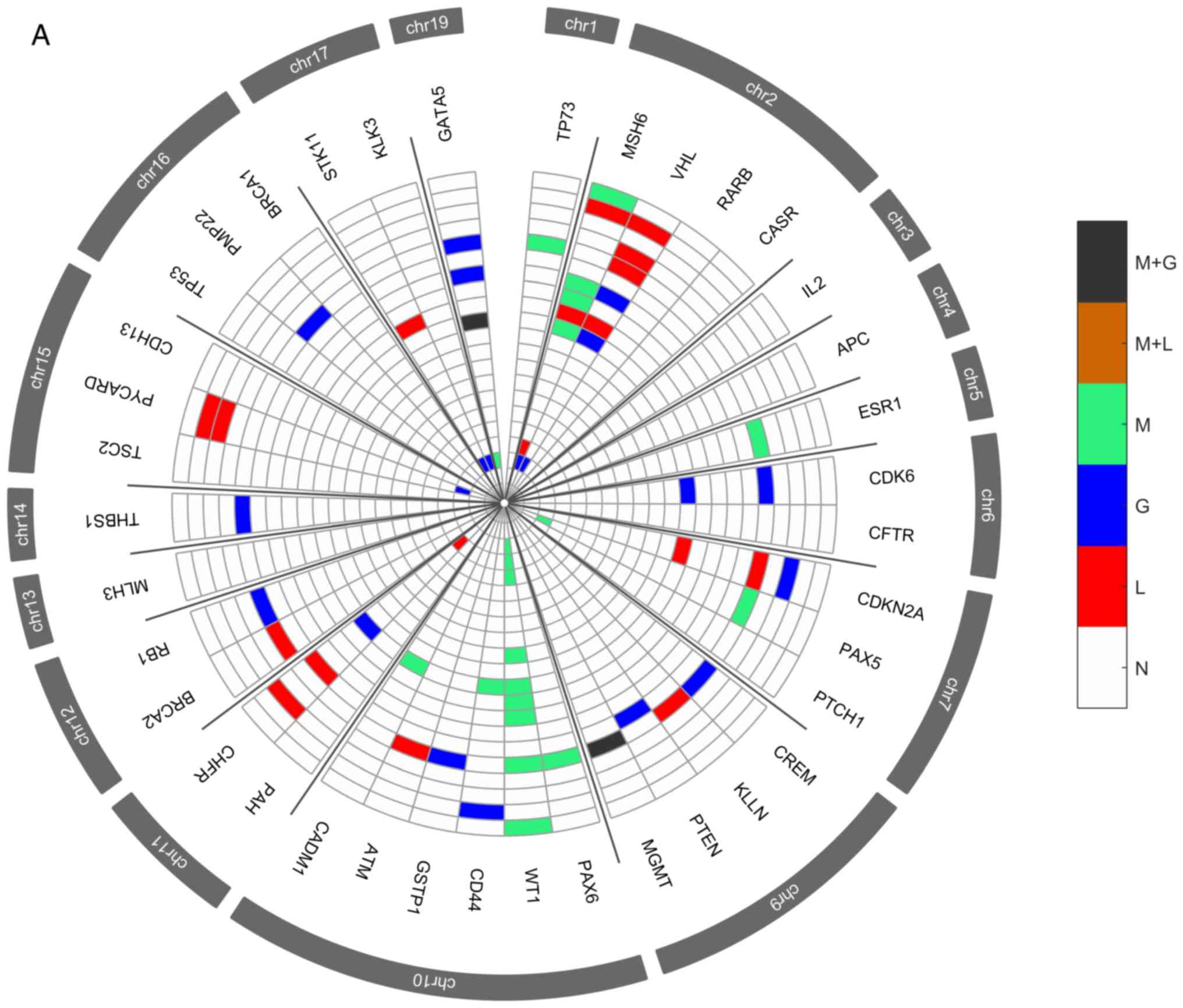 | Figure 4.Radar chart with analyzed genes for
methylation and the respective copy number alterations (A) in all
patients with non-tumor upper aerodigestive tract cancer. M + G,
methylation and copy number gains; M + L, methylation and copy
number losses; M, methylation; G, gains; L, losses; N, Normal.
