Introduction
Breast cancer became the primary cause of mortality
among women worldwide in 2017 (1).
According to the different subtypes of breast carcinoma, there are
four key treatments of breast cancer, including surgery,
chemotherapy, endocrine therapy and radiotherapy (2). However, prognosis remains poor due to
increasing resistance to apoptosis and recurrence (3). Therefore, novel treatments that are
less toxic and more sensitive are urgently required.
Erastin is a small molecular compound that inhibits
the solute carrier family 7 member 5 inhibiting the
cystine/glutamate antiporter (system xc-) and induces ferroptosis
by binding to voltage-dependent anion-selective channel protein
(VDAC)2 or VDAC3, causing mitochondrial oxidative injury (4). Erastin also binds to solute carrier
family 7 member 5 (SLC7A5), which interferes with cystine uptake
via the SLC3BA2/SLC7A11 complex in trans to deplete glutathione
(GSH) (5). In contrast to other
forms of cell death, ferroptosis is a unique type of programmed
cell death that has two major characteristics; lipid peroxide
accumulation and iron dependency (6). Previous studies have demonstrated that
ferroptosis is often accompanied by autophagy and can be inhibited
by autophagy inhibitors (7–9). As an inducer of ferroptosis, erastin
has been shown to induce ferroptosis in oncogenic RAS mutation cell
lines and in other cancer cells, including liver cancer (10), acute lymphoblastic leukemia (11) and rhabdomyosarcoma (12). Although erastin activates ferroptosis
in triple-negative breast cancer cells by suppressing the
expression of glutathione peroxidase 4 and upregulating the
expression of cysteine dioxygenase (13), understanding is limited regarding the
effect of erastin treatment or the mechanism of erastin in other
types of breast cancer cells.
Ferroptosis is considered to be a type of reactive
oxygen species (ROS)-dependent regulated necrosis that is
accompanied by intracellular iron accumulation (14). Iron exists as Fe2+ and
Fe3+ in cells. Free Fe2+ catalyzes the
formation of hydroxyl radicals and hydroxide from hydrogen
peroxide, which is termed the Fenton reaction (15). During the catalytic cycle of the
Fenton reaction, Fe3+ can be recycled to reproduce
Fe2+ via superoxide radicals. Additionally,
Fe2+ catalyzes the lipid peroxidation of unsaturated
fatty acids (15). When ROS levels
exceed the antioxidant capacity of cells, it causes oxidative
stress, which triggers oxidative damage to proteins, DNA and lipids
(15). Previous studies have
reported that autophagy accelerates ferroptosis by: i) generating
lysosomal ROS (16); ii)
accumulating labile iron via nuclear receptor coactivator
4-mediated ferritinophagy (17);
iii) promoting lipid peroxidation via Ras-related protein
Rab-7-mediated lipophagy (18); iv)
promoting GSH depletion via beclin1-mediated system xc-inhibition
(19); v) promoting lysosomal cell
death via signal transducer and activator of transcription
3-mediated cathepsin B release (20); and iv) contributing to
chaperone-mediated autophagy glutathione peroxidase 4 degradation
via heat shock protein 90-mediated lysosome-associated membrane
protein 2A stability (21). Although
numerous studies have investigated the mechanism of erastin in
ferroptosis-associated pathways, to the best of our knowledge, the
relationship between intracellular iron levels and erastin-induced
autophagy remains unclear in breast cancer cells.
The present study therefore investigated the changes
of iron levels in erastin-induced autophagy and aimed to elucidate
its underlying mechanism using human MCF-7 and MDA-MB-231 cell
lines.
Materials and methods
Reagents
Erastin, 3-methyladenine (3-MA) and bafilomycin A1
(Baf-A1) were purchased from Selleck Chemicals. Anti-autophagy
related (ATG)12 (cat. no. 4180) was purchased from Cell Signaling
Technology, Inc. Deferoxamine (DFO), anti-p62 (cat. no. ab56416)
and anti-ATG5 (cat. no. ab221604) were purchased from Abcam.
Anti-beclin 1 (cat. no. B6061) and anti-microtubule-associated
proteins 1A/1B light chain 3B (LC3B) (cat. no. l7543) were obtained
from Sigma-Aldrich; Merck KGaA. Anti-β-actin (cat. no. AF5001), and
horseradish peroxidase-conjugated anti-mouse IgG (cat. no. A0216)
and anti-rabbit IgG (cat. no. A0208) were purchased from Beyotime
Institute of Biotechnology.
Cell lines and culture
Human MCF-7 and MDA-MB-231 cell lines were purchased
from the Shanghai Institute of Cell Biology at the Chinese Academy
of Sciences. All cells were cultured in DMEM (SH 30243.01; Hyclone;
Cyvita) supplemented with 10% heat-inactivated fetal bovine serum
(FB15015; http://zn.clarkbio.com/Clark), 2 mmol/l glutamine,
penicillin (100 U/ml) and streptomycin (100 µg/ml). Cells treated
with 5 mmol/l 3-MA, 1.5 µmol/l Baf-A1 or 500 µmol/l DFO for 1 h
prior to erastin treatment which lasted for 24 h with different
concentrations (MCF-7 cells were treated with 40 and 80 µmol/l;
MDA-MB-231 cells were treated with 20 and 40 µmol/l) at 37°C and 5%
CO2 in a humid environment and used for experimentation
when entering the mid-log phase. A light microscope was used at
magnification, ×400 to observe cellular changes.
Cell viability assay
MCF-7 (6×103 cells/well) and MDA-MB-231
(8×103 cells/well) breast cancer cells were seeded into
96-well microplates, cultured at 37°C for 24 h and treated with 5
mmol/l 3-MA, 1.5 µmol/l Baf-A1 or 500 µmol/l DFO for 1 h prior to
erastin treatment which lasted for 24 h with two values separately
(MCF-7 cells were treated with 40 and 80 µmol/l; MDA-MB-231 cells
were treated with 20 and 40 µmol/l) at 37°C and 5% CO2
in a humid environment. Cellular viability was assessed by
performing an MTT assay (DMSO was used to dissolve the purple
formazan), the results of which were expressed as a ratio to the
absorbance of control cells at 490 nm. Absorbances were measured at
490 nm using an automatic multi-well spectrophotometer (Bio-Rad
Laboratories, Inc.). The IC50 values were calculated
using GraphPad Prism 6 software (GraphPad Software, Inc.), and the
mean IC50 value of four experiments was presented.
Lactate dehydrogenase (LDH) release
cell death assay
The LDH cytotoxicity assay kit (cat. no. C0017;
Beyotime Institute of Biotechnology) was used to determine the cell
death rate. According to the manufacturer's protocol, the
absorbance of each sample was measured at 490 nm using an automatic
multi-well spectrophotometer. The cell death ratio was calculated
using the following formula: Cell death ratio (%)=
(Asample-Acontrol/Amax-Acontrol)
×100%. Where Asample is the sample absorbance value,
Acontrol is the absorbance value of the control group
and Amax is the absorbance value of the positive
group.
Iron assay
Intracellular Fe2+ levels were determined
using an iron colorimetric assay kit (cat. no. K390) purchased from
BioVision, Inc. According to the manufacturer's instructions, MCF-7
and MDA-MB-231 breast cancer cells were collected and separated
into untreated, erastin-treated and DFO-pretreated groups before
the cells were added to iron assay buffer, homogenized on ice and
centrifuged at 16,000 × g for 10 min at 4°C to obtain the
supernatant. Samples (50 µl/well) were then incubated with 50 µl
assay buffer in a 96-well microplate for 30 min at 25°C. Samples
were subsequently incubated with 100 µl iron probe in the dark for
60 min at 25°C and assessed with a microplate reader at a
wavelength of 593 nm. Absorbance values were calibrated to a
standard concentration curve to calculate the concentration of
iron. The results are expressed as a ratio to the concentration of
control cells.
Transfection of small interfering RNA
(siRNA)
Transfection with ATG5 siRNA (5 µg/µl) was performed
using Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. ATG5
siRNA (5′-GACGUUGGUAACUGACAAATT-3′) and scrambled siRNA
(5′-UUCUCCGAACGUGUCACGUTT-3′) were purchased from Shanghai
GenePharma Co., Ltd. MCF-7 and MDA-MB-231 cells were separately
seeded into a 6-well plate at 6×104 and 8×104
cells per well overnight. The following day, cells were transfected
with ATG5-specific siRNAs and scrambled siRNAs for 48 h. The
efficiency of transfection was determined by western blotting.
Gel electrophoresis and western
blotting
MCF-7 and MDA-MB-231 breast cancer cells treated
with eastin alone, 5 mmol/l 3-MA, 1.5 µmol/l Baf-A1, ATG5 siRNA or
500 µmol/l DFO for 1 h prior to erastin were collected and
homogenized as described previously (22). Homogenates were centrifuged at 1,000
× g for 10 min at 4°C to obtain the supernatant, and the protein
content of the supernatant was determined using BCA Protein assay
kit (Beyotime Institute of Biotechnology). Equal quantities of
protein including anti-ATG5 (55 kd), anti-ATG12 (55 kd), anti-P62
(62 kd), anti-Beclin1 (69 kd) and anti-LC3B (14 and 16 kd) were
electrophoresed on 8-12% sodium dodecyl sulfate-polyacrylamide gels
based on the molecular weight of the target protein and transferred
to PVDF membranes (EMD Millipore). The membranes were then blocked
with 5% skimmed milk in PBS for 2 h at room temperature and
incubated overnight at 4°C with primary antibodies, including
anti-ATG5 (1:1,000), anti-ATG12 (1:1,000), anti-P62 (1:1,000),
anti-Beclin-1 (1:1,000) and anti-LC3B (1:1,000). The membranes were
washed with PBS-0.1% Tween-20 (PBS-T) buffer for 30 min at 25°C
prior to incubation with horseradish peroxidase-conjugated goat
anti-rabbit IgG (1:1,000) or anti-mouse IgG (1:1,000) at 25°C for 2
h. Membranes were subsequently washed with PBS-T buffer and
immunoreactive proteins were visualized on a chemi-luminescence
developer (Chemiscope 5300; Clinx Science Instruments, Co., Ltd.)
with an enhanced chemiluminescence reagent (P10300; NCM Biotech).
Densitometry was performed using ImageJ software v1.46 (National
Institutes of Health).
Statistical analysis
All data was obtained from at least four independent
experiments and are expressed as the mean ± standard deviation.