Radar chart with analyzed genes for methylation and the respective
copy number alterations (B) in patients with non-tumor upper
aerodigestive tract cancer with relapses or metastasis. M + G,
methylation and copy number gains; M + L, methylation and copy
number losses; M, methylation; G, gains; L, losses; N, Normal.
Radar chart with analyzed genes for methylation and the respective
copy number alterations (D) in all patients with tumor upper
aerodigestive tract cancer. M + G, methylation and copy number
gains; M + L, methylation and copy number losses; M, methylation;
G, gains; L, losses; N, Normal. Radar chart with analyzed genes for
methylation and the respective copy number alterations (C) in
patients with non-tumor upper aerodigestive tract cancer without
relapses or metastasis. M + G, methylation and copy number gains; M
+ L, methylation and copy number losses; M, methylation; G, gains;
L, losses; N, Normal. Radar chart with analyzed genes for
methylation and the respective copy number alterations (E) in
patients with tumor upper aerodigestive tract cancer with relapses
or metastasis. M + G, methylation and copy number gains; M + L,
methylation and copy number losses; M, methylation; G, gains; L,
losses; N, Normal. Radar chart with analyzed genes for methylation
and the respective copy number alterations (F) in patients with
tumor upper aerodigestive tract cancer without relapses or
metastasis. M + G, methylation and copy number gains; M + L,
methylation and copy number losses; M, methylation; G, gains; L,
losses; N, Normal. |
In the non-tumor samples, the most frequent
methylated genes were WT1 and MSH6 genes (Fig. 4A-C). In the tumor samples, the most
frequent methylated genes were WT1, GATA5 and MSH6
genes (Fig. 4D-F). Besides, in the
non-tumor samples few genetic and epigenetic alterations were
observed, some of those appear in both tumor and non-tumor samples,
which may be indicative of dissemination of cells with malignant
features.
Only WT1 gene presented in one patient both
copy number loss and methylation in the tumor sample. Also, in
tumor samples, GATA5, MSH6, ESR1 and MGMT genes
presented both copy number gain and methylation (Fig. 4D). While in non-tumor samples,
MGMT and GATA5 presented both copy number gain and
methylation (Fig. 4A).
After a two step genes reduction process, a
statistical model linking upper aerodigestive tract
recurrence/metastasis development and the identified genetic and
epigenetic profile was build using a logistic regression. The
developed model comprises WT1 gene promoter methylation and
CNA of VHL and THBS1 genes. The model obtained does
not present statistical significance (χ2(5)-8.876; P=0.114) but the
value of explained variance (RNaguelkerke2) is 0.510 and the accuracy
(78.9%) is higher than obtained by chance (63.2%). Univariate
analysis of the independent variables shows that when WT1 is
methylated both in tumor and non-tumor tissues the chance of
relapses/metastasis is disminished with odds-ratio equal to 0.33
(P=0.346) and 0.286 (P=0.367), respectively. Regarding CNA, the
alteration of the VHL gene reduces the chance of
relapses/metastasis (OR=0.167; P=0.173) whereas the alteration of
THBS1 gene increases the change of relapses/metastasis
(OR=5.5; P=0.146).
Predictive model using genomic and
epigenetic data
Taking into account the results obtained by
array-CGH and MS-MLPA techniques, the logistic model was adjusted
in order to comprise the previously selected genes as well as the
3p chromosome arm. The model obtained was assessed with a
leave-one-out method in order to use all the cases but one as the
training set. The accuracy to predict the leave-out case was 77.8%
that compares to 61.1% if it was by chance. The sensitivity and
specificity were 81.8 and 71.4%, respectively. The mean accuracy in
the training set was 99.4% and the sensitivity and specificity were
99.5 and 99.1%, respectively.
Discussion
Identification of genome-wide high resolution DNA
copy number alterations through array-CGH has been applied to a
wide range of tumors including upper aerodigestive tract (2,13–17),
being reported commom alterations in multiple chromosome arms, such
as losses of 3p and 8p, and gains of 3q, 5p and 8q. However, the
pivotal chromosomal alterations and genes that play a central role
in upper aerodigestive tract cancer development and progression as
well as in recurrence and metastasis development are not still
fully understood. The integration of specific biological markers
with a role in the clinical management of these tumors together
with the TNM staging and histopathological grading need to be
explored to improve the diagnosis, prognosis and target therapies
design.
In the present study we observed several copy number
gains and losses in all chromosomes, which revealed the great
genomic complexity that underlies upper aerodigestive tract
carcinomas. Gains of chromosomes 3q, 7p, 8, 9q, 11q, 12p,17q and
18p and losses in chromosome 3p, 9p, 11p and Y are the most
frequently altered in our cohort (Fig.
1), where are mapped several known and novel putative oncogenes
and tumor suppressor genes that can provide a good basis for
functional studies with potential to design novel drug targets.
Losses in 3p showed to be predictive of reduced risk of
relapse/metastasis development during follow-up of our upper
aerodigestive tract cancer patients, after the treatment of primary
tumor (Figs. 2 and 3). In agreement with this finding, previous
published studies had demostrated that 3p losses are an early event
that often occur in potential malignant lesions, being included in
the Califano genetic progression model for HNSCC (18). The tumor-suppressor genes mapped at
3p responsible for head and neck oncogenesis remains unclear
(19).
Additionally, to the genomic characterization of the
upper aerodigestive tract carcinoma through array-CGH technology
highlighting specific chromosomal alterations, we also identified
several genetic and epigenetic alterations both in the tumor and
non-tumor tissue samples by MS-MLPA technique (Fig. 4). Some genetic and epigenetic
alterations were observed in both tumor and non-tumor samples,
sugesting already the presence of cells with molecular malignant
features even in tissues without phenotipic manifestation.
Considering that in the case of the studied samples, the patients
are exposed to the risk factors, namely the carcinogenic action of
tobacco smoke often in synergy with alcohol, affecting a large area
of tissue, which could be expected that genetic and epigenetic
alterations can occur in tumor and macroscopically non-tumor tissue
(7). The genetic alterations more
frequently altered in tumor samples were loss of VHL
(3p25.3), CDKN2A (9p21.3), CREM (10p11.21),
ATM (11q22.3) and CADM1 (11q23) genes and gain at
GATA5 (20q13.33), CDK6 (7q21.2), CD44 (11p13),
PTCH1 (9q22.32), RB1 (13q14.2), THBS1 (15q14)
and TSC2 (16p13.3) genes (Fig.
4). In the tumor samples, the most frequent methylated genes
were WT1, GATA5 and MSH6 genes (Fig. 4). Tumors encompass a heterogeneous
set of cells with different genetic, epigenetic and phenotypic
characteristics that can differentially lead to progression,
metastasis and drug resistance (20). Nevertheless, cancer treatment is
still carried out considering the tumors as a homogenous disease.