Statistical analyses were performed with Microsoft Excel 2010
(Microsoft Corporation) and GraphPad Prism 6 software (GraphPad
Software, Inc.). Statistical comparisons were made using one-way
ANOVA with Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Erastin inhibits viability and induces
breast cancer cell death
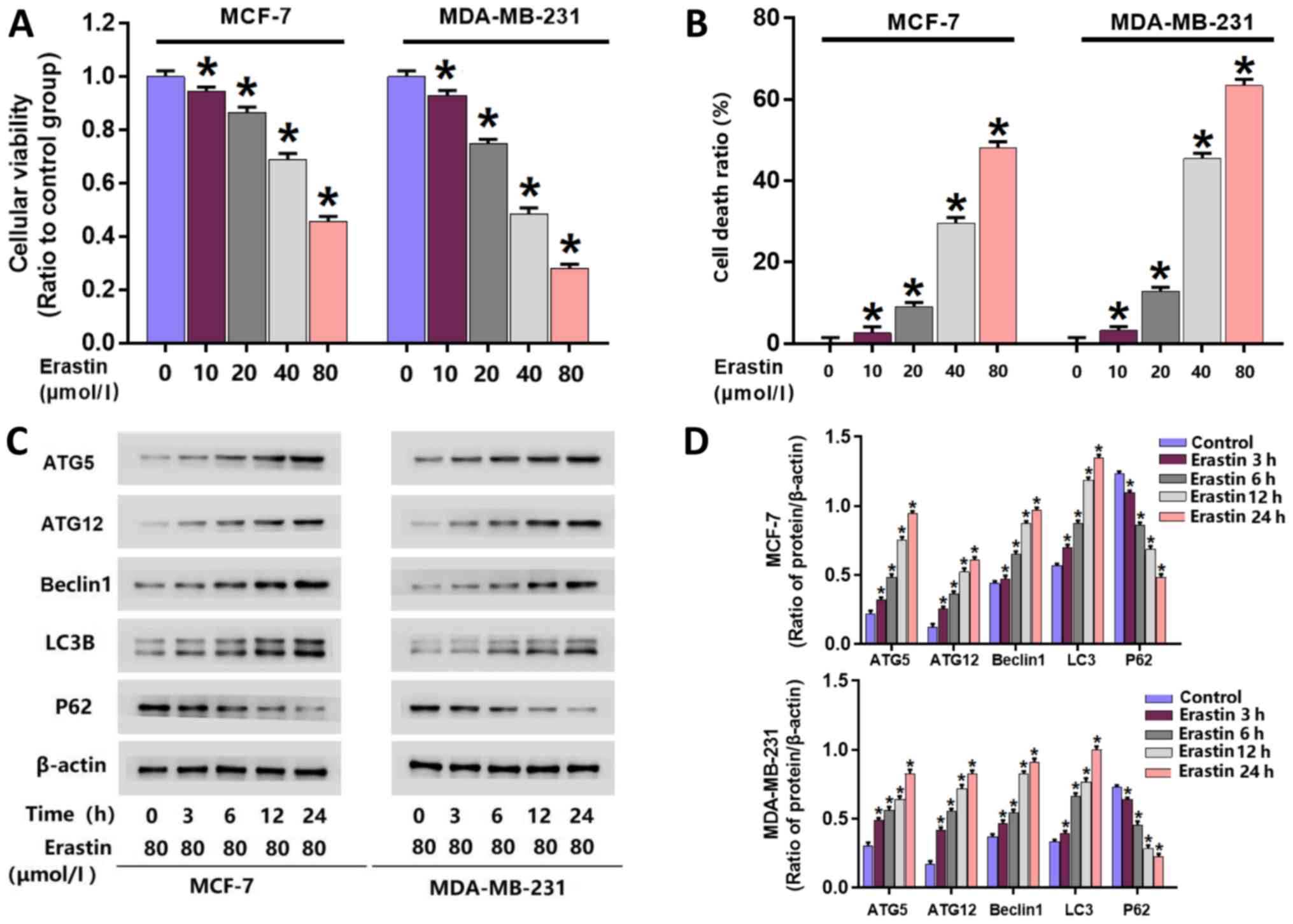
To investigate the toxic effect of erastin on breast
cancer cells, an MTT assay was performed to examine the viability
of erastin-treated MCF-7 and MDA-MB-231 cells. Following treatment
with 10, 20, 40 and 80 µmol/l erastin for 24 h, cell viability was
significantly reduced in MCF-7 and MDA-MB-231 cells compared with
untreated cells (Fig. 1A). The
IC50 values of erastin in MDA-MB-231 and MCF-7 cells at
24 h were 40 and 80 µmol/l, respectively. Therefore, erastin at
these concentrations was used for subsequent experiments.
A LDH release assay was performed to examine
erastin-induced death in MDA-MB-231 and MCF-7 cells. MDA-MB-231 and
MCF-7 cells were treated with erastin at their respective
IC50 concentrations for 3, 6, 12 and 24 h. The results
at 24 h demonstrated significant increases in cellular death in
each cell line following treatment with 10, 20, 40 and 80 µmol/l
erastin compared with untreated cells (Fig. 1B). These data indicated that erastin
inhibited the viability of breast cancer cells and triggered breast
cancer cell death
Erastin upregulates the expression of
autophagy-associated proteins
To further investigate whether erastin activates
autophagy in breast cancer cells, western blotting was performed to
analyze the expression of autophagy-associated proteins. Following
erastin treatment, the expression of autophagy-associated proteins
in breast cancer cells, including beclin-1, ATG5, ATG12 and LC3B,
were all significantly increased. Additionally, p62 levels, as a
substrate of autophagy, were significantly downregulated in
erastin-treated cells (Fig. 1C and
D).