Nowadays, technologies for interrogating at a single level, the
whole genome, transcriptome, epigenome and proteome have suffered
great progress allowing to study the intratumor heterogeneity in
individual tumors as well as to understand the function and effect
of specific cell populations on tumorigenesis, namely, which
features could promote tumor initiation, progression or drug
resistance (20). So, the great
molecular and clinical behavior heterogeneity of upper
aerodigestive tract tumors hampers to predict the tumor progression
using only the available set of clinical markers; therefore, the
development of a prognosis predictive model is a novel and
promising strategy to increase the upper aerodigestive tract cancer
survival rate and improve the quality of life of the patients,
allowing the implementation of precision medicine. The identified
genomic and epigenetic signature was used to build a predictive
statistical model of recurrence and metastasis development that
comprises the 3p chromosomal region and WT1, VHL and
THBS1 genes. Methylation of the WT1 gene was
observed in the higher number of the patients of our cohort, either
in tumor and non-tumor samples, which are in agremment with our
previous work in oral squamous cell carcinoma (11). In the 3p chromosome, we verified that
specifically the VHL (3p25.3) gene, a tumor suppressor gene
with functions related to regulation of genes and control of cell
division (21), has a role in our
predictive model. A correlation between VHL loss and
epithelial-mesenchymal transition in oral squamous cell carcinoma
has been suggested, affecting the prognosis of the patients
(22). Additionally, gains at
THBS1 (15q14) gene were observed in our cohort. This gene is
described with a role in stimulation of cancer cell migration and
expression of matrix metalloproteases, promoting oral squamous cell
invasion (23). Altogether, these
results suggest a specific set of chromosomes and genes that seem
to have an important role in the development and prediction of
relapse/metastasis in the upper aerodigestive tract cancer. This
model can help not only in these patients management as well as in
the design of targeted therapies. So, our results may improve
selection of patients for existing therapies as well as for the
development of novel therapies. Improvements in therapies targeting
the VHL pathway, namely the VHL-HIF-VEGF axis has been translated
into development of therapies with improved clinical response
(24,25). Moreover, the upregulation of THBS1,
was associated with chemotherapy resistance in breast cancer
patients, being shown that THBS1 mediate chemoresistance through
the integrin β1/mTOR pathway, which suggest that therapies
targeting integrin β1/mTOR pathway may be a promising strategy to
overcome chemotherapy resistance (26). Additionally, WT1 protein emerges as a
promising tumor antigen for the development of universal cancer
vaccines for adjuvant treatment against residual disease and cancer
relapses (27). Thus, the
identification of prognostic biomarkers to stratify cancer patients
into distinct subgroups of clinical outcomes as well as with
predictive value for response to novel target therapies is
vital.
It is also important to stress some limitations of
this study, namely the reduced number of patients analyzed as well
as the fact that our cohort presented a relatively reduced clinical
follow-up time (range from 6 to 46 months), so, some patients with
a molecular profile similar to those with relapse/metastasis could
be incorrectly classified only because the patients were not
followed up enough time to be diagnosed with recurrence/metastasis.
In the future, this predictive model should be tested in larger
cohorts of distinct populations of upper aerodigestive tract
carcinoma. Further studies and larger follow-up times should be
performed to validate the clinical application of this model in the
management of these patients.
The clinical application of this genomic and
epigenetic predictive model is promising since it is possible to
identify newly diagnosed upper aerodigestive tract cancer patients
with risk of development of recurrence/metastasis and, in this
sense, monitor them closely, avoiding or performing early detection
of the recurrences and even provide more aggressive and
personalized treatment in order to reduce the morbidity and
mortality associated with this disease. We also highlighted in this
study some chromosomal regions and genes that can be good
candidates for targeted therapy studies.
Taking into account that upper aerodigestive tract
cancer has a poor overall survival with tendency to recur, this
predictive genomic and epigenetic model for recurrence and
metastasis development may pave the way to a more practical and
individualized patient management and targeted drug design. Further
studies in large cohorts are needed to validate the clinical
application of these potential biomarkers and molecular model in
the prediction of relapses and metastases development.
Acknowledgements
Not applicable.