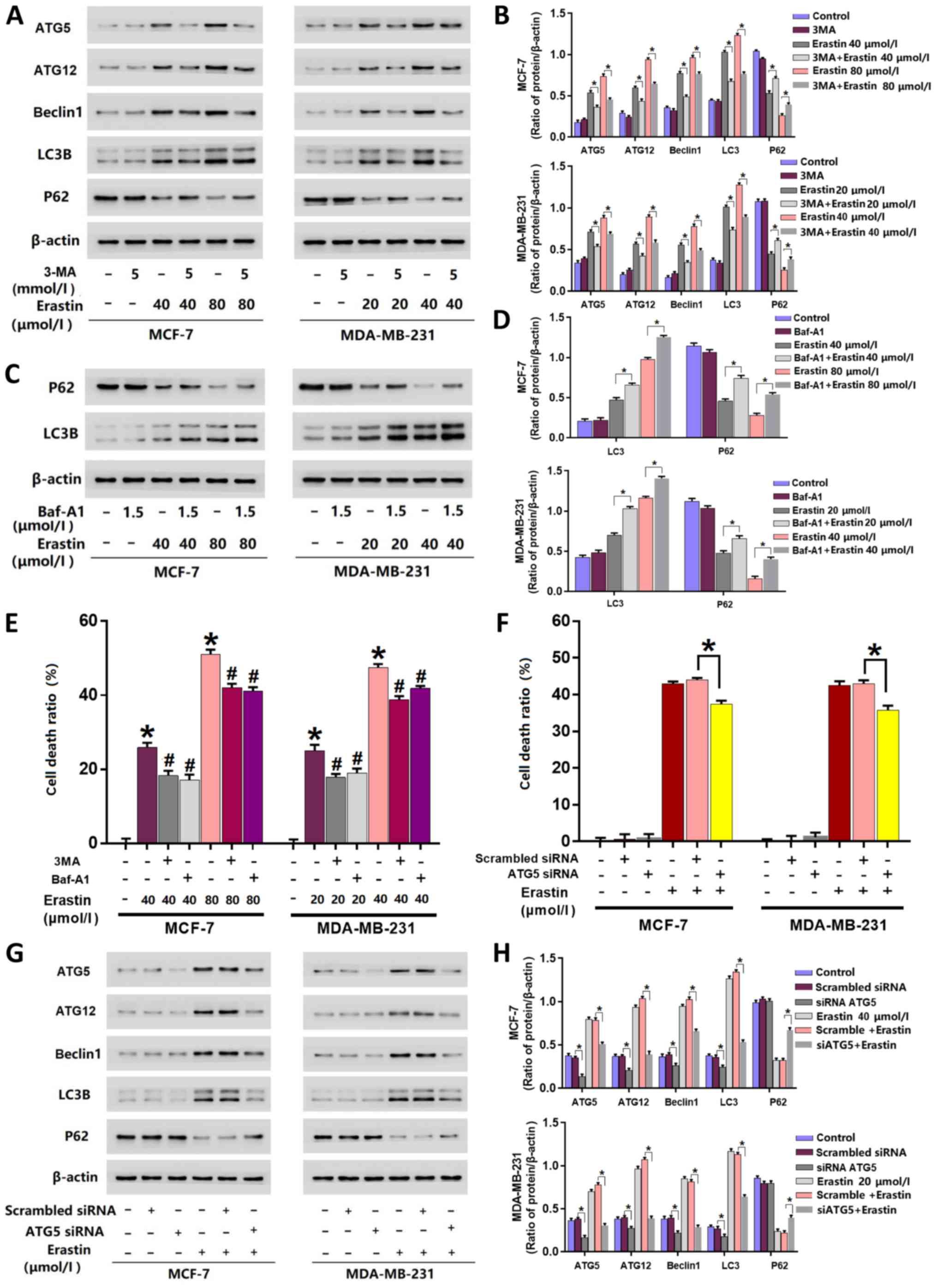
Inhibition of autophagy prevents
erastin-induced death in breast cancer cells
3MA and Baf-A1 are often used to inhibit autophagy
activation (5). 3MA inhibits
autophagy initiation (23), and
Baf-A1 disturbs the fusion of autophagosomes with lysosomes
(24). The inhibitory effects of 3MA
or Baf-A1 on autophagy activation are also decided by their
concentrations and treatment times. Higher concentrations of 3MA or
Baf-A1 and longer incubation times lead to obvious changes in the
baseline level of autophagy-related proteins (23,24). In
the present study, the cells were incubated with 5 mmol/l 3MA and
1.5 µmol/l Baf-A1 for 25 h, which did not exhibit marked inhibition
on the baseline levels of autophagy-related proteins (Fig. 2A-D). To further elucidate erastin
activated autophagy, cells were treated with 5 mmol/l 3-MA for 1 h
prior to erastin treatment. Cells were additionally pre-treated
with 1.5 µmol/l Baf-A1, an inhibitor of autophagy, for 1 h prior to
erastin treatment. The results of the LDH release assay revealed
that cells pre-treated with 3-MA or Baf-A1 were significantly more
resistant to erastin-induced death compared with cells only treated
with erastin (Fig. 2E).
Inhibition of autophagy inhibits the
erastin-induced expressional upregulation of autophagy-associated
proteins
As aforementioned, the results of the current study
demonstrated that autophagy-associated proteins (beclin1, ATG5,
ATG12, LC3B and P62) were affected by erastin. However, 3-MA
treatment prevented cell death and inhibited the upregulation of
autophagy-associated proteins induced by erastin. Western blotting
revealed that the expression levels of beclin1, ATG5, ATG12 and
LC3B were significantly downregulated, while P62 was significantly
upregulated when cells were pre-treated with 3-MA compared with
those only treated with erastin (Fig. 2A
and B). Baf-A1 treatment demonstrated similar effects to that
of 3-MA at a concentration of 1.5 µmol/l on the expression of P62.
However, in contrast to 3-MA, the main effect of Baf-A1 is to
inhibit the fusion of autophagosome and autolysosome (24), which resulted in a significant
increase of LC3B in cells pre-treated with Baf-A1 compared with
cells only treated with erastin (Fig. 2C
and D). Therefore, it was unnecessary in the present study to
detect the expression of ATG5, ATG12 and Beclin-1.
Knockdown of ATG5 with siRNA prevents
erastin-induced breast cancer cell death
To further investigate erastin induced autophagic
death in breast cancer cells, ATG5 was knocked-down with siRNA. The
results demonstrated that ATG5-knockdown significantly decreased
the cell death ratio of erastin-treated cells (Fig. 2F) and significantly inhibited the
erastin-induced changes of autophagy-related proteins (Fig. 2G and H).
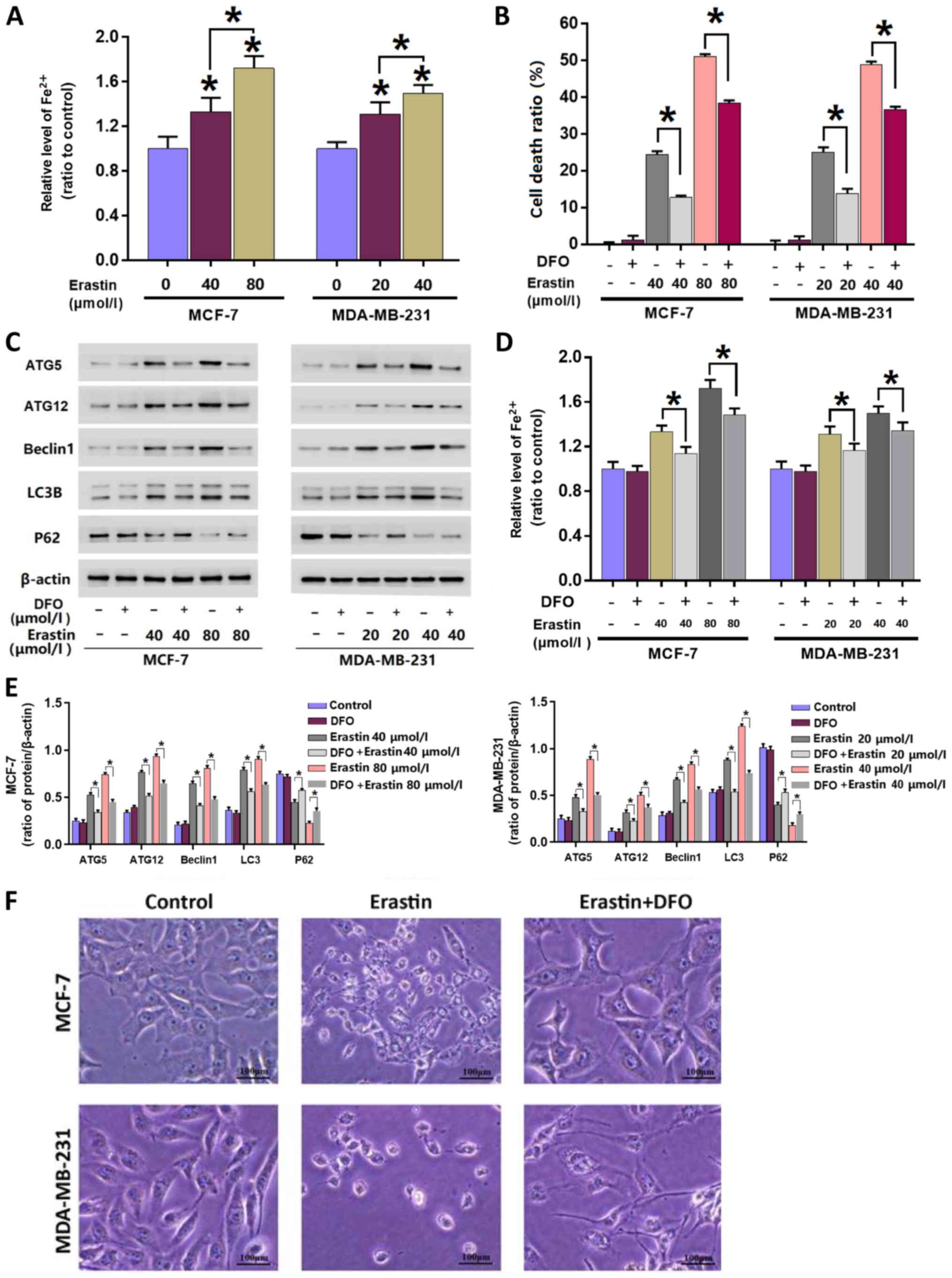
Mitigation of erastin-induced iron
level increases inhibits erastin-induced autophagy
To investigate whether the levels of intracellular
iron are intrinsically associated with erastin-induced autophagy,
an iron assay was performed to assess intracellular iron
accumulation after treatment with erastin. The results revealed
that when compared with the control group, iron levels
significantly increased in cells incubated with erastin for 24 h.