Funding
Funding was received from HEALTHY AGING 2020 (grant
no. CENTRO-01-0145-FEDER-000012).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
IPR performed the aCGH experiments and wrote the
manuscript. FC performed the statistical analysis. MR performed the
MS-MLPA experiments. FM, AM and JM collected the samples and
performed the critical interpretation of the data from the clinical
point of view. IMC and JBM contributed to conception and design of
the study and critically revised the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Committee on
Ethics in Research of the Faculty of Medicine of the University of
Coimbra. All patients provided their written consent to participate
in the study after being informed about the research purposes,
following the regulations in the Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Canning M, Guo G, Yu M, Myint C, Groves
MW, Byrd JK and Cui Y: Heterogeneity of the head and neck squamous
cell carcinoma immune landscape and its impact on immunotherapy.
Front Cell Dev Biol. 7:522019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ribeiro IP, Caramelo F, Esteves L, Menoita
J, Marques F, Barroso L, Miguéis J, Melo JB and Carreira IM:
Genomic predictive model for recurrence and metastasis development
in head and neck squamous cell carcinoma patients. Sci Rep.
7:138972017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
de Visscher AV and Manni JJ: Routine
long-term follow-up in patients treated with curative intent for
squamous cell carcinoma of the larynx, pharynx, and oral cavity.
Does it make sense? Arch Otolaryngol Head Neck Surg. 120:934–939.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
de Miguel-Luken MJ, Chaves-Conde M and
Carnero A: A genetic view of laryngeal cancer heterogeneity. Cell
Cycle. 15:1202–1212. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Torrente MC, Rodrigo JP, Haigentz M Jr,
Dikkers FG, Rinaldo A, Takes RP, Olofsson J and Ferlito A: Human
papillomavirus infections in laryngeal cancer. Head Neck.
33:581–586. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thariat J, Vignot S, Lapierre A, Falk AT,
Guigay J, Van Obberghen-Schilling E and Milano G: Integrating
genomics in head and neck cancer treatment: Promises and pitfalls.
Crit Rev Oncol Hematol. 95:397–406. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kujawski M, Rydzanicz M, Sarlomo-Rikala M,
Gabriel A and Szyfter K: Chromosome alterations reflect clonal
evolution in squamous cell carcinoma of the larynx. Med Sci Monit.
8:BR279–BR282. 2002.PubMed/NCBI
|
|
8
|
American Joint Committee on Cancer
(AJCC)-TNM staging classification. 7th. Springer; New York, NY:
2010
|
|
9
|
Ribeiro IP, Marques F, Caramelo F, Ferrão
J, Prazeres H, Julião MJ, Rifi W, Savola S, de Melo JB, Baptista IP
and Carreira IM: Genetic imbalances detected by multiplex
ligation-dependent probe amplification in a cohort of patients with
oral squamous cell carcinoma-the first step towards clinical
personalized medicine. Tumour Biol. 35:4687–4695. 2014.PubMed/NCBI
|
|
10
|
Ribeiro IP, Marques F, Caramelo F, Pereira
J, Patrício M, Prazeres H, Ferrão J, Julião MJ, Castelo-Branco M,
de Melo JB, et al: Genetic gains and losses in oral squamous cell
carcinoma: Impact on clinical management. Cell Oncol (Dordr).