Furthermore, when the concentration of erastin was increased from
40 to 80 µmol/l, iron levels significantly increased further. The
data indicated that erastin-induced an increase in intracellular
irons in each breast cancer cell line in a concentration- and
incubation time-dependent manner (Fig.
3A).
To elucidate the importance of iron accumulation,
cells were treated with iron chelator DFO at 500 µmol/l for 1 h and
subsequently incubated with erastin for 24 h. Similarly with 3MA
and Baf-A1, the dose of DFO used in the current study was 500
µmol/l, which did not show any inhibitory effect on cellular
viability or the baseline level of autophagy-related proteins
(Fig. 3B-E). The results
demonstrated that DFO significantly inhibited the erastin-induced
increase of intracellular Fe2+ iron in MDA-MB-231 and
MCF-7 cells (Fig. 3D). Additionally,
the LDH release assay demonstrated that erastin-induced breast
cancer cell death was significantly inhibited in the presence of
DFO (Fig. 3B). Consistently,
observation under a light microscope demonstrated that DFO
pre-treatment markedly reversed the erastin-induced cellular
changes (Fig. 3F).
To determine the relationship between autophagy and
erastin-induced increases of intracellular iron, western blotting
was performed to assess the changes of autophagy-associated
proteins in each breast cancer cell line following pre-treatment
with DFO. The results demonstrated that DFO significantly inhibited
the erastin-induced changes in the autophagy-associated protein
expression levels (Fig. 3C and E).
These data suggested that DFO treatment inhibited erastin-induced
autophagy by mitigating erastin-induced iron levels.
Discussion
The results of the current study demonstrated that
erastin inhibited the viability of breast cancer cells, triggered
breast cancer cell death and increased intracellular iron levels in
a time or dose-dependent manner. In erastin-treated cells, the
protein expression of autophagy-associated beclin1, ATG5, ATG12 and
LC3B was increased, while levels of P62 were decreased. However,
these effects on LC3B and P62 could be inhibited by 3-MA
pretreatment and ATG5-knockdown via siRNA. The mitigation of
erastin-induced iron levels via DFO pre-treatment inhibited
erastin-induced autophagy-associated protein alterations and
mitigated cell death. Taken together, the results indicated that
erastin induces autophagic breast cancer cell death by improving
intracellular iron levels.
The relationship between iron and breast cancer is
complex. Huang et al (25)
hypothesized that iron deficiency contributes to the high
recurrence of breast cancer in premenopausal women via increased
serum vascular endothelial growth factor concentrations.
Additionally, it was predicted that iron load may serve a role in
the incidence of breast cancer in postmenopausal women (25). The present data demonstrated that
iron overload induced by erastin exhibited a good antitumor effect
via inducing autophagic death in breast cancer cells. Breast cancer
develops in women as a result of multiple factors, which include
diet, socioeconomic status and genetic mutations (25). Therefore, the relationship between
iron and breast cancer requires further investigation. Ferroptosis
is a novel type of regulated cell death that is accompanied by
intracellular iron accumulation (14). Activating ferroptosis to eliminate
breast cancer cells has emerged as a potential therapeutic
approach. Erastin is an inducer of ferroptosis that has been
reported to efficiently induce death in various types of cancer
cells, such as liver cancer (10)
and rhabdomyosarcoma cells (12).
Consistently, the present study demonstrated that erastin inhibited
the viability of breast cancer cells and induced cell death in
MCF-7 and MDA-MB-231 cells in a dose-dependent manner.
A previous study has reported that erastin induces
ferroptosis in triple-negative MDA-MB-231 breast cancer cells
(13). The current study
demonstrated that erastin activated autophagy in triple-negative
MDA-MB-231 and estrogen receptor-positive MCF-7 breast cancer cell
lines. Beclin1, ATG5, ATG12, LC3B and p62 are key autophagic marker
proteins whose levels reflect the occurrence of autophagy (8). In the present study, western blotting
revealed that protein expression levels of Beclin1, ATG5, ATG12 and
LC3B were increased, while p62 levels were decreased in a
time-dependent manner following erastin treatment.
Autophagy-inhibition using 3-MA, Baf-A1 and ATG5-knockdown via
siRNA inhibited the erastin-induced effects on autophagy-related
proteins and cell death.