37:29–39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ribeiro IP, Caramelo F, Marques F,
Domingues A, Mesquita M, Barroso L, Prazeres H, Julião MJ, Baptista
IP, Ferreira A, et al: WT1, MSH6, GATA5 and PAX5 as epigenetic oral
squamous cell carcinoma biomarkers-a short report. Cell Oncol
(Dordr). 39:573–582. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ribeiro IP, Caramelo F, Esteves L,
Oliveira C, Marques F, Barroso L, Melo JB and Carreira IM: Genomic
and epigenetic signatures associated with survival rate in oral
squamous cell carcinoma patients. J Cancer. 9:1885–1895. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jarmuz-Szymczak M, Pelinska K,
Kostrzewska-Poczekaj M, Bembnista E, Giefing M, Brauze D,
Szaumkessel M, Marszalek A, Janiszewska J, Kiwerska K, et al:
Heterogeneity of 11q13 region rearrangements in laryngeal squamous
cell carcinoma analyzed by microarray platforms and fluorescence in
situ hybridization. Mol Biol Rep. 40:4161–4171. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Järvinen AK, Autio R, Haapa-Paananen S,
Wolf M, Saarela M, Grénman R, Leivo I, Kallioniemi O, Mäkitie AA
and Monni O: Identification of target genes in laryngeal squamous
cell carcinoma by high-resolution copy number and gene expression
microarray analyses. Oncogene. 25:6997–7008. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giefing M, Martin-Subero JI, Kiwerska K,
Jarmuz M, Grenman R, Siebert R and Szyfter K: Characterization of
homozygous deletions in laryngeal squamous cell carcinoma cell
lines. Cancer Genet Cytogenet. 184:38–43. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Giefing M, Zemke N, Brauze D,
Kostrzewska-Poczekaj M, Luczak M, Szaumkessel M, Pelinska K,
Kiwerska K, Tönnies H, Grenman R, et al: High resolution ArrayCGH
and expression profiling identifies PTPRD and PCDH17/PCH68 as tumor
suppressor gene candidates in laryngeal squamous cell carcinoma.
Genes Chromosomes Cancer. 50:154–166. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cancer Genome Atlas Network: Comprehensive
genomic characterization of head and neck squamous cell carcinomas.
Nature. 517:576–582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Califano J, van der Riet P, Westra W,
Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B,
Koch W and Sidransky D: Genetic progression model for head and neck
cancer: Implications for field cancerization. Cancer Res.
56:2488–2492. 1996.PubMed/NCBI
|
|
19
|
Lee DJ, Schonleben F, Banuchi VE, Qiu W,
Close LG, Assaad AM and Su GH: Multiple tumor-suppressor genes on
chromosome 3p contribute to head and neck squamous cell carcinoma
tumorigenesis. Cancer Biol Ther. 10:689–693. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lawson DA, Kessenbrock K, Davis RT,
Pervolarakis N and Werb Z: Tumour heterogeneity and metastasis at
single-cell resolution. Nat Cell Biol. 20:1349–1360. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim WY and Kaelin WG: Role of VHL gene
mutation in human cancer. J Clin Oncol. 22:4991–5004. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang S, Zhou X, Wang B, Zhang K, Liu S,
Yue K, Zhang L and Wang X: Loss of VHL expression contributes to
epithelial-mesenchymal transition in oral squamous cell carcinoma.
Oral Oncol. 50:809–817. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pal SK, Nguyen CT, Morita KI, Miki Y,
Kayamori K, Yamaguchi A and Sakamoto K: THBS1 is induced by TGFB1
in the cancer stroma and promotes invasion of oral squamous cell
carcinoma. J Oral Pathol Med. 45:730–739. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Patel PH, Chadalavada RS, Chaganti RS and
Motzer RJ: Targeting von Hippel-Lindau pathway in renal cell
carcinoma. Clin Cancer Res. 12:7215–7220. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Beroukhim R, Brunet JP, Di Napoli A, Mertz
KD, Seeley A, Pires MM, Linhart D, Worrell RA, Moch H, Rubin MA, et
al: Patterns of gene expression and copy-number alterations in
von-hippel lindau disease-associated and sporadic clear cell
carcinoma of the kidney. Cancer Res. 69:4674–4681. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang T, Srivastava S, Hartman M, Buhari
SA, Chan CW, Iau P, Khin LW, Wong A, Tan SH, Goh BC and Lee SC:
High expression of intratumoral stromal proteins is associated with
chemotherapy resistance in breast cancer. Oncotarget.
7:55155–55168. 2016.PubMed/NCBI
|
|
27
|
Van Driessche A, Berneman ZN and Van
Tendeloo VF: Active specific immunotherapy targeting the Wilms'
tumor protein 1 (WT1) for patients with hematological malignancies
and solid tumors: Lessons from early clinical trials. Oncologist.
17:250–259. 2012. View Article : Google Scholar : PubMed/NCBI
|