Ferroptosis is a type of programmed cell death
induced by iron-dependent lipid peroxidation (26). Iron, as a promoter of cell growth and
proliferation, fulfils an important role in human diseases, such as
Parkinson's (27). An abnormal
increase of intracellular iron in the absence of erastin has also
been reported to induce cell death (3). Fang et al (28) used ferric ammonium citrate (FAC) and
a membrane-permeable ferric 8-hydroxyquinoline complex (Fe-8HQ),
which are two types of iron agents, to improve intracellular iron
levels. It was identified that either FAC or Fe-8HQ induced death
in various types of cells in a dose-dependent manner. Another study
reported by Nakamura et al (29) demonstrated that iron overload could
improve intracellular ROS levels, which then contribute to cell
death via activating the MAPK signaling pathway. The Fenton
reaction, which recycles Fe2+, is a key step in
ferroptosis that produces ROS, contributing to cell death (15). Therefore, improving iron levels may
suppress tumor growth and enhance the anticancer activity of
ferroptosis inducers (27). However,
the mechanism of erastin-induced cell death in breast carcinoma
remains unclear. In the present study, the iron assay revealed that
intracellular iron accumulates in breast cancer cells following
treatment with erastin.
Erastin induces breast cancer cell death and
increases intracellular iron levels (6). However, to the best of our knowledge,
the relationship between erastin-induced autophagic cell death and
iron levels in breast carcinoma is unknown. Previously, autophagy
has been demonstrated to contribute to ferroptosis via increasing
intracellular iron level by degradation of nuclear receptor
coactivator 4, which is an endogenous inhibitor of ferritin
(7). However, it remains unknown
whether iron plays a role in autophagy activation. Thus, the
present study used breast cancer cells and investigated the role of
iron in erastin-induced lethal autophagy. The current study
therefore pre-treated cells with an iron chelator (DFO) prior to
erastin treatment. As a result, the accumulation of intracellular
iron was inhibited and cell death was decreased. The results
further revealed that DFO inhibited the expression of
autophagy-related proteins affected by erastin. The results
demonstrated that the mitigation of erastin-induced irons levels
inhibited erastin-induced autophagy. The current results indicated
that abnormal improvement of intracellular iron could activate
autophagic cell death in breast cancer cells.
Despite not elucidating the precise mechanism by
which iron levels inhibit erastin-induced autophagy, previous
studies have provided results supporting those of the current
study. Numerous studies have demonstrated that iron excess and
oxidative stress may lead to ROS accumulation, which damages cells
and activates autophagy (21,30,31).
Excessive autophagy contributes to ferroptosis by regulating
cellular iron equilibria (30). A
previous study reported that erastin induces autophagy by depleting
GSH, which increases lipid ROS generation (32,33).
Therefore, iron improvement could activate autophagy via
ROS-related pathways, such as AMPK/mTOR (29). Mitochondria are regarded as the
location where ROS, as a crucial messenger, are produced via
electron transmission chain (34).
Furthermore, some pro-death factors, such as cytochrome C, nuclease
endo G and apoptosis inducing factor, are also located within
mitochondria, and can induce cell death after being released when
mitochondria are impaired (34).
Although mitochondria are also the location of energy generation;
glycolysis is the primary pathway of ATP generation in cancer cells
(34). However, whether the
autophagy activated by erastin could inhibit the function of
mitochondria remains to be investigated further. In the present
study, DFO inhibited erastin-induced autophagy and improved
intracellular iron levels. Thus, it was concluded that ROS serves
an important role in the relationship between iron level
improvement and erastin-induced autophagy.
In conclusion and to the best of our knowledge, the
current study demonstrated for the first time that erastin triggers
autophagy in breast cancer cells by improving intracellular iron
levels.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
MXL, PFG and DS designed the research and analyzed
the data. MXL, XZW and SL performed the experiments. CH, CCW, LW
and XYW were responsible for acquisition of data, analysis and
interpretation of data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Choi J, Gyamfi J, Jang H and Koo JS: The
role of tumor-associated macrophage in breast cancer biology.
Histol Histopathol. 33:133–145. 2018.PubMed/NCBI
|
|
2
|
Fisusi FA and Akala EO: Drug combinations
in breast cancer therapy. Pharm Nanotechnol. 7:3–23. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nedeljković M and Damjanović A: Mechanisms
of chemotherapy resistance in triple-negative breast cancer-how we
can rise to the challenge. Cells. 8:9572019. View Article : Google Scholar
|
|
4
|
Maldonado EN, Sheldon KL, DeHart DN,
Patnaik J, Manevich Y, Townsend DM, Bezrukov SM, Rostovtseva TK and
Lemasters JJ: Voltage-Dependent anion channels modulate
mitochondrial metabolism in cancer cells: Regulation by free
tubulin and erastin. J Biol Chem. 288:11920–11929. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W and
Wang J: Molecular mechanisms of ferroptosis and its role in cancer
therapy. J Cell Mol Med. 23:4900–4912. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dixon SJ, Lemberg KN, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao M, Monian P, Pan Q, Zhang W, Xiang J
and Jiang X: Ferroptosis is an autophagic cell death process. Cell
Res. 26:1021–1032. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou B, Liu J, Kang R, Klionsky DJ,
Kroemer G and Tang D: Ferroptosis is a type of autophagy-dependent
cell death. Semin Cancer Biol. 14 (Suppl):S1044–S1579. 2019.
|
|
9
|
Kang R and Tang D: Autophagy and
ferroptosis-What's the connection? Curr Pathobiol Rep. 5:153–159.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Y, Zhu G, Liu Y, Wu Q, Zhang X, Bian
Z, Zhang Y, Pan Q and Sun F: O-GlcNAcylated c-jun antagonizes
ferroptosis via inhibiting GSH synthesis in liver cancer. Cell
Signal. 63:1093842019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dächert J, Schoeneberger H, Rohde K and
Fulda S: RSL3 and erastin differentially regulate redox signaling
to promote smac mimetic-induced cell death. Oncotarget.
7:63779–63792. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dächert J, Ehrenfeld V, Habermann K,
Dolgikh N and Fulda S: Targeting ferroptosis in rhabdomyosarcoma
cells. Int J Cancer. 15:510–520. 2019.
|
|
13
|
Yu M, Gai C, Li Z, Ding D, Zheng J, Zhang
W, Lv S and Li W: Targeted exosomes-encapsulated erastin induced
ferroptosis in the triple negative breast cancer cells. Cancer Sci.
110:3173–3182. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vanden Berghe T, Linkermann A,
Jouan-Lanhouet S, Walczak H and Vandenabeele P: Regulated necrosis:
The expanding network of non-apoptotic cell death pathways. Nat Rev
Mol Cell Biol. 15:135–147. 2014. View
Article : Google Scholar
|
|
15
|
Doll S and Conrad M: Iron and ferroptosis:
A still ill-defined liaison. IUBMB Life. 69:423–434. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
JZ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schroeder B, Schulze RJ, Weller SG,
Sletten AC, Casey CA and McNiven MA: The small GTPase Rab7 as a
central regulator of hepatocellular lipophagy. Hepatology.
61:1896–1907. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu
J, Xie Y, Liu J, Klionsky DJ, Kroemer G, et al: AMPK-Mediated BECN1
phosphorylation promotes ferroptosis by directly blocking system
Xc(−) activity. Curr Biol. 28:2388–2399. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brown CW, Amante JJ, Goel HL and Mercurio
AM: The α6β4 integrin promotes resistance to ferroptosis. J Cell
Biol. 216:4287–4297. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M,
Shan B, Pan H and Yuan J: Chaperone-Mediated autophagy is involved
in the execution of ferroptosis. Proc Natl Acad Sci USA.
116:2996–3005. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou Z, Lu B, Wang C, Wang Z, Luo T, Piao
M, Meng F, Chi G, Luo Y and Ge P: RIP1 and RIP3 contribute to
shikonin-induced DNA double-strand breaks in glioma cells via
increase of intracellular reactive oxygen species. Cancer Lett.
390:77–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pan H, Wang Y, Na K, Wang Y, Wang L, Li Z,
Guo C, Guo D and Wang X: Autophagic flux disruption contributes to
ganoderma lucidum polysaccharide-induced apoptosis in human
colorectal cancer cells via MAPK/ERK activation. Cell Death Dis.
10:4562019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Choi J, Jo M, Lee E, Oh YK and Choi D: The
role of autophagy in human endometrium. Biol Reprod. 86:702012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang X: Does iron have a role in breast
cancer? Lancet Oncol. 9:803–807. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Z, Ding Y, Wang X, Lu S, Wang C, He
C, Wang L, Piao M, Chi G, Luo Y and Ge P: Pseudolaric acid B
triggers ferroptosis in glioma cells via activation of Nox4 and
inhibition of Xct. Cancer Lett. 428:21–33. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang S, Luo J, Zhang Z, Dong D, Shen Y,
Fang Y, Hu L, Liu M, Dai C, Peng S, et al: Iron and magnetic: New
research direction of the ferroptosis-based cancer therapy. Am J
Cancer Res. 8:1933–1946. 2018.PubMed/NCBI
|
|
28
|
Fang S, Yu X, Ding H, Han J and Feng J:
Effects of intracellular iron overload on cell death and
identification of potent cell death inhibitors. Biochem Biophys Res
Commun. 503:297–303. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakamura T, Naguro I and Ichijo H: Iron
homeostasis and iron-regulated ROS in cell death, senescence and
human diseases. Biochim Biophys Acta Gen Subj. 1863:1398–1409.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Torii S, Shintoku R, Kubota C, Yaegashi M,
Torii R, Sasaki M, Suzuki T, Mori M, Yoshimoto Y, Takeuchi T and
Yamada K: An essential role for functional lysosomes in ferroptosis
of cancer cells. Biochem J. 473:769–777. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Byun YJ, Kim SK, Kim YM, Chae GT, Jeong SW
and Lee SB: Hydrogen peroxide induces autophagic cell death in C6
glioma cells via BNIP3-mediated suppression of the mTOR pathway.
Neurosci Lett. 18:131–135. 2009. View Article : Google Scholar
|
|
32
|
Sun Y, Zheng Y, Wang C and Liu Y:
Glutathione depletion induces ferroptosis, autophagy, and premature
cell senescence in retinal pigment epithelial cells. Cell Death
Dis. 9:7532018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mancilla H, Maldonado R, Cereceda K,
Villarroel-Espindola F, Montes de Oca M, Angulo C, Castro MA, Slebe
JC, Vera JC, Lavandero S and Concha II: Glutathione depletion
induces spermatogonial cell autophagy. J Cell Biochem.
116:2283–2292. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang Y, Karakhanova S, Hartwig W, D'Haese
JG, Philippov PP, Werner J and Bazhin AV: Mitochondria and
mitochondrial ROS in cancer: Novel targets for anticancer therapy.
J Cell Physiol. 231:2570–2581. 2016. View Article : Google Scholar : PubMed/NCBI
